

Abstract
Aberrant Shh signaling promotes tumor growth in diverse cancers. The importance of Shh signaling is particularly evident in medulloblastoma and basal cell carcinoma (BCC), where inhibitors targeting the Shh pathway component Smoothened (Smo) show great therapeutic promise. However, the emergence of drug resistance limits long-term efficacy and the mechanisms of resistance remain poorly understood. Using new medulloblastoma models, we identify two distinct paradigms of resistance to Smo inhibition. Sufu mutations lead to maintenance of the Shh pathway in the presence of Smo inhibitors. Alternatively activation of the RAS/MAPK pathway circumvents Shh pathway-dependency, drives tumor growth and enhances metastatic behavior. Strikingly, in BCC patients treated with Smo inhibitor, squamous cell cancers with RAS/MAPK activation emerged from the antecedent BCC tumors. Together these findings reveal a critical role of RAS/MAPK pathway in drug resistance and tumor evolution of Shh pathway-dependent tumors.
Keywords: Medulloblastoma, Hedgehog pathway, Basal Cell carcinoma, Drug resistance, Smo inhibition, RAS, MAPK, FGF
INTRODUCTION
Sonic Hedgehog (Shh) signaling plays a critical role in growth and patterning during development, and aberrant activation of Shh signaling is implicated in several cancers (1). Germline mutations that activate Shh signaling predispose to basal cell carcinoma (BCC) and medulloblastoma (MB) in humans and mice, whereas somatic mutations in Shh components are frequently observed in such tumors (2, 3).
Signaling is initiated when Shh, a secreted protein, binds its receptor, Patched (Ptch). In the absence of Shh, Ptch suppresses activity of Smoothened (Smo). When Shh binds Ptch, Smo initiates a signaling cascade which inactivates the tumor suppressor Suppressor of Fused (Sufu), and activates Gli transcription factors. Gli target genes include Gli1, Ptch, , Nmyc and Cyclin D1 (Ccnd1). Small-molecule antagonists of Smo, including Vismodegib (GDC-0449, Roche), Sonidegib (LDE225, Novartis), and XL-139 (BMS/Exelixis), provide promising targeted therapy for BCC and medulloblastoma (1, 4–7), and are in clinical trials or in use for these indications.
Despite initial success of Smo inhibitors, long-term efficacy is limited by pre-existing or acquired drug resistance (8, 9). Studies of other cancers indicate that both cell-autonomous mutations and microenvironment-derived factors contribute to therapeutic resistance (10). Amplification of Gli2 and point mutations in Smo that prevent drug binding have been reported to cause resistance in preclinical and clinical studies (4, 5, 11). Increased activation of PI3K, aPKC-ι/λ, or cell cycle components may also contribute to resistance (5, 12, 13). Additional mechanisms of resistance are likely to arise in clinical practice, and must be understood to develop more effective therapeutic strategies for Shh-dependent tumors.
To date, the absence of reliable in vitro systems for growing and maintaining Shh-dependent tumors has been a major impediment for studying these cancers (14). Here, we report an approach for generating stable MB cell lines that are tumorigenic and retain key characteristics of Shh-subtype MB. Using these models, we identify two paradigms of resistance to Smo inhibitors. Loss of Sufu reactivates the Shh pathway downstream of Smo and thereby causes acquired therapeutic resistance. In a second scenario, activation of RAS/MAPK pathway overrides oncogenic addiction to Shh signaling and enables proliferation of resistant tumors with enhanced metastatic behavior. In human cancers, MAPK pathway activation is increased in metastatic MB tumor cells. Strikingly, the MAPK pathway also becomes activated after Vismodegib treatment as Shh-dependent basal cell cancer transitions to squamous cell cancer resistant to Smo inhibitors. Together, these results indicate that reactivation of the Shh pathway or interactions between Shh and MAPK pathways can alter tumor behavior and therapeutic responses. Therefore, future treatments must consider these distinct mechanisms of tumor evolution.
METHODS
Detailed description is in Supplemental Materials.
Animals
All experimental procedures were done in accordance with the National Institutes of Health guidelines and approved by the Dana-Farber Cancer Institutional Animal Care and Use Committee. Ptch+/− mice (2) (Jackson Laboratory). nu/nu mice (Charles River Laboratories).
Human Studies
All human subjects work was reviewed by the Institutional Review Board Committees of Brigham and Women’s Hospital and Dana-Farber Cancer Institute, University of Calgary, and Stanford University for appropriate use, that informed consent was obtained from all subjects when required, and appropriate waiver of consent requirements was obtained for minimal risk studies.
SMB Cell Culture
SMB cells were cultured as neurospheres in DMEM/F12 media (2% B27, 1% Pen/Strep). SMB(GF) cells were generated by culturing parental SMB cells for ≥3 weeks with the above media supplemented with EGF, bFGF (20 ng/mL each), 0.2% Heparin.
Cell Survival Assays
SMB cells in 96-well plates (3 × 104 cells/well) were incubated for 72 hrs in LDE225, Vismodegib, LEQ506 or ATO, or for 120 hrs in BKM120, BEZ235, PD325901 or CI-1040. Viability was measured using CellTiter 96 Aqueous One Solution (Promega), and calculated as percentage of control (DMSO-treated).
Gene Copy Number Analysis
Genomic DNA was extracted with DNeasy Blood and Tissue kit (Qiagen). Genomic copy number for Sufu was determined by qPCR with custom-designed primers using 5 ng of genomic DNA/reaction. Copy number was calculated as described in supplemental information.
Immunohistochemistry, Immunocytochemistry, and Immunoblotting
Human medulloblastoma and matched metastases were stained with hematoxylin and eosin (H&E), or with anti- pERK1/2 (Cell Signaling; 1:400), visualized using Envision Plus Detection kit (DAKO). Human skin tumors were immunostained with: anti-Keratin14(ab7800 Abcam); anti-Gli1 (C-18 Santa Cruz); anti-pERK (#9101 Cell Signaling. Immunoreactivity was visualized with Alexa-Fluor secondary antibodies and confocal microscopy (Leica SP8). Staining Antibodies: Ki67 (Leica Microsystems, 1:400), Nestin (Abcam, 1:400), Tuj1 (Covance, 1:400), GFP (Aves Labs, 1:1000), and Zic (made in house, 1:400) (15). Immunoblot antibodies: pAKT (S473), AKT, pERK1/2 (T202/Y204), ERK1/2, pS6, S6, pan-Ras, Gli1, Sufu, p53, cleaved Caspase-3, Nmyc, Flag tag (Cell Signaling, 1:1000), Actin (Sigma, 1:10,000), HA-tag (Millipore, 1:1000), Gli2 (Aviva, 1:1000), c-MYC (Santa Cruz, 1:1000), V5-tag (Invitrogen, 1:1000).
Transplantation and in vivo Treatment
5 × 106 cells in 100 μL were injected subcutaneously in flank of nu/nu mice (6–8 weeks old). Tumor volumes (V=0.5 × A × B2) were measured twice/week. When tumors reached 150 mm3, animals were randomly grouped for treatment with vehicle or LDE225 (diphosphate salt in 0.5% methylcellulose, 0.5% Tween 80, at 80 mg /kg by oral gavage once daily). Mice with tumors ≥2,000 mm3 were euthanized. For orthotopic tumors, 1 × 106 cells in 2 μL were injected into cerebella of nu/nu mice (6–8 weeks old). Animals were sacrificed when symptomatic.
Skin Tumor Sequencing
Sequencing of clinical samples was performed under IRB-approval at Stanford University. Medically qualified patients 18 years or older with advanced BCCs were enrolled and informed consent was obtained for tumor sequencing (protocol #18325). Tissue samples were stored in RNALater at −20°C (Ambion). DNA was isolated using DNeasy Blood & Tissue kit. Capture libraries were constructed from 2 μg DNA from BCC and normal skin using Agilent SureSelect XT Human All Exon V4 kit. Enriched exome libraries were multiplexed and sequenced on Illumina HiSeq 2500 platform to generate 100-bp paired-end reads. Sequencing reads were aligned to human reference genome sequence (hg19) using Burrows-Wheeler Aligner (BWA). SAM to BAM conversion and marking of PCR duplicates were performed using Picard tools (version 1.86), followed by local realignment around indels and base quality score recalibration using the Genome Analysis Toolkit (GATK) (v2.3.9). Mean target coverage was 114X over coding regions. Somatic SNVs and indels were called using GATK. Variants were annotated for standard quality metrics and for presence in dbSNP138.
ACCESSION NUMBERS
Microarray data accession number GSE69359.
RESULTS
SMB cell lines maintain features of Shh-subtype MB
Studies of Shh-dependent tumors have been limited by the lack of appropriate tumor cell lines. To establish new Shh pathway-dependent tumor cell lines, we propagated tumor cells from spontaneous MB of Ptch+/− mice as neurospheres (2). To retain Shh pathway-dependency, we omitted EGF and bFGF from the media as these growth factors can promote differentiation of granule cell precursors (GCPs), cells of origin for Shh-subtype MB (16, 17). Several tumor samples tested (7 of 35, or 20%) grew for many passages and were designated Shh-subtype MB (SMB) lines. The three most stable lines, SMB21, SMB55 and SMB56, were used in this study.
SMB lines are tumorigenic and retain key characteristics of in vivo Shh-subtype MB. SMB cells are small (~5 μm in diameter) and exhibit a morphology similar to cerebellar GCPs. These tumor-derived cells express MB markers, including stem cell/progenitor marker Nestin, neuronal marker Tuj1, cerebellar marker Zic1, cell proliferation marker Ki67, and Math1(Atoh1), a hallmark of Shh-subtype MB (Fig. 1A and B). Importantly, SMB cells exhibit constitutively activated Shh signaling, as Shh signaling components and target genes Gli1, Gli2, Boc, Ccnd1, Nmyc and SFRP1, are highly expressed in SMB cells and in primary MB from Ptch+/− mice (Fig. 1B). To compare SMB cells to human MB, we performed gene expression analysis of SMB cells, in vivo primary MB, and cerebellum of P6 and adult mice. We compared these profiles to signature profiles of human MB (WNT, SHH, Group C, Group D). (18). Similarity to each subtype for each sample is defined by the “signature score”, which quantitatively measures similarity of gene expression patterns to predefined signature genes. Subtype signature score indicates that SMB cells and in vivo primary MB both closely resembled Shh-subtype MB (Fig. 1C). We utilized a second algorithm, agreement of differential expression (AGDEX) (19), to compare SMB cells to human MB. AGDEX analysis also indicates that both Ptch+/− primary MB and SMB cells exhibit the highest agreement with human SHH subtype (Supplementary Fig. S1F). Notably, activated Shh signaling in SMB cells is exquisitely sensitive to Smo inhibitors, as demonstrated by reduced Gli1 mRNA and protein following treatment with Smo inhibitor LDE225 (Fig. 1D, E). Importantly, proliferation and survival of SMB cells also depend on active Shh signaling as demonstrated in cell survival assays with three different Smo inhibitors LDE225, Vismodegib, and LEQ506 (20). These inhibitors reduce cell number in all SMB lines, and increase apoptosis as assessed by activated Caspase-3 (Fig. 1E, F; Supplementary Fig. S1A).
Figure 1. SMB cells maintain key features of Shh-subtype MB.
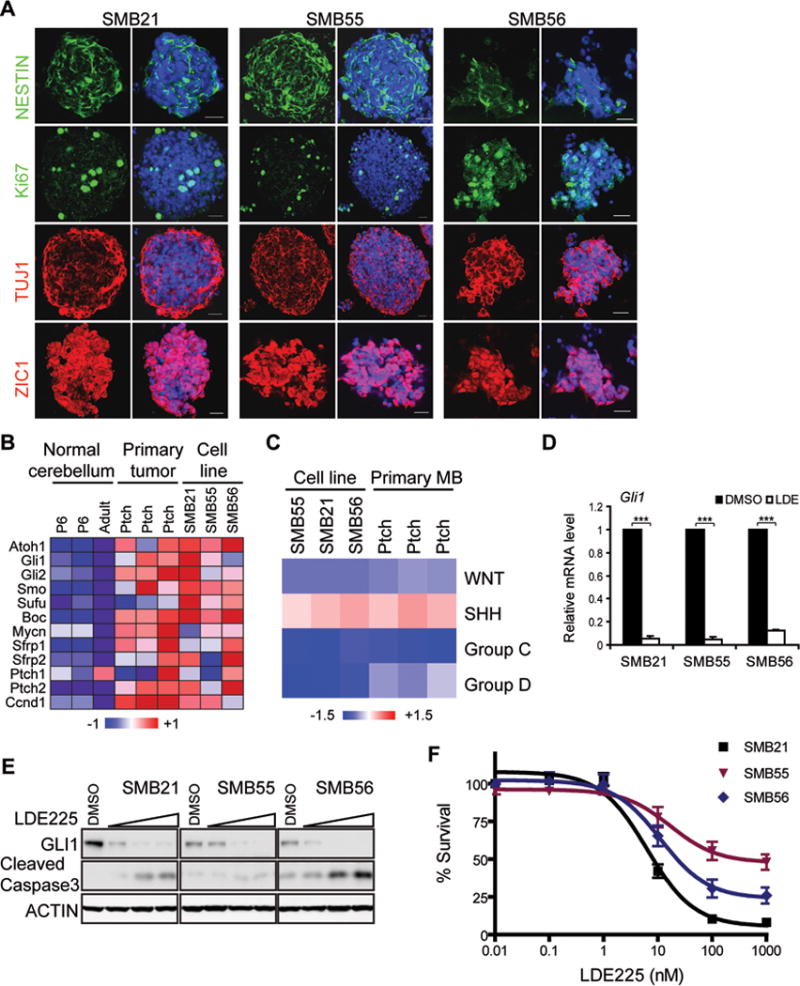
(A) Immunostaining of SMB cells with anti-Nestin, Ki67, Tuj1 Zic1. DAPI in blue. Scale bar: 20 μm.
(B) Microarray analysis reveals Shh-subtype signature in SMB lines and MB from Ptch+/− mice, compared to adult and P6 cerebellum.
(C) Expression of SMB cells, in vivo primary MB, P6 and adult cerebellum, analyzed using signature profiles of human MBs (WNT, SHH, Group C, Group D). Similarity to each subtype for each sample defined by “signature score” and normalized by normal mouse samples.
(D) Quantitative RT-PCR (qRT-PCR) analysis of Gli1 in SMB cells treated with DMSO or 1 μM LDE225 for 24 hrs. mean ± s.d., n = 3, *** p < 0.0001, Student’s t-test.
(E) Dose dependent inhibition of Shh signaling by LDE225 (0, 5, 50, 500 nM, 48 hrs). Shh signaling assessed by immunoblot for Gli1; Apoptosis assessed by cleaved Caspase3.
(F) Survival analysis of SMB cells treated with indicated LDE225 concentrations (72 hrs) (mean ± s.e.m., n = 6).
SMB cells, even after more than 20 passages in culture, initiate tumors in vivo when transplanted into nude mice, either subcutaneously or as orthotopic xenografts in the brain (Supplementary Fig. S1B). Transplanted SMB cells exhibit typical Shh-subtype histology (Supplementary Fig. S1C–E). Recent sequencing studies of large cohorts of MB patients revealed that p53 is among the most frequently mutated genes in Shh-subtype MB (10–20%) (21, 22). To evaluate p53 status in our SMB cells, we first sequenced coding exons of p53. Y233C and C138R point mutations were detected in SMB21 and SMB55 cells, respectively, but no mutations were detected in SMB56 cells. However other alterations can impinge on p53 activity. All three SMB lines showed elevated levels of p53 protein compared to wild-type murine neural progenitor cells, and p53 expression in SMB cells did not change in response to gamma irradiation (Supplementary Fig. S1G), indicating that the p53 signaling axis is dysregulated in all SMB lines. Together, these data indicate that our culturing protocol can establish Shh-subtype cell lines with dysregulated p53, and these lines can be used to study Shh-subtype MB in vitro and in vivo and provide a platform for rapid, large-scale functional study and drug screening.
SMB cell lines provide a model to study drug resistance
A major concern with Smo inhibitors or other targeted therapies is the emergence of drug resistance. To determine whether SMB cell lines can help address this challenge, we asked if the effects of known resistance mechanisms, such as Smo mutants (D477G, L225R, S391N) and Gli2 overexpression, can be recapitulated in SMB cells. To achieve stable expression of exogenous genes, we adapted the piggyBac transposon system to integrate exogenous genes into the SMB genome (Supplementary Fig. S2A). SMB21 cells stably expressing GFP, Smo(WT), Smo(D477G), Smo(L225R), Smo(S391N), Gli2, or Gli2ΔN, constitutively active Gli2 lacking the amino-terminal repressor domain (23), were tested for sensitivity to LDE225 in a cell survival assay. While cells expressing GFP or Smo(WT) remain sensitive to Smo inhibitors LDE225, LEQ506 or Vismodegib, cells expressing Smo mutants, Gli2, or Gli2ΔN are resistant to Smo inhibitors (Fig. 2A; Supplementary Fig. S2B–D). Consistent with previous studies, cells expressing Smo mutants or Gli2ΔN exhibit constitutively activated Shh signaling even in the presence of LDE225, as demonstrated by Gli1 expression (Fig. 2B).
Figure 2. SMB cells provide a model to study resistance to Smo inhibition.

(A) Survival analysis of SMB21 cells expressing GFP (control), Smo mutants, or Gli2ΔN treated with indicated LDE225 concentrations (72 hrs) (mean ± s.e.m., n = 4)
(B) Shh signaling was analyzed by immunoblot for Gli1 in SMB21 cells expressing GFP, HA-tagged wild-type or mutant Smo, treated with DMSO or 1 μM LDE225 for 24 hrs.
(C) SMB21 or SMB21 cells expressing Smo(WT) retain LDE225 responsiveness in vivo; SMB21 cells expressing Smo(D477G) or Gli2ΔN initiate resistant tumors. Tumor volume over time. Mean ± s.e.m., n = 5
To test resistance of engineered SMB cells in vivo, SMB21 parental, Smo(WT), Smo(D477G) and Gli2ΔN cells were subcutaneously transplanted into nude mice. While SMB21 parental and Smo(WT)-expressing cells form tumors responsive to LDE225 in vivo, cells expressing Smo(D477G) or Gli2ΔN generate tumors resistant to LDE225 (Fig. 2C). Together, these results indicate that mutations that confer clinical resistance to LDE225 are effective in SMB cells, demonstrating that these lines provide attractive systems for studying drug resistance.
Identification of novel routes for circumventing Smo inhibition
To identify novel routes through which medulloblastoma cells can escape Smo inhibition, we used SMB cells to test three groups of candidates. The first group includes key Shh pathway components downstream of Smo: Sufu, Gli1, Gli3, and Nmyc. We used the piggyBac transposon system to overexpress Gli1, Gli3 or Nmyc, or used shRNA to knockdown Sufu. Unlike Gli2, expression of Gli1, Gli3 or Nmyc in SMB cells cannot bypass Smo inhibition, reactivate Shh signaling, or promote proliferation and survival in cells treated with LDE225 (Supplementary Fig. S2D, F). In contrast, shRNA knockdown of Sufu reactivates Shh signaling and confers robust resistance to LDE225 in SMB cells (Fig. 3A, B). Consistent with the key role of Sufu, several of the resistant tumors that arose spontaneously from subcutaneously implanted SMB cells following treatment with LDE225 showed drastic reduction of Sufu protein levels compared to sensitive tumors not exposed to LDE225 (Fig. 3C), and many exhibited genomic loss of Sufu (Fig. 3D). To identify possible treatment for tumors that are resistant due to Sufu loss, we tested arsenic trioxide (ATO), a FDA-approved drug shown to antagonize Gli action (24). ATO inhibits proliferation of SMB cells and Sufu knockdown cells with an effective dose similar to previous studies (Fig. 3E) (24). Together these data indicate that Gli inhibitors can treat intrinsic Shh pathway activation.
Figure 3. Loss of Sufu confers resistance to Smo inhibition.
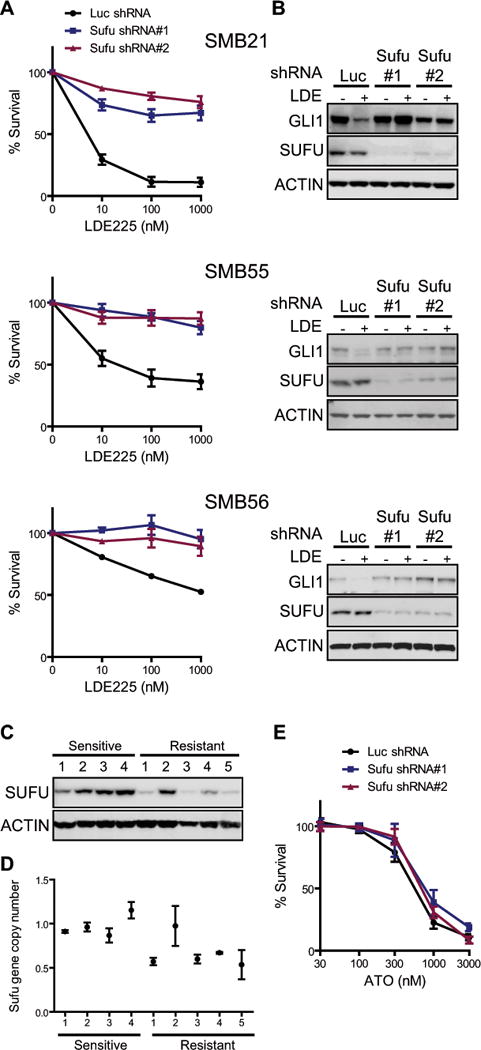
(A) Relative survival for SMB21, SMB55, SMB56 with shRNA knockdown of Sufu treated with indicated LDE225 concentrations (72 hrs) (mean ± s.e.m., n = 3).
(B) ShRNA knockdown of Sufu causes constitutive activation of Shh signaling. DMSO or 1 μM LDE225 for 24 hrs. Immunoblot for Gli1.
(C) Subcutaneously grafted SMB21 cells initially responded to LDE225, but developed resistance after 40 days. Sufu immunoblot of vehicle (n = 4) and LDE225 (n = 5) treated tumors showed drastically reduced Sufu protein in resistant tumors (#1,3,4,5).
(D) Genomic copy number of Sufu in sensitive and resistant tumors determined by quantitative PCR.
(E) Relative survival for SMB21 and Sufu knockdown cells treated with ATO (72 hrs) (mean ± s.e.m., n = 3).
In addition to Shh pathway-specific components, we tested molecules that are key drivers of WNT and Group 3 (MYC) MB subtypes (25). Neither expression of wild-type nor constitutively activated form of CTNNB1, CTNNB1(S33Y), nor overexpression of stabilized MYC, MYC(T58A), confer resistance to Smo inhibitors (Supplementary Fig. S2F–I). Thus, oncogenic mutations critical for other MB subtypes cannot alter subtype identity and thereby confer resistance. Recent studies indicate that WNT and Shh-subtype MB have distinct cellular origins, as Shh-subtype MB originate from GCPs and WNT subtype from dorsal brainstem cells (26). Thus the cellular context may explain why signaling pathways driving other MB subtypes fail to confer Smo-inhibitor resistance.
The third group of candidates tested encompassed genes involved in RTK/RAS signaling, as RTK/RAS signaling is implicated in development of normal cerebellar GCPs and MB (27, 28). Expression of HRAS(G12V) or BRAF(V600E), but not PI3KCA(H1047R) or AKT(Myristoylated), induced resistance to LDE225 in SMB cells (Fig. 4A–C; Supplementary Fig. S3A, C). As expected, both HRAS(G12V) and BRAF(V600E) activate the MAPK pathway in SMB cells, as demonstrated by increased phosphorylation of ERK (Fig. 4A’; Supplementary Fig. S3B, D). HRAS(G12V) and BRAF(V600E) also cause resistance to other Smo inhibitors LEQ506 and Vismodegib, indicating that this effect is generalizable for Smo antagonists (Supplementary Fig. S3E, F). SMB(HRAS) cells subcutaneously transplanted in nude mice were resistant to treatment with Smo inhibitors (Fig. 4D). Furthermore, MAPK activation is greater in SMB tumors that spontaneously develop resistance to Smo inhibitors following treatment with LDE225 than in vehicle treated, sensitive tumors (Fig. 4E). Taken together these data indicate that activation of RAS/MAPK provides a novel way for cells to evade Smo inhibition.
Figure 4. RAS/MAPK signaling confers resistance to Smo inhibitor.
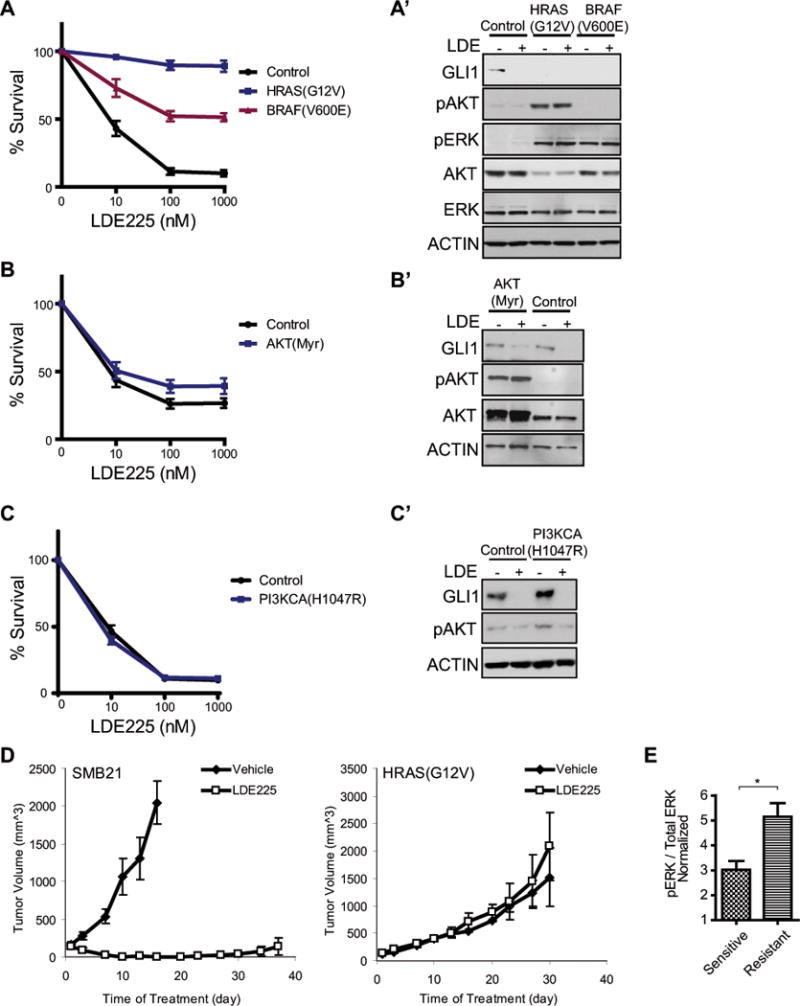
(A–C) Relative survival for SMB21 cells expressing candidate genes treated with LDE225 (72 hrs). HRAS(G12V), BRAF(V600E), not PIK3CA(H1047R) or myristoylated AKT, confer resistance. (mean ± s.e.m., n > 5).
(A’) Elevated phospho-Erk in SMB21 cells expressing HRAS(G12V) or BRAF(V600E).
(B’–C’) Elevated phospho-AKT in SMB21 cells expressing PIK3CA(H1047R) or AKT(Myr).
(D) SMB21(HRAS) cells initiate resistant tumors in vivo. Tumor volume over time. Mean ± s.e.m., n = 5. (Experiments were performed concurrently with Fig. 2C, the same SMB21 control shown here and Fig 2C.)
(E) Phospho-Erk in sensitive and resistant tumors from engrafted SMB21 cells mean ± s.e.m, * p < 0.05, unpaired student t-test.
Surprisingly, HRAS(G12V) does not confer resistance by reactivating Shh signaling downstream of Smo (Fig. 5A). Instead, HRAS(G12V) suppresses Shh signaling in SMB cells, as expression of multiple Shh pathway targets and components are down-regulated in SMB(HRAS) cells (Fig. 5A–C; Supplementary Fig. S4A–D). Notably, Math1(Atoh1), a hallmark of Shh-subtype MB that is not a direct target of SHH signaling, is decreased in SMB(HRAS) cells. These results suggest that HRAS renders SMB cells independent of Shh-signaling for growth, and thereby causes resistance.
Figure 5. FGF/RAS/RAF signaling suppresses Shh-signaling.
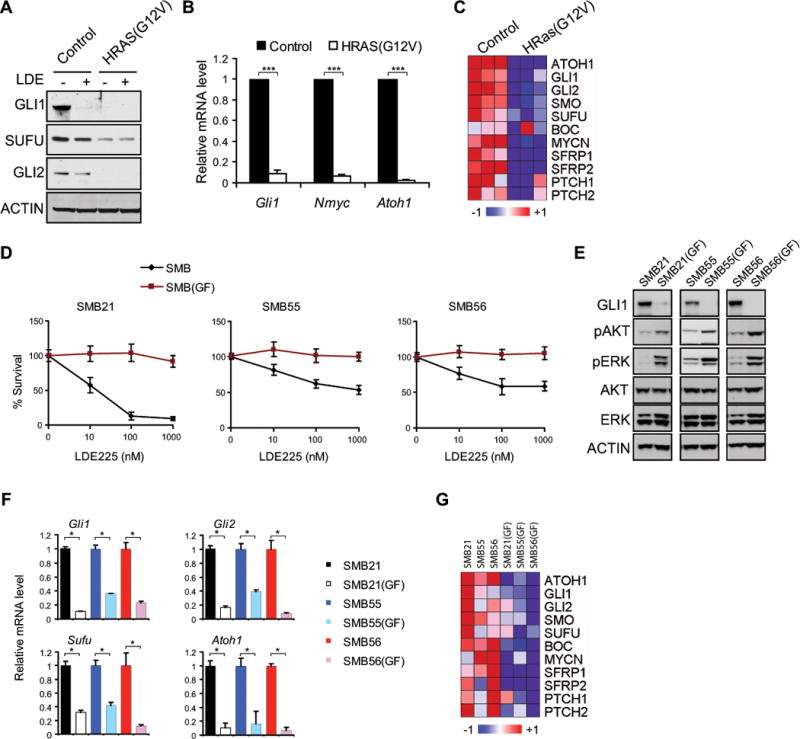
(A–C) Shh-pathway targets and components decrease in SMB(HRAS) cells, by immunoblot, qRT-PCR and microarrays. mean ± s.d., n = 3, *** p < 0.0005, Student’s t-test.
(D) Growth factors (GF) induce Shh-signaling independence in SMB cells. SMB cells cultured with or without GF for 2 weeks, then treated with LDE225 (72 hrs) (Relative survival; mean ± s.e.m., n = 3 ~ 8).
(E) GF treatment activates AKT and MAPK signaling.
(F–G) GF treatment suppresses Shh-pathway targets and components, qRT-PCR and microarray analysis of SMB and SMB(GF) cells Means ± s.d., n = 3, * p < 0.05, Student’s t-test.
RAS is normally regulated by upstream growth factors and receptor tyrosine kinases, and so receptor activation might mimic the effects of HRAS in SMB cells. During normal cerebellar GCP development, growth factors (GF), such as bFGF, antagonize Shh-pathway activity and promote differentiation (29, 30). When SMB cells were exposed to growth factors that are common components of stem cell media (bFGF and EGF; 20 ng/mL) both PI3K-AKT and RAS-RAF-MAPK pathways were activated, Shh signaling was suppressed, and SMB cells became resistant to LDE225 (Fig. 5D–G). We tested the growth factors individually, and found that bFGF, not EGF, suppresses Shh signaling and causes resistance (Supplementary Fig. S4E, F). Together, these results demonstrate that sustained activation of FGF/RAS/RAF signaling enables Shh-subtype MB to grow in a Shh pathway-independent manner.
FGF/RAS-mediated resistance to Smo inhibitors is reversible
To investigate whether FGF/RAS/MAPK signaling is required for SMB cells to both develop and maintain resistance to Smo inhibitors, we removed oncogenic HRAS from SMB(HRAS) cells using lentiviral delivered Flp to cleave FRT sites within the transposon (Supplementary Fig. S5A, B). Removal of RAS decreases Erk phosphorylation and restores Shh signaling activity and susceptibility to Smo inhibitors (Fig. 6A–C). Similarly, cells resistant to Smo inhibitors due to prolonged bFGF treatment regained Shh signaling activity and susceptibility to Smo inhibitors when bFGF was removed from media for prolonged time periods (Fig. 6D, E). Together these data indicate that prolonged activation of FGF/RAS/MAPK signaling both initiates and maintains Shh-signaling independence and resistance to Smo inhibitors.
Figure 6. FGF/RAS-dependent resistance is reversible.
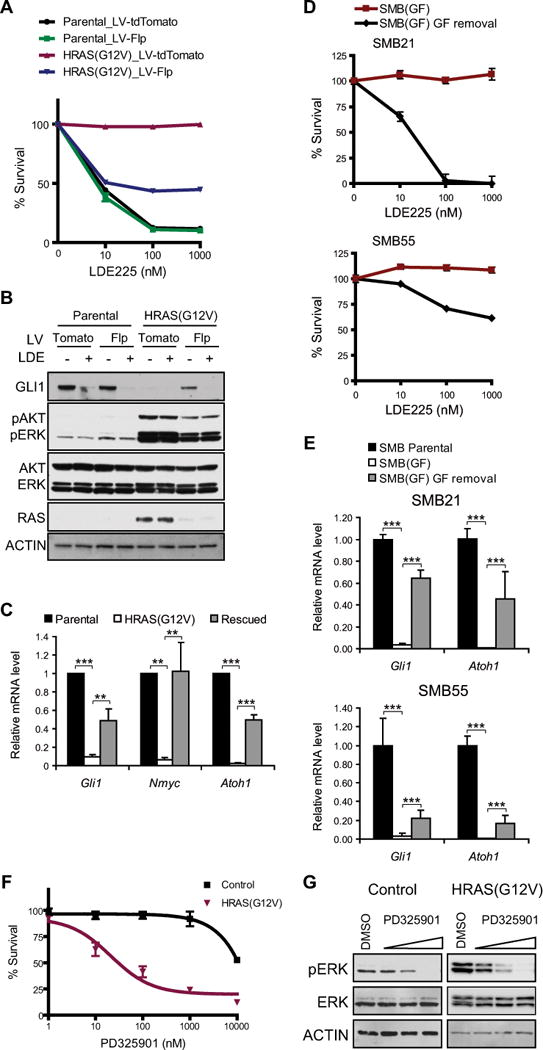
(A) Removal of HRAS(G12V) restores LDE225 sensitivity. SMB(HRAS) cells were infected with lentivirus expressing Flp or tdTomato (control), then assayed by relative survival at indicated LDE225 concentrations (mean ± s.e.m., n = 3).
(B–C) Shh signaling assayed by Immunoblot and qRT-PCR. mean ± s.d., n = 3, ** p < 0.01, *** p < 0.001, one-way ANOVA, Bonferroni correction.
(D–E) SMB21(GF) and SMB55(GF) cells were cultured in media without GF for 3 weeks. (D) Relative survival was then analyzed in indicated LDE225 concentrations (72 hrs). (mean ± s.e.m., n > 9). (E) qRT-PCR analysis of Atoh1, Gli1. mean ± s.d., n = 6, *** p < 0.001, one-way ANOVA with Bonferroni.
(F) Relative survival for SMB21 and SMB21(HRAS) cells treated with MEK inhibitor PD325901 (120 hrs) (mean ± s.e.m., n = 4).
(G) MEK inhibitor PD325901 (0, 10, 100, 1000 nM for 24 hrs) reduce phospho-Erk analyzed by immunoblot.
To identify therapeutic approaches for treating SMB cells resistant to Smo inhibitors, we tested PI3K and MEK pathway inhibitors. While HRAS cells showed similar sensitivity as SMB parental cells to PI3K inhibitors BEZ235 and BKM120, they were much more sensitive to MEK inhibitors CI-1040 and PD325901. Thus MEK inhibitors provide therapeutic treatment for resistant tumors driven by activation of FGF/RAS/RAF signaling (Fig. 6F, G; Supplementary Fig. S5C–E, Fig. S6).
RAS/MAPK activation alters characteristics of Shh pathway-dependent tumors
Morphologically, SMB(HRAS) cells exhibit a dramatically different appearance from SMB parental cells. SMB parental cells are small, with little cytoplasmic material, whereas SMB(HRAS) cells appear larger with extended cellular processes (Supplementary Fig. S7). Immunohistochemical characterization revealed that SMB(HRAS) cells are proliferative, as indicated by Ki67, and poorly differentiated, as indicated by Nestin, a stem cell/progenitor marker (Fig. 7A).
Figure 7. RAS activation alters characteristics of Shh pathway-dependent tumors.
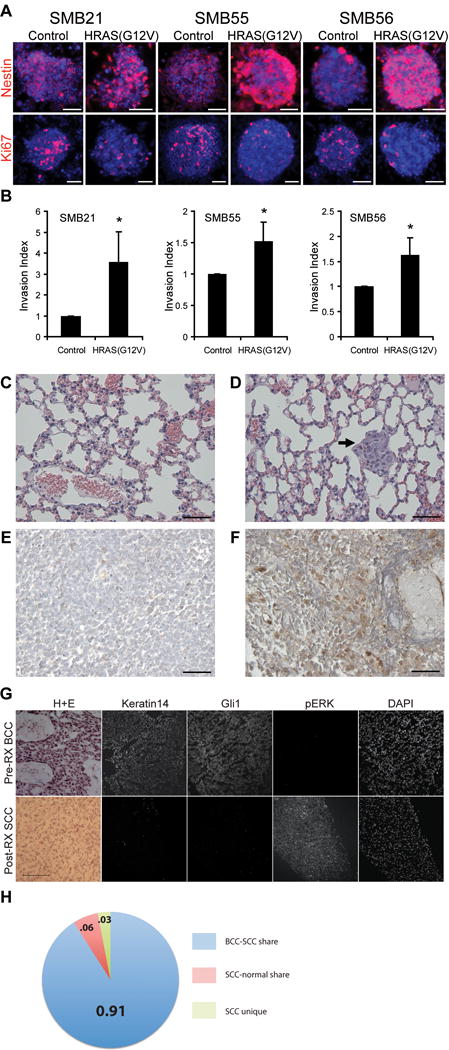
(A) SMB and SMB(HRAS) cells stained with anti- Nestin, Ki67; DAPI in blue. Scale bar: 50 μm.
(B) Cell invasion of SMB(HRAS) or SMB cells assessed using Matrigel-coated transwell apparatus. Invasion measured at 18 hrs, and normalized to SMB cells. (Mean ± s.d., n = 3, * p < 0.05, Student’s t-test)
(C–D) H&E-stained lung sections from animals engrafted with SMB (C) or SMB(HRAS) cells (D). Arrow: metastasis. Metastases detected in 2 of 9 HRAS mice; 0 out of 8 SMB parental mice. Scale bar: 50 μm.
(E–F) Phospho-ERK in primary and metastatic samples from human Shh-subtype MB. Phospho-ERK high in metastatic, frontal lobe tumor (F), but not in matched primary cerebellar tumor (E). Scale bar: 50 μm.
(G) Pre-treatment BCC with characteristic H+E, Keratin14, and Gli1 with an absence of phospho-ERK immunoreactivity. SCC tumor that developed at the same location after Vismodegib displayed a spindle-like morphology characteristic of SCC, and lacked keratin14 and Gli1 immunostaining, but was positive for phospho-ERK. Scale bar: 100 μm.
(H) Sequencing suggests shared lineage for post-treatment resistant SCC and pretreatment BCC. BCC and SCC share most genetic variations (n=1248, ratio=0.91); SCC-normal share (n=84, ratio=0.06); SCC unique (n=43, ratio=0.03).
The striking morphological differences in SMB(HRAS) cells suggests they may be more motile than parental cells. Indeed these cells were more invasive when tested in a matrigel invasion assay (Fig. 7B). Interestingly, when SMB(HRAS) cells were subcutaneously injected in nude mice, they initiated resistant tumors, and also generated lung metastases in 2 of 9 mice. Metastases, were never found in 8 mice injected with SMB cells (Fig. 7C, D). Together, these data indicate that activation of RAS/MAPK increases tumor invasiveness.
Clinical observations from a rare set of matched primary and metastatic lesions from an individual patient provide additional evidence that RAS activation promotes metastasis in MB, as the primary lesion exhibits low level of MAPK activation, whereas the metastatic frontal lobe lesion exhibits robust MAPK activation (Fig. 7E, F). In human primary Shh-subtype medulloblastoma, most areas are predominantly negative for MAPK activation (Supplementary Fig. S8, Table S2); however, in one tumor, cancer cells in the perivascular niche were strikingly positive for ERK phosphorylation (Supplementary Fig. S8D) while in a desmoplastic/nodular MB, ERK phosphorylation was elevated in perinodular regions (Supplementary Fig. S8B). Both tumors exhibited regions with high FGF immunostaining (Supplementary Fig. S8B, D). Cells with MAPK activation within human Shh-subtype medulloblastomas may generate resistant tumors when challenged by Smo inhibitors.
Vismodegib treatment of BCC engenders RAS/MAPK-dependent tumors
Smo inhibitor Vismodegib is approved for clinical use for patients with advanced BCC, but is not yet approved for MB. Among BCC patients, approximately 21% that initially respond subsequently develop tumors resistant to this inhibitor (31). In some cases, post-treatment resistant tumors exhibit characteristics of squamous cell carcinoma (32–36). We analyzed three patients who developed squamous cell carcinoma at the site of the antecedent BCC tumor following treatment with Vismodegib. Post-treatment resistant tumors display low level of Gli1 and high level of phospho-ERK, suggesting upregulated RAS/MAPK and down-regulated Shh signaling (Fig. 7G). To determine whether SCCs that develop following Vismodegib are derived from the antecedent BCC in the same location, we analyzed patient-matched normal tissue or blood, and pre- and post-relapse tumor samples by exome sequencing with germline and dbSNP variants removed. Tumors we assayed initially responded to Vismodegib before acquiring resistance to the inhibitor. Strikingly, 91% of genetic variations (n=1248) in the SCC sample were shared between pre-treatment BCC and post-treatment SCC, while only 3% and 6% of somatic genetic variations (n=43 and n=84) were unique to the SCC or shared with patient-matched normal sample respectively, suggesting that the SCC arose from the BCC (Fig 7H). These results suggest that tumors can evolve from Shh pathway-dependent BCC to RAS/MAPK pathway-dependent SCCs and thereby develop resistance to Smo inhibitors.
DISCUSSION
The studies presented here introduce a set of Shh pathway-dependent MB cell lines (SMB), and identify two distinct mechanisms of therapeutic resistance to Smo inhibitors. Loss of Sufu drives resistance to Smo inhibition by activating downstream Shh signaling. Alternatively, activation of RAS/MAPK signaling, either due to new mutations or to microenvironmental factors, constitutes a novel mechanism of resistance that circumvents Shh pathway dependence in a growing tumor.
SMB cells as a model for Shh-subtype MB
Cell lines that faithfully model the cancer from which they are derived provide important tools for studying disease mechanism and discovering novel therapies. Investigation of MB biology has been limited by the lack of stable lines that are tumorigenic and Shh pathway-dependent. Most established MB lines, are adherent cell cultures maintained in serum-containing media, and do not depend on the Shh pathway for growth and survival (6, 14). Current protocols can only culture freshly isolated tumor cells for a short time before key characteristics of Shh-subtype MB are lost. Here, we present Shh pathway-dependent MB lines (SMB) that can be used as effective and faithful in vitro models to study Shh-subtype MB.
The protocol that enabled development of SMB lines is a modified version of neural stem cell culture methods. Key aspects include growing cells as non-adherent spheres and eliminating serum, EGF and bFGF from media. Generation of tumor spheres is commonly used to enrich for cancer stem-like cells (37). Indeed high grade gliomas can be perpetuated as neurospheres by maintaining cells in neural stem cell media with EGF and bFGF (37, 38). However, these conditions do not maintain tumorigenicity and Shh pathway-dependency of MB cells (39), as FGF signaling has an antagonistic effect on Shh signaling (29, 40, 41). Instead, MB neurospheres from Ptch+/− mice cultured without exogenous EGF or bFGF generate Shh-subtype MB cell lines (SMB) that are tumorigenic, maintain markers of Shh-subtype MB and remain dependent on Shh pathway activity even after many passages in vitro.
A previous study isolated rare lines from medulloblastomas of Ptch+/−;p53−/− mice (42). We observe distinct modes of p53 signaling dysregulation in each SMB line. Because all three lines are derived from Ptch+/−;p53+/+ mice, mutations or inactivation of p53 signaling may have developed during primary MB tumorigenesis in each individual animal. In human MB, the importance of p53 has become increasingly apparent. Among human medulloblastoma, p53 mutations were detected in 13% to 21% of Shh-subtype MBs (21, 22). We conclude that SMB cells offer a faithful model for investigating the Shh pathway in MB, and facilitate high-throughput drug testing and large-scale functional screens. In the future, a similar approach might enable generation of human Shh pathway-dependent MB lines.
Mechanisms of resistance to Smo inhibition
Several Smo inhibitors show promise in preclinical and clinical studies. Vismodegib was the first drug of this class approved by the FDA to treat BCCs. In one study, 6 of 28 BCC patients treated with Vismodegib developed resistance to Smo inhibitors (31). Here we demonstrate that Sufu mutation can occur after treatment with Smo inhibitors, and cause secondary resistance by activating Shh signaling downstream of Smo. As loss of Sufu generates medulloblastomas that never respond to Smo inhibition (21, 43), this finding provides proof of principle that clinically relevant resistance mechanisms can be studied in SMB cells.
Data that overexpression of Gli1 or Nmyc does not confer resistance to Smo inhibition may seem surprising. However, Gli2 is the primary activator of the Shh signaling pathway in GCP development and MB, whereas Gli1 is not essential (44, 45). In preclinical and clinical settings, amplification and constitutive activation of Gli2 generates Shh-subtype MB resistant to Smo inhibition (5, 12, 21). In a recent study of human Shh subtype MB (n=133), 10 cases of Gli2 amplification were identified, but no cases of Gli1 amplification were seen (21). Therefore, our results with overexpression of Gli transcription factors are consistent with clinical observations. In contrast, Nmyc amplification is reported in Shh-subtype MB that do not respond to Smo inhibition (21). While we cannot exclude the possibility that higher expression achieved by other means might confer resistance, our results suggest that other genetic changes may be needed in conjunction with Nmyc for tumors to grow in the presence of Smo inhibitors.
An important finding of this study is identification of a Shh pathway-independent mechanism of resistance. RAS-mediated resistance involves shifting oncogenic addiction to a second oncogenic pathway. De novo mutations or a microenvironment with abundant growth factor can stimulate RAS/MAPK signaling, eliminate Shh pathway-dependency, and cause resistance in MB. Indeed, in human MB, components of RTK/RAS/MAPK pathway are often overexpressed (27), and epigenetic inactivation of RAS association domain family 1A (RASSF1A) tumor suppressor gene is frequently observed (46, 47). Although de novo RAS/RAF mutations have not been detected in primary human MB, such mutations might be favored following treatment with Smo inhibitors (28), as mutations that confer resistance are often only detected following treatment with targeted therapies (11, 48). Strikingly in our studies, xenografts with spontaneous resistance to Smo inhibitors display activation of ERK signaling in vivo. Thus, our data indicate that growth factor stimulation, genetic or epigenetic changes affecting the RAS/RAF/MEK pathway should be assessed in patients that develop resistance to Smo inhibitors.
In addition to intrinsic mutations in tumor cells, tumor microenvironment may alter drug efficacy (49). Our study suggests that microenvironments with abundant FGF could provide protective niches for cells exposed to Smo inhibitors. We show that in human MB, ERK activation occurs in locations adjacent to blood vessels or in perinodular spaces. Recent work suggested that stromal production of placental growth factor (PlGF) in human MB promotes cancer cell survival by activating the MAPK cascade (50). Thus paracrine PlGF-mediated RAS/MAPK signaling could also attenuate efficacy of Smo inhibitors.
Cross-talk between Shh and FGFR/RAS signaling has been widely recognized during organogenesis in multiple tissues (51, 52). Depending on biological context, interactions between FGF/RAS and Shh pathways can be synergistic or antagonistic. In cerebellar GCPs and Ptch+/− MB cells, acute bFGF treatment suppresses Shh signaling and proliferation, and concomitantly promotes cell differentiation (29, 41). Similarly, oncogenic RAS can block Shh signaling in NIH3T3 cells and pancreatic cancer models (53). Here we again observe an antagonistic relationship between Shh and FGF/RAS signaling in SMB cells. Importantly however, this process does not promote terminal differentiation; instead tumor cells remain proliferative and tumorigenic.
RAS/MAPK signaling in metastasis and tumor evolution
Strikingly, RAS/MAPK activation alters multiple characteristics of Shh-dependent tumors. Morphologic and transcriptional profiles of SMB(HRAS) cells differ from SMB cells; while SMB cells are small with classic MB histology, SMB(HRAS) cells display an extended morphology, are more invasive in vitro and more metastatic in vivo. Comparison of Shh-subtype primary MB and matched metastatic lesions from the same person, reveal high level of phosphorylated ERK1/2 in metastases. Consistent with our findings, ectopic expression of Eras (embryonic stem cell-expressed Ras), which is structurally similar to RAS oncoprotein (54), increases leptomeningeal metastases in models of Shh-subtype MB, and these metastatic cells differ genetically and epigenetically from primary tumor cells (55, 56). Together, these data indicate that RAS activation results in resistance to Smo inhibitors and alters tumor characteristics.
Clinical studies of resistant BCC also suggest a role of RAS/MAPK in tumor evolution. Several studies have reported occurrences of squamous cell carcinomas (SCC) during treatment of BCC with Vismodegib (32–36). Sequencing of matched pre- and post-treatment skin tumor samples supports the hypothesis that BCC tumors activate RTK/RAS/MAPK signaling and generate SCCs under selective pressure by Smo inhibition (57). Thus, our findings indicate a novel scenario of resistance by which Shh pathway-dependent tumor cells evolve and escape Shh signaling dependence. Future studies are required to assess the prevalence of this resistance mechanism in patients.
Supplementary Material
Acknowledgments
Studies supported by grants from NIH (CA142536 to RAS, ARO4786 and 5ARO54780 to AEO, HG4069 supports WL), Novartis Pharmaceuticals, the Emerald Foundation, Alex’s Lemonade Stand Foundation, Autism Speaks Foundation and American Brain Tumor Association.
Footnotes
COMPETING FINANCIAL INTERESTS
JFK is a Novartis employee. AEO and AC are clinical investigators for Genentech and Novartis.
AUTHOR CONTRIBUTIONS
X.Z., T.P., K.J.O., P.Z., E.P., S.K.D., R.A.S. designed studies, performed experiments.
J.A.C., J.F.K. provided human MB data.
S.X.A., R.W., A.C., J.L., A.E.O. provided data for SCC/BCC.
X.Z., T.P., E.P., R.A.S. analyