

Abstract
Purpose
Primary central nervous system lymphoma (PCNSL) is an aggressive non-Hodgkin lymphoma confined to the CNS. Whether there is a PCNSL-specific genomic signature and, if so, how it differs from systemic diffuse large B-cell lymphoma (DLBCL) is uncertain.
Experimental design
We performed a comprehensive genomic study of tumor samples from 19 immunocompetent PCNSL patients. Testing comprised array-comparative genomic hybridization and whole exome sequencing.
Results
Biallelic inactivation of TOX and PRKCD were recurrently found in PCNSL but not in systemic DLBCL, suggesting a specific role in PCNSL pathogenesis. Additionally, we found a high prevalence of MYD88 mutations (79%) and CDKN2A biallelic loss (60%). Several genes recurrently affected in PCNSL were common with systemic DLBCL, including loss of TNFAIP3, PRDM1, GNA13, TMEM30A, TBL1XR1, B2M, CD58, activating mutations of CD79B, CARD11 and translocations IgH-BCL6. Overall, BCR/TLR/NF-κB pathways were altered in >90% of PNCSL, highlighting its value for targeted therapeutic approaches. Furthermore, integrated analysis showed enrichment of pathways associated with immune response, proliferation, apoptosis, and lymphocyte differentiation.
Conclusions
In summary, genome-wide analysis uncovered novel recurrent alterations, including TOX and PRKCD, helping to differentiate PCNSL from systemic DLBCL and related lymphomas.
Keywords: PCNSL, array-based comparative genomic hybridization, whole exome sequencing, TOX, PRKCD, NF-kB signaling pathway
Introduction
Primary central nervous system lymphoma (PCNSL) is an aggressive variant of non-Hodgkin lymphoma confined to the central nervous system. Most PCNSL (90%) are part of the immune-privileged site-associated diffuse large B-cell lymphomas (IPDLBCLs). IPDLBCL consist of late–germinal center or post–germinal center lymphoid cells that show very distinct characteristics that separate them from nodal DLBCLs. IPDLBCL are now recognized as a distinct entity in the World Health Organization lymphoma classification(1).
Unlike nodal DLBCL, only a limited number of genetic studies have been performed in PCNSL, partly due to the lack of available tissue specimens since most cases are diagnosed by stereotactic biopsy. In addition to the low amount of available tissue for genomic analysis, the source of biological samples is often limited to formalin-fixed and paraffin-embedded (FFPE) specimens. In recent years, an improved understanding of PCNSL biology has emerged, largely from the use of modern high-throughput molecular techniques in FFPE specimens(2–5).
Several recurrent chromosomal abnormalities and mutations have been identified, such as mutations in PRDM1(6) and CARD11(7), translocations of IgH-BCL6(8), deletions of 6p21, 6q, 8q12, 9p21 and gains of 7q, 11q and chromosome 12(3, 4, 9, 10). Recently, the first whole exome sequencing (WES) study was reported on 4 PCNSL patients, showing recurrent mutations in MYD88, PIM1 and TBL1XR1(5). Gene expression profiling studies suggested the existence of three gene “signatures” associated with PCNSL that resemble the classification proposed in systemic DLBCL(11). However, unique molecular features have been found in PCNSL suggesting implication of the brain microenvironment in neoplastic proliferation and CNS tropism for tumoral B cells(12, 13).
Nevertheless, because of the paucity of genomic data little is known of how PCNSL differs from systemic DLBCL with respect to its molecular features and pathogenesis. To determine if there is a specific CNS signature we report a comprehensive genomic study in tumor samples from 19 PCNSL patients that combined array-based comparative genomic hybridization (aCGH), whole exome sequencing, mate-pair whole genome sequencing, targeted sequencing and fluorescence in situ hybridization (FISH). Using this combinatorial approach we found a complex karyotype and uncovering novel recurrent alterations, including loss/deletion of TOX and PRKCD, helping to differentiate PCNSL from systemic DLBCL and related lymphomas.
Patients and Methods
Tumor samples
Samples from 19 immunocompetent (HIV- and EBV-) PCNSL were retrieved from the Mayo Clinic Tumor Registry (IRB approval 08-001933) and the University of Virginia (IRB approval 14225). A review of clinical histories confirmed that each case was newly diagnosed, was confined to the CNS, had no occult disease by standard staging(14), and that each patient had no apparent immunodeficiency. Clinical annotations including pathology, treatment, follow-up and survival data of the cohort is shown in Supplementary Table S1.
Five-micron sections were cut from each FFPE block and placed on clean glass slides. Slides were stained with hematoxylin and eosin to confirm the presence of sufficient tumor rich tissue (>75%) without significant hemorrhage, necrosis or artifact. Confirmatory immunohistochemistry using antibodies directed against CD20 and CD3 was performed on the second and third slide of each patient set. All confirmed cases were then screened for Epstein-Barr virus utilizing in situ hybridization probes that recognize EBV-encoded RNA.
Molecular tests
DNA was obtained either from frozen tissue or from FFPE tissues. In a subset of 3 samples with frozen tissue available, the amount of DNA recovered was not enough for performing molecular techniques, so a whole genome amplification step was added (see below). In 9 out of 19 samples, we identified biopsies that included tumor-free tissue. These tissues were used to extract normal DNA for validation sequencing. The source of DNA and the molecular techniques performed in each sample are described in Supplementary Table S2.
DNA isolation
DNA was obtained from frozen tissue in 7 cases using Gentra Puregene Core A kit (Qiagen) according to manufacturer’s recommendations. In the remaining 12 cases, FFPE samples were deparaffinized using 3 xylene washes for 5 minutes each. Xylene was washed out with decreasing series of ethanol (100%, 95%, 70%, 50%, 30%, 0%) and finally washed 3 times in 1mM EDTA (pH 8.0) for 5 minutes each. The tissue was pelleted and washed 2 times with PBS (pH=7.5). Samples were incubated overnight with 180 ml buffer ATL and 20 ml proteinase K. DNA was obtained using the MicroDNA extraction kit according to the manufacturer’s recommendations (MicroDNA kit, Qiagen).
In three samples with insufficient DNA available, amplification of the whole genome was performed using the illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare) following the manufacturer’s recommendations. DNA concentration was measured by flourometric method (Qubit).
aCGH
aCGH was performed in 18 cases (Supplementary Table S2) using the Human Genome 244A and the Sureprint G3 microarrays (Agilent Technologies). The digestion, labeling and hybridization steps were done as previously described(4, 15, 16). Briefly, 1.2 ug of tumor and reference DNA were independently fragmented with Bovine DNaseI (Ambion) for 12 minutes at room temperature. DNA samples from a pool of 9 female, lymphoblastoid cell lines from the Coriell repository were used as the normal reference in the hybridization experiments. Tumor and reference samples were labeled with Alexa 5 and Alexa 3 dyes, respectively. Labeled reactions were cleaned up and hybridized at 65°C for 40 hours. Microarrays were scanned in a DNA Microarray Scanner and features were extracted with Feature Extraction software (Agilent).
Extracted data was analyzed using Nexus software (Biodiscovery). Copy-number abnormalities (CNA) were calculated using RANK segmentation algorithm, a modified version of the circular binary segmentation algorithm. For CNA detection, a significant threshold was set at 2.4E-5 with a minimum number of probes per segment set at 2 (244K array format), 3 (Sureprint G3 in fresh tumor samples) or 10 (Sureprint G3 in FFPE samples). To identify and eliminate the germ line copy number variations (CNV) from the study, we created a CNV database including the copy-number (using platform SNP6.0) and sequencing studies available in The Centre for Applied Genomics data portal (TCAG) as well as our findings in 10 HapMap samples run by Sureprint G3 arrays(16). Microarray data is deposited in GEO dataset under accession GSE28952.
Whole exome end-paired sequencing
Exome capture was done utilizing the solution-phase hybrid Sure Select 50 MB Capture kit (Agilent Technologies). Next, 100 bp paired-end DNA libraries were prepared and 4 samples were run per lane on the HiSeq2000 sequencer. An automated workflow for exome-seq data analysis was developed, which includes read quality control, read alignment, and mutation detection. Paired-end reads were aligned to hg19 using Novoalign (Novocraft Technologies). Quality of sequencing chemistry was evaluated using FastQC. After alignment, PCR duplication rates and percent reads mapped on target were used to assess the quality of the sample preparations. Realignment and recalibration steps were done based on the Best Practice Variant Detection v3 recommendations implemented in The Genome Analysis Toolkit (GATK)(17). Duplicate marking was used to flag those reads in the alignment file –without removing them – and afterwards the SNV callers do not consider reads with the duplicate flag for the analysis. Germline variant calling (both single nucleotide and small insertions and deletions) was also called through GATK. Somatic single nucleotide variations (SNVs) were genotyped using SomaticSniper(18), whereas insertions and deletions were called by GATK Somatic Indel Detector. Each variant in coding regions was functionally annotated by snpEFF and PolyPhen-2(19) to predict biological effects. The variants were annotated using our TREAT workflow(20) whether or not the gene is associated with disease or phenotypes, and any associated pathways. We removed variants found in the 1000 genomes project, the HapMap sequencing project and the BGI – Danish Sequencing Project. In addition, we removed variants present in the dbSNP dataset unless these mutations were also present in the COSMIC database. Variants of significant interest were visually inspected using IGV(21).
Validation Sequencing
Targeted Sequencing
All the coding regions of MYD88 were sequenced using semiconductor technology (IonTorrent PGM, Life Technologies) following manufacturer’s recommendations(22). All the coding regions were amplified in 150 bp amplicons using customized oligos (Ion Ampliseq designer, Life Technologies) and multiplex PCR with 10ng of input DNA per reaction. DNA libraries preparation and enrichment were done using the IonOneTouch and IonOneTouch ES automated systems, respectively. Samples were sequenced using the 316™ chip, with an average of 2,005× depth of coverage per nucleotide. Data was analyzed using IonReporter software (Life Technologies).
Sanger sequencing
All mutations identified by WES in CARD11, CD79B, PRKCD, TNFAIP3 were validated using PCR amplification followed by Sanger sequencing. In each case, tumor and normal counterpart samples were sequenced in order to confirm the somatic origin of the mutations. List of primers used for PCR and sequencing are listed in Supplementary Table S3.
Interphase FISH
Interphase FISH was performed on sections of FFPE (N=9) or frozen tumor samples (N=2) as described previously(23). The 11 cases were screened for IGH-BCL6 translocations. For IGH-BCL6 we used a two-color break-apart probe for BCL6 (BAP; Abbott Molecular) and a homebrew two-color dual fusion IGH-BCL6 probe composed of a SpectrumGreen labeled probe for 14q32 and a SpectrumOrange-labeled probe for 3q27.3 (BACS RP11-88P6, RP11-211G3, and CTD2522K3). For PRKCD and TOX, homebrew probes were used; PRKCD: G248P87977G3/WI2-1696N05; TOX: G248P80762C4/WI2-1217E08 (Genome Reference Consortium). Control probes were used in both cases: CEP3 (SpectrumOrange) with PRKCD (SpectrumGreen) and LSI C-MYC (SpectrumOrange) with TOX (SpectrumGreen). Three patients were screened for each, PRKCD and TOX. In each sample, at least 50 tumor cells were scored. Samples were scored as positive when at least 20 cells with a recognized abnormal signal pattern were present(24). The list of cases analyzed and the respective results for each region are shown in Supplementary Table S4.
Gene expression analysis
Gene expression levels of PRKCD and TOX were analyzed using publicly available datasets. Expression level across different tumor cell lines was obtained from the Broad-Novartis Cancer Cell Line Encyclopedia.
Functional gene set enrichment
Enrichment of functional gene classes within frequently abnormal genes was performed using the DAVID Functional Annotation tool.
Statistical Analysis
Overall survival (OS) was estimated using Kaplan Meier method. Differences between survival curves were estimated by the log-rank test. Results were considered significant when P < 0.05.
Results
Analysis of chromosomal abnormalities shows a complex karyotype in PCNSL
In order to identify genetic abnormalities in PCNSL we screened 18 patient samples by aCGH (Figure 1). PCNSL cases showed a complex genome with a median of 21 CNA per patient (range 10–47). Overall, 16 chromosomal abnormalities were found in >25% of cases (10 deletions and 6 gains). The most common abnormality was the deletion of 9p21.3 (CDKN2A and CDKN2B), which was found in 83% of cases. Multiple abnormalities were commonly found in chromosome 6. Deletion of 6p21 (HLA genes) was identified in 9 of 18 cases (50%), losses of 6q21 (PRDM1) and 6q23 (TNFAIP3) in 10 cases each (55%), and deletion of 6q14.1 (TMEM30A) in 8 cases (44%). Of the remaining lesions found in >25% of cases, 6 encompassed single or few genes, including losses of 3p21 (PRKCD), 3q26.32 (TBL1XR1), 8q12 (TOX), 12q24.31 (BCL7A, SETD1 and 9 other genes) and gains of 3p14 (FOXP1) and 3q27 (BCL6)(Figure 1). BCL6 and FOXP1 were characterized by breakpoints inside the gene or near 5′ of the gene, suggesting their involvement in unbalanced chromosomal translocations. Other recurrent abnormalities were gains of 7q21–q31 (39%), 11q (28%), 12q (50%) and 19q13 (33%) and loss of 10p15.3-p14 (33%). Gain of 12q was mutually exclusive with deletion 10p15.3-p14 and gains of 3p14 (FOXP1) and 11q, which were found alone or in combination in all 9 cases without gain of 12q (P = 0.0004) (Supplementary Figure S1). Furthermore, losses of 3p21.31 (PRKCD) and 6q23 (TNFAIP3) were rare events in cases with 12q. The complete list of CNA is listed in Supplementary Table S5.
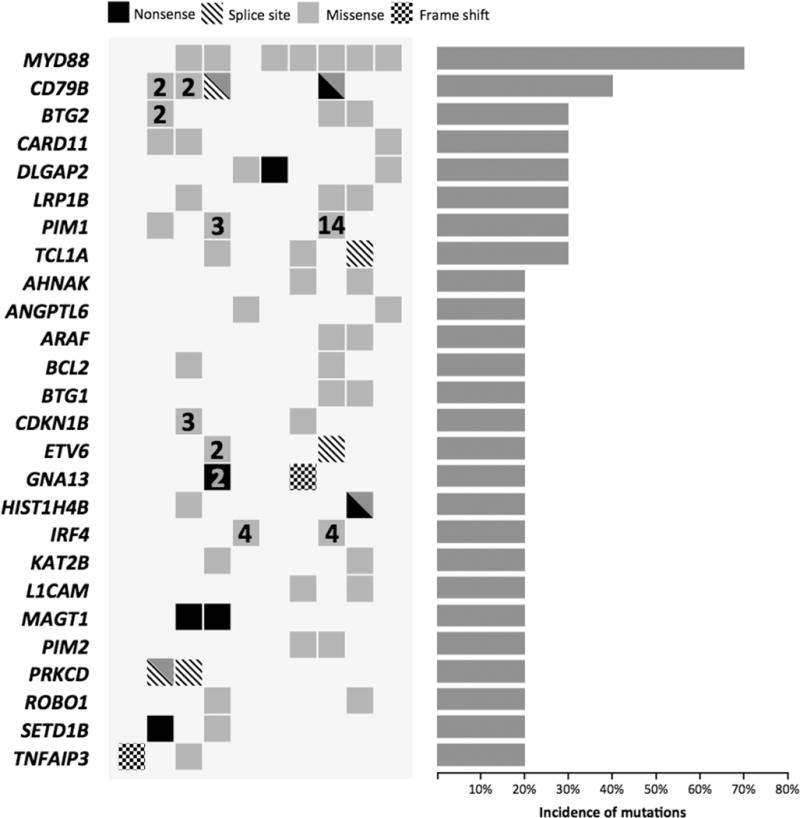
Figure 1.

Overview of copy number abnormalities identified in the cohort of PCNSL analyzed. Chromosomes 1 to Y are represented from left to right. Dark gray blocks represent chromosome gains, whereas light gray blocks represent chromosome losses. The amplitude in each abnormal region represents the incidence of each copy- number abnormality in the studied cohort. Genes found in recurrent minimal aberrant regions (>25%) are indicated.
Next, we focused on the analysis of homozygous deletions (HD) and high-copy gain, which are likely to harbor tumor suppressor genes and oncogenes, respectively. Overall, 13 HD and 4 high copy level gain were found; 55% of cases analyzed show HD of CDKN2A whereas another 21% showed monoallelic deletion of the gene. Recurrent HD were also found in HLA genes in 22% (4/18), PRDM1, TMEM30A, TOX and CD58 (in 2 cases each; 11%). Another 34 genes, including B2M, ETV6, TNFAIP3 and TNFRSF10A, were found deleted in individual cases (Table 1). Conversely, high-level amplifications were rare in PCNSL. Overall, only four regions were identified having 2 or more extra copies: 1p31.1-p22.2 (BCL10 and other 32 genes), 7p22.3-p22.2 (CARD11 and other 36 genes), 12q14.1-q21.33 and 18q21.2 (TCF4), in one sample each.
Table 1.
Chromosomal regions implicated either in homozygous deletions (HD) or >2 extra copies (Amplification). Note: In regions larger than 1 Mb, only the most relevant genes were listed (numbers in parentheses indicate the total number of genes included in those regions).
| Chr | Cytoband | Start_bp | End_bp | Size (Mb) | Change | Number of cases (%) | Genes |
|---|---|---|---|---|---|---|---|
| 1 | p35.2 | 30438023 | 31187531 | 0.75 | HD | 1 (5) | MATN1, LAPTM5, SDC3, PUM1, SNORD103A |
| 1 | p13.1 | 116859897 | 117098412 | 0.24 | HD | 2 (11) | CD58, IGSF3 |
| 4 | q34.3 | 178684938 | 181419558 | 2.73 | HD | 1 (5) | LINC01098 |
| 6 | p21.32 | 32716055 | 32728687 | 0.01 | HD | 4 (22) | HLA |
| 6 | q14.1 | 76013320 | 76023847 | 0.01 | HD | 2 (11) | TMEM30A |
| 6 | q21 | 106645690 | 106658114 | 0.01 | HD | 2 (11) | PRDM1 |
| 6 | q21 | 108337759 | 108342883 | 0.01 | HD | 1 (5) | SEC63 |
| 6 | q26 | 161563675 | 161586174 | 0.02 | HD | 1 (5) | AGPAT4 |
| 7 | p15.1 | 30094975 | 30096933 | 0.002 | HD | 1 (5) | PLEKHA8 |
| 8 | p21.3 | 22934625 | 23145415 | 0.21 | HD | 1 (5) | TNFRSF10B, TNFRSF10C, TNFRSF10D, TNFRSF10A |
| 8 | q12.1 | 59996016 | 60127073 | 0.13 | HD | 2 (11) | TOX |
| 9 | p21.3 | 21961516 | 21988776 | 0.08 | HD | 10 (56) | CDKN2A, CDKN2B |
| 12 | p13.2 | 11787795 | 11985797 | 0.2 | HD | 1 (5) | ETV6 |
| 15 | q15.3 – q21.1 | 42663648 | 43310674 | 0.65 | HD | 1 (5) | SPG11, PATL2, B2M, TRIM69, c150rf43, SORD, DUOX2, DOUXA1, DOUX1, SHF |
| 16 | q12.2 | 51781157 | 51921599 | 0.14 | HD | 1 (5) | CHD9 |
| 17 | q24.1 | 60452046 | 60482005 | 0.03 | HD | 1 (5) | GNA13 |
| 19 | q13.12 | 42005151 | 42137239 | 0.13 | HD | 1 (5) | ZNF790, ZNF345, ZNF829, ZNF568 |
| X | q27.1 | 138466967 | 138747086 | 0.28 | HD | 1 (5) | F9, MCF2, APT11C |
| X | q28 | 150511563 | 151661046 | 1.15 | HD | 1 (5) | PASD1, PRRG3, FATE1, CNGA2, MAGEA4, GABRE, MAGEA5, MAGEA10, GABRA3, GABRQ |
| 5 | q22.1 | 110445097 | 110474082 | 0.03 | Amplification | 1 (5) | WDR36 |
| 7 | p12.2 | 50114207 | 50502559 | 0.39 | Amplification | 1 (5) | c7orf72, IKZF1, FIGNL1, DDC |
| 18 | q21.2 | 51053466 | 51141361 | 0.09 | Amplification | 1 (5) | TCF4 |
| 1 | p31.1 – p22.2 | 82724188 | 88291462 | 5.57 | Amplification | 1 (5) | BCL10 (37) |
| 7 | p22.3 | 178408 | 4945057 | 4.77 | Amplification | 1 (5) | CARD11 (33) |
| 19 | p13.3 | 1567089 | 1584543 | 0.02 | Amplification | 1 (5) | TCF3 |
| 7 | p11.2 | 55464898 | 55602703 | 0.14 | Amplification | 1 (5) | LANCL2, VOPP1 |
| 16 | q24.1 | 84486886 | 84587286 | 0.1 | Amplification | 1 (5) | IRF8 |
Finally, BCL6 translocations were identified in 2 of 11 PCNSL cases analyzed. Both cases showed IGH-BCL6 fusion (Supplementary Figure S2A and Supplementary Table S4).
Amplified genomic DNA and DNA extracted from FFPE blocks were suitable for WES
We analyzed 10 PCNSL by WES with an average of 65.6 million mapped reads in capture regions. After duplication marking and removal, we obtained a median of 80% of the exome covered by 30× depth or more. In 3 cases, DNA was extracted from FFPE tissue. In the remaining 7 cases, DNA was obtained from frozen tissue. In 3 out of these 7 cases there was not enough input genomic DNA and a whole genome amplification step was added before proceeding (Supplementary Table S2). Sequencing data generated from amplified DNA and from FFPE samples showed a lower coverage than the data generated from high molecular weight genomic DNA, but still showed satisfactory quality control measurements and average fold coverage for the analysis (Supplementary Table S6). After data filtering, we found a mean non-synonymous mutation rate of 3.3 mutations per Mb (range 0.8 to 6.1 mutations per Mb). The complete list of mutated gene is shown in Supplementary Table S7.
Integrated mutation and copy-number data identified PCNSL-specific abnormalities
By analyzing mutation data (Figure 2) and integrating mutation and copy number data (Figure 3) we identified a group of genes recurrently activated and/or biallelically mutated in PNCSL that has not been previously identified in nodal DLBCL, including PRKCD and TOX. Protein kinase C delta (PRKCD) emerged as a potentially relevant gene in PCNSL. We identified mutations affecting splice donor sites in 2 out of 10 cases analyzed by WES. One of these cases had an additional missense mutation (p.W608S). Sanger sequencing confirmed all mutations. Next, the somatic origin of the mutations was confirmed by the lack of them in normal tissue sequenced from both cases. Furthermore, both cases also showed monoallelic deletion of PRKCD, thus confirming the biallelic loss of the gene (Figure 4). Additionally, we identified focal monoallelic deletions (<1Mb) involving PRKCD in another 3 out of 18 cases analyzed by aCGH (Figure 1B and Supplementary Figure S2B). DNA was not available for sequencing these 3 cases. Mutations in PRKCD have not been found in nodal DLBCL. Furthermore, COSMIC dataset (Sanger Institute) only shows PRKCD mutations in one sample (p.A163T; acute lymphoblastic leukemia) out of 1058 hematological malignancies analyzed (0.01%), thus suggesting that PRKCD impairment is specific to PCNSL. Using public datasets, we analyzed the expression level of PRKCD across cell lines established from different types of cancer. Using the Broad-Novartis dataset we found that DLBCL shows the highest expression of PRKCD across all entities.
Figure 2.
Recurrent mutations in PCNSL. The columns of the heatmap represent individual patients. Type of mutation found is color-coded. In cases with multiple mutations in the same gene, a combination of colors (more than one type of mutations) or numbers indicating the number of mutations found (more than one mutation of the same type) was used. Right: Histogram showing the incidence of mutations found in each gene.
Figure 3.
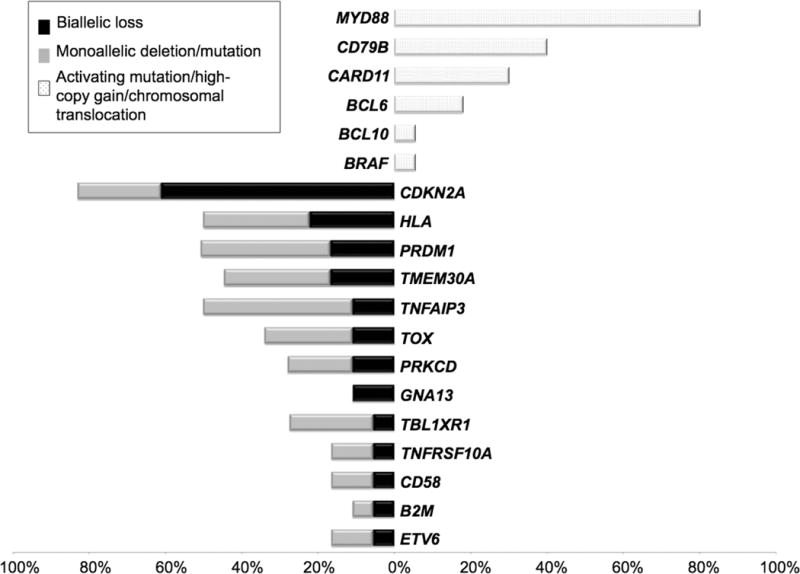
Integration of copy-number and sequencing data shows a set of recurrent abnormalities. List of genes that were affected with activating mutations/translocations/high copy gain (top) or biallelic loss (bottom).
Figure 4.
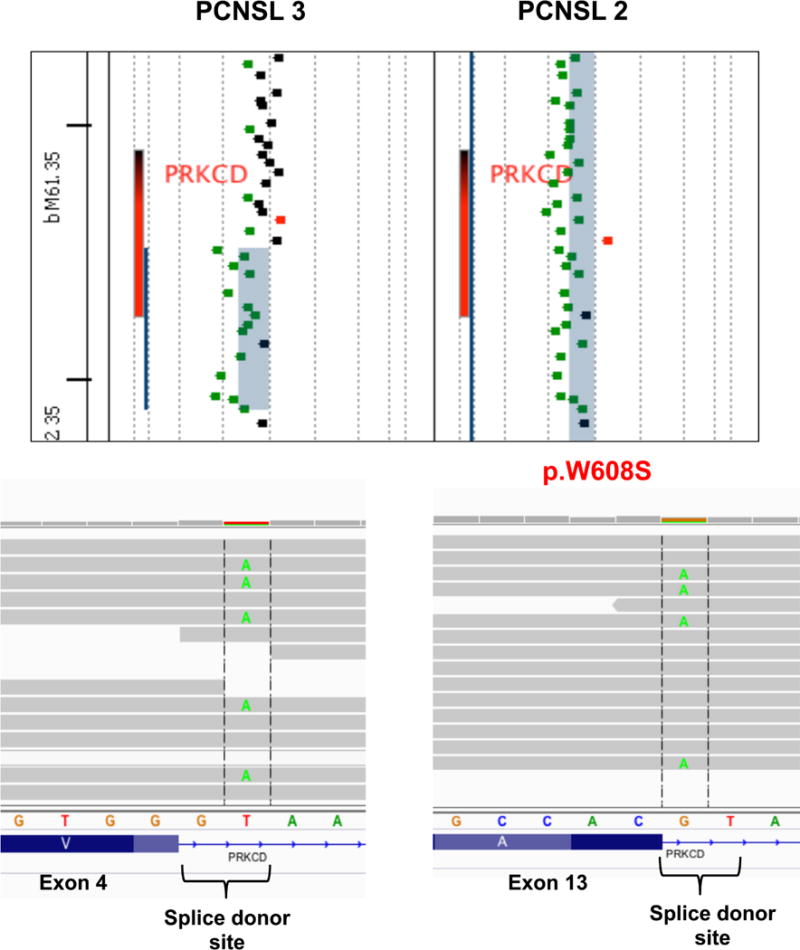
Protein Kinase C delta (PRKCD) is recurrently inactivated in PCNSL. Concomitant PRKCD monoallelic deletions (top figure) and mutations affecting both alleles (bottom figures) were recurrently found in PCNSL.
Another interesting finding was the recurrent abnormalities affecting thymocyte selection-associated high mobility group box (TOX) gene seen only in PCNSL. Thus, 11% of PCNSL showed homozygous deletion of TOX and another 17% of cases showed focal monoallelic deletions in the gene (Figure 5 and Supplementary Figure S2C). In comparison, no homozygous deletions affecting TOX were identified across 754 cell lines analyzed, including 127 lines originated from hematopoietic and lymphoid tissues (Source: Cancer Genome Project dataset, Sanger Institute).
Figure 5.
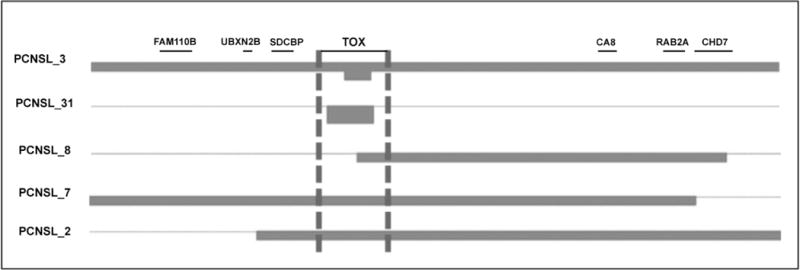
Homozygous (top 2 samples) and focal heterozygous deletions (bottom 3 samples) affecting TOX were recurrently found in PCNSL.
On the other hand, we found recurrent mutations in several genes that are linked to DLBCL pathogenesis, principally the high prevalence of MYD88 activating mutations, found mutated in 70% of cases by WES (Figure 2). We additionally screened another 4 PCNSL cases for MYD88 mutations by targeted sequencing. Overall, mutations were found in 11 of 14 cases analyzed (79%). The mutations were found in the residue L265P, with the exception of two mutations, affecting the residue 217 (V217F) and the residue 232 (M232T), respectively. The prevalence of MYD88 mutations found is nearly 2-fold higher than what has been reported in nodal ABC DLBCL.
Other affected genes shared with nodal DLBCL were mutations on CD79B, PIM1 (40%), BTG2, CARD11 (30% each), GNA13, TNFAIP3 (20% each) and BRAF (10%). Three of 4 cases with CD79B mutations also have mutations affecting MYD88 (Figure 2). All mutations found in CD79B, CARD11 and TNFAIP3 as well as its somatic origin were confirmed using Sanger sequencing.
Genomic data integration highlighted the high frequency of genetic lesions affecting a variety of signaling pathways that share their ability to induce the NF-κB transcription complex, including the B-cell receptor, CD40 and Toll-like receptor pathways. Overall, we found components of these pathways to be altered in 93% of PCNSL. Furthermore, our data showed the recurrent impairment of networks associated with proliferation, immune response, regulation of apoptosis, and lymphocyte differentiation and activation.
Abnormalities in 6q21 are associated with worse outcome
In order to determine if there was an association between the identified recurrent abnormalities and survival we compared the OS in patients with and without abnormalities found in >20% of cases. Deletion of 6q21 (PRDM1) was associated with shorter OS in univariate analysis (P = 0.001; Supplementary Figure S3). However, this observation needs to be considered with cautious, especially considering the small size and heterogeneous treatment received by the cohort under analysis.
Discussion
Unlike systemic DLBCLs, only a small number of genetic studies have been performed in PCNSL(2–5, 7, 10, 11, 13), mainly because of the limited amount and source of material for study. Most PCNSL biopsies are obtained by stereotactic surgery and then placed in FFPE blocks. Therefore, the use of samples in systematic genomic studies has been challenging because the yield of DNA obtained from this source is low and the quality poor. We have previously shown that DNA samples obtained from FFPE tissues can be used for array-based genomic analysis in PCNSL(4). FFPE specimens are valuable material for PCNSL research, especially in retrospective studies with long follow-up data. Thus, the data provided in our study encouraged the future interrogation of large archival PCNSL tissues by WES.
PCNSL are recognized as a distinct entity by the WHO classification, showing characteristics that separate them from nodal DLBCLs(1). However, only a few studies have been focused on better understanding the genomic landscape of PCNSL and the comparison with the better-defined nodal DLBCL. For example, recent studies have shown that PCNSL shares several abnormalities with DLBCL, including translocations of IgH-BCL6, deletions of 9p21 and mutations in MYD88 and CARD11(3–5, 7, 25–27). Montesinos-Rongen et al. performed a comparative gene expression analysis and proposed that PCNSL are a post-germinal center disease associated with the activated B cell (ABC)-DLBCL type in origin(11). Other gene expression studies have found similarities between entities but also showed pathways unique to PCNSL(2, 9, 10, 13). In this study we aimed to perform a comprehensive genomic study in PCNSL to better elucidate the genomic landscape of the disease.
By integrating copy number and sequencing data we found a highly complex karyotype with a subset of PCSNL-specific alterations, thus reinforcing the idea that IP-DLBCLs are a pathogenetically distinct group. One of the novel and interesting findings is the recurrent abnormalities identified in PRKCD; no other hematological malignancy shows recurrent abnormalities in this gene. PRKCD, a pro-apoptotic protein kinase, has been implicated in a wide variety of cellular processes, such as growth, differentiation, secretion, apoptosis and tumor development(28–31). In addition, numerous studies in humans and mice have shown that loss of PRKCD is associated with defective B-cell apoptosis, B-cell hyperproliferation and autoimmune disorders(32–35). Furthermore, the decreased expression of PRKCD via the negative regulation of MiR-181a confers resistance of cervical cancer to radiation therapy inhibiting irradiation-induced apoptosis and decreasing G2/M block(36). The study by Ke et al. study shows that MiR-181a contributes to radio-resistance by targeting PRKCD, leading to inhibition of radiation-induced apoptosis(36). Radio-insensitive cervical cancer specimens had higher expression of MiR-181a, compared with radio-sensitive cases. MiR-181a negatively regulates the expression of PRKCD via targeting its 3′-untranslated region. These results suggest that lack of PRKCD expression might be a biomarker for insensitivity of radiation treatment in cervical cancer but perhaps these results can be extrapolated to other cancers.
Another relevant finding was the identification of recurrent biallelic alterations of TOX. This gene plays a key role in T cell development(37, 38). The absence of TOX prevents CD4 lineage T cell development, including natural killer T (NKT) and T regulatory (T reg) CD4 T cell sub-lineages, but has only modest effects on CD8 T cell development(39). Furthermore, TOX deficient mice (TKO) lacked lymph nodes and had a significant decrease in the frequency and size of Peyer’s patches(40). A recent study provides data that support an important role of TOX expression in B-cell biology(41). TOX was identified as one of the top differentially expressed genes between memory B cells (IgG+ CD27+) and marginal zone B-cells (IgD+ CD27+). More specifically, it was shown that TOX is >10X under-expressed in MZB compared with memory B-cells. As yet there is no known role of TOX in PCNSL, the recurrent alterations of this gene lead to speculate that TOX may contribute to its pathogenesis. Interestingly, our analysis suggested that loss of TOX is associated with shorter OS. Further studies, in larger cohorts of PCNSL with robust age, performance status, co-morbidity and treatment data, will be required to confirm this finding.
Moreover, we identified that almost all PCNSL cases (~90%) harbor mutations leading to activation of the NF-κB signaling pathway such as activating mutations of MYD88, CARD11, CD79, and deletions of TNFAIP3 and TBL1XR1. These data suggest that the activation of NF-κB signaling pathway is a key driver of lymphomagenesis in PCNSL, an immune-privileged site. Inside the pathway, the high prevalence of MYD88 activating mutations is remarkable. MYD88 has become a central gene of B-cell pathogenesis, with mutations found in ABC-DLBCL, being ubiquitous in Waldenström’s Macroglobulinemia and to a lesser extent in marginal zone lymphoma and chronic lymphocytic leukemia (42–48). Furthermore, three recent studies have been performed in PCNSL showing numbers ranging from 38 to 75% of cases mutated(5, 25, 49).
In summary, we identified a related genomic landscape between PCNSL and ABC-DLBCL and confirmed the existence of a specific molecular signature in PCNSL characterized by recurrent alterations in PRKCD and TOX. Furthermore, this comprehensive genomic study identified targetable pathways in the disease. PCNSL is an aggressive malignancy and better and more specific treatments are needed. The identification of mutations affecting the BCR, TLR and NF-κB signaling pathways in most of the PCNSL cases, highlights the potential value of these pathways to be targeted in PCNSL.
Statement of Translational Relevance.
Primary Central Nervous System Lymphoma (PCNSL) is an aggressive brain cancer characterized by a perivascular accumulation of malignant cells with lymphoid characteristics. Without translatable knowledge of the mechanisms of PCNSL development and progression treatment falls on conventional managements such as radiotherapy and chemotherapy. These treatments are intense, and the quality of survival is often poor.
This study revealed a previously unappreciated genetic complexity in immunocompetent PCNSL. We identified a related genomic landscape between PCNSL and ABC-DLBCL but confirmed the existence of a specific molecular signature in PCNSL, reinforcing the idea that PCNSL is a different entity. Furthermore, this comprehensive genomic study identified targetable pathways in the disease. The identification of mutations affecting the BCR, TLR and NF-κB signaling pathways in most of the PCNSL cases, highlights the potential value of these pathways as treatment targets in PCNSL.
Acknowledgments
E.B. is a recipient of the Marriott Specialized Workforce Development Awards in Individualized Medicine, The Henry Predolin Foundation Career Development Award and the George Haub Family Career Development Award Fund in Cancer Research.
Funding: This work was supported by the Mayo Clinic SPORE in Brain Cancer (P50 CA108961; PI: BP O’Neill), the Iowa/Mayo Lymphoma SPORE (P50 CA97274; PI: Weiner), the George Haub Family Career Development Award Fund in Cancer Research and the Henry Predolin Foundation.
Footnotes
Conflict of Interest Disclosures
There are no conflicts of interest.
Authorship Contributions
E.B., B.P.O., R.F. designed the study; E.B., S.V.W., J.O., J.A.S., B.E. performed the research; E.B., Y.W.A., J.E., A.K.S. analyzed data; E.M., D.S., M.B.L., R.V., R.T., T.E.W. identified cases and collected clinical data, E.B. wrote the paper; all authors reviewed and gave final approval of the manuscript.
References
- 1.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th. Lyon, France: IARC Press; 2008. [Google Scholar]
- 2.Richter J, Ammerpohl O, Martin-Subero JI, Montesinos-Rongen M, Bibikova M, Wickham-Garcia E, et al. Array-based DNA methylation profiling of primary lymphomas of the central nervous system. BMC Cancer. 2009;9:455. doi: 10.1186/1471-2407-9-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwindt H, Vater I, Kreuz M, Montesinos-Rongen M, Brunn A, Richter J, et al. Chromosomal imbalances and partial uniparental disomies in primary central nervous system lymphoma. Leukemia. 2009;23:1875–84. doi: 10.1038/leu.2009.120. [DOI] [PubMed] [Google Scholar]
- 4.Braggio E, McPhail ER, Macon W, Lopes MB, Schiff D, Law M, et al. Primary central nervous system lymphomas: a validation study of array-based comparative genomic hybridization in formalin-fixed paraffin-embedded tumor specimens. Clin Cancer Res. 2011;17:4245–53. doi: 10.1158/1078-0432.CCR-11-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Aguilar A, Idbaih A, Boisselier B, Habbita N, Rossetto M, Laurenge A, et al. Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin Cancer Res. 2012;18:5203–11. doi: 10.1158/1078-0432.CCR-12-0845. [DOI] [PubMed] [Google Scholar]
- 6.Courts C, Montesinos-Rongen M, Brunn A, Bug S, Siemer D, Hans V, et al. Recurrent inactivation of the PRDM1 gene in primary central nervous system lymphoma. J Neuropathol Exp Neurol. 2008;67:720–7. doi: 10.1097/NEN.0b013e31817dd02d. [DOI] [PubMed] [Google Scholar]
- 7.Montesinos-Rongen M, Schmitz R, Brunn A, Gesk S, Richter J, Hong K, et al. Mutations of CARD11 but not TNFAIP3 may activate the NF-kappaB pathway in primary CNS lymphoma. Acta neuropathologica. 2010;120:529–35. doi: 10.1007/s00401-010-0709-7. [DOI] [PubMed] [Google Scholar]
- 8.Montesinos-Rongen M, Zuhlke-Jenisch R, Gesk S, Martin-Subero JI, Schaller C, Van Roost D, et al. Interphase cytogenetic analysis of lymphoma-associated chromosomal breakpoints in primary diffuse large B-cell lymphomas of the central nervous system. J Neuropathol Exp Neurol. 2002;61:926–33. doi: 10.1093/jnen/61.10.926. [DOI] [PubMed] [Google Scholar]
- 9.Booman M, Szuhai K, Rosenwald A, Hartmann E, Kluin-Nelemans H, de Jong D, et al. Genomic alterations and gene expression in primary diffuse large B-cell lymphomas of immune-privileged sites: the importance of apoptosis and immunomodulatory pathways. J Pathol. 2008;216:209–17. doi: 10.1002/path.2399. [DOI] [PubMed] [Google Scholar]
- 10.Sung CO, Kim SC, Karnan S, Karube K, Shin HJ, Nam DH, et al. Genomic profiling combined with gene expression profiling in primary central nervous system lymphoma. Blood. 2011;117:1291–300. doi: 10.1182/blood-2010-07-297861. [DOI] [PubMed] [Google Scholar]
- 11.Montesinos-Rongen M, Brunn A, Bentink S, Basso K, Lim WK, Klapper W, et al. Gene expression profiling suggests primary central nervous system lymphomas to be derived from a late germinal center B cell. Leukemia. 2008;22:400–5. doi: 10.1038/sj.leu.2405019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubenstein JL, Fridlyand J, Shen A, Aldape K, Ginzinger D, Batchelor T, et al. Gene expression and angiotropism in primary CNS lymphoma. Blood. 2006;107:3716–23. doi: 10.1182/blood-2005-03-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tun HW, Personett D, Baskerville KA, Menke DM, Jaeckle KA, Kreinest P, et al. Pathway analysis of primary central nervous system lymphoma. Blood. 2008;111:3200–10. doi: 10.1182/blood-2007-10-119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreri AJ. How I treat primary CNS lymphoma. Blood. 2011;118:510–22. doi: 10.1182/blood-2011-03-321349. [DOI] [PubMed] [Google Scholar]
- 15.Braggio E, Keats JJ, Leleu X, Van Wier S, Jimenez-Zepeda VH, Valdez R, et al. Identification of copy number abnormalities and inactivating mutations in two negative regulators of nuclear factor-kappaB signaling pathways in Waldenstrom’s macroglobulinemia. Cancer Res. 2009;69:3579–88. doi: 10.1158/0008-5472.CAN-08-3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kay NE, Eckel-Passow JE, Braggio E, Vanwier S, Shanafelt TD, Van Dyke DL, et al. Progressive but previously untreated CLL patients with greater array CGH complexity exhibit a less durable response to chemoimmunotherapy. Cancer Genet Cytogenet. 2011;203:161–8. doi: 10.1016/j.cancergencyto.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–7. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pek JW, Kai T. DEAD-box RNA helicase Belle/DDX3 and the RNA interference pathway promote mitotic chromosome segregation. Proc Natl Acad Sci U S A. 2011;108:12007–12. doi: 10.1073/pnas.1106245108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nature biotechnology. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–52. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 23.Cataldo KA, Jalal SM, Law ME, Ansell SM, Inwards DJ, Fine M, et al. Detection of t(2;5) in anaplastic large cell lymphoma: comparison of immunohistochemical studies, FISH, and RT-PCR in paraffin-embedded tissue. The American journal of surgical pathology. 1999;23:1386–92. doi: 10.1097/00000478-199911000-00009. [DOI] [PubMed] [Google Scholar]
- 24.Remstein ED, Dogan A, Einerson RR, Paternoster SF, Fink SR, Law M, et al. The incidence and anatomic site specificity of chromosomal translocations in primary extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) in North America. The American journal of surgical pathology. 2006;30:1546–53. doi: 10.1097/01.pas.0000213275.60962.2a. [DOI] [PubMed] [Google Scholar]
- 25.Montesinos-Rongen M, Godlewska E, Brunn A, Wiestler OD, Siebert R, Deckert M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta neuropathologica. 2011;122:791–2. doi: 10.1007/s00401-011-0891-2. [DOI] [PubMed] [Google Scholar]
- 26.Cady FM, O’Neill BP, Law ME, Decker PA, Kurtz DM, Giannini C, et al. Del(6)(q22) and BCL6 rearrangements in primary CNS lymphoma are indicators of an aggressive clinical course. J Clin Oncol. 2008;26:4814–9. doi: 10.1200/JCO.2008.16.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montesinos-Rongen M, Schafer E, Siebert R, Deckert M. Genes regulating the B cell receptor pathway are recurrently mutated in primary central nervous system lymphoma. Acta neuropathologica. 2012;124:905–6. doi: 10.1007/s00401-012-1064-7. [DOI] [PubMed] [Google Scholar]
- 28.Mischak H, Goodnight JA, Kolch W, Martiny-Baron G, Schaechtle C, Kazanietz MG, et al. Overexpression of protein kinase C-delta and -epsilon in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. The Journal of biological chemistry. 1993;268:6090–6. [PubMed] [Google Scholar]
- 29.Pessino A, Passalacqua M, Sparatore B, Patrone M, Melloni E, Pontremoli S. Antisense oligodeoxynucleotide inhibition of delta protein kinase C expression accelerates induced differentiation of murine erythroleukaemia cells. The Biochemical journal. 1995;312(Pt 2):549–54. doi: 10.1042/bj3120549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, et al. Proteolytic activation of protein kinase C delta by an ICE/CED 3-like protease induces characteristics of apoptosis. The Journal of experimental medicine. 1996;184:2399–404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu Z, Hornia A, Jiang YW, Zang Q, Ohno S, Foster DA. Tumor promotion by depleting cells of protein kinase C delta. Molecular and cellular biology. 1997;17:3418–28. doi: 10.1128/mcb.17.6.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuehn HS, Niemela JE, Rangel-Santos A, Zhang M, Pittaluga S, Stoddard JL, et al. Loss-of-function of the protein kinase C delta (PKCdelta) causes a B-cell lymphoproliferative syndrome in humans. Blood. 2013;121:3117–25. doi: 10.1182/blood-2012-12-469544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belot A, Kasher PR, Trotter EW, Foray AP, Debaud AL, Rice GI, et al. Protein kinase cdelta deficiency causes mendelian systemic lupus erythematosus with B cell-defective apoptosis and hyperproliferation. Arthritis and rheumatism. 2013;65:2161–71. doi: 10.1002/art.38008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyamoto A, Nakayama K, Imaki H, Hirose S, Jiang Y, Abe M, et al. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature. 2002;416:865–9. doi: 10.1038/416865a. [DOI] [PubMed] [Google Scholar]
- 35.Limnander A, Depeille P, Freedman TS, Liou J, Leitges M, Kurosaki T, et al. STIM1, PKC-delta and RasGRP set a threshold for proapoptotic Erk signaling during B cell development. Nature immunology. 2011;12:425–33. doi: 10.1038/ni.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ke G, Liang L, Yang JM, Huang X, Han D, Huang S, et al. MiR-181a confers resistance of cervical cancer to radiation therapy through targeting the pro-apoptotic PRKCD gene. Oncogene. 2013;32:3019–27. doi: 10.1038/onc.2012.323. [DOI] [PubMed] [Google Scholar]
- 37.Aliahmad P, O’Flaherty E, Han P, Goularte OD, Wilkinson B, Satake M, et al. TOX provides a link between calcineurin activation and CD8 lineage commitment. J Exp Med. 2004;199:1089–99. doi: 10.1084/jem.20040051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilkinson B, Chen JY, Han P, Rufner KM, Goularte OD, Kaye J. TOX: an HMG box protein implicated in the regulation of thymocyte selection. Nature immunology. 2002;3:272–80. doi: 10.1038/ni767. [DOI] [PubMed] [Google Scholar]
- 39.Aliahmad P, Kaye J. Development of all CD4 T lineages requires nuclear factor TOX. J Exp Med. 2008;205:245–56. doi: 10.1084/jem.20071944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aliahmad P, de la Torre B, Kaye J. Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nature immunology. 2010;11:945–52. doi: 10.1038/ni.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Descatoire M, Weller S, Irtan S, Sarnacki S, Feuillard J, Storck S, et al. Identification of a human splenic marginal zone B cell precursor with NOTCH2-dependent differentiation properties. J Exp Med. 2014;211:987–1000. doi: 10.1084/jem.20132203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–9. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–5. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 45.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med. 2012;367:826–33. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 47.Varettoni M, Arcaini L, Zibellini S, Boveri E, Rattotti S, Riboni R, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom’s macroglobulinemia and related lymphoid neoplasms. Blood. 2013;121:2522–8. doi: 10.1182/blood-2012-09-457101. [DOI] [PubMed] [Google Scholar]
- 48.Xu L, Hunter ZR, Yang G, Zhou Y, Cao Y, Liu X, et al. MYD88 L265P in Waldenstrom macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013;121:2051–8. doi: 10.1182/blood-2012-09-454355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kraan W, Horlings HM, van Keimpema M, Schilder-Tol EJ, Oud ME, Scheepstra C, et al. High prevalence of oncogenic MYD88 and CD79B mutations in diffuse large B-cell lymphomas presenting at immune-privileged sites. Blood cancer journal. 2013;3:e139. doi: 10.1038/bcj.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]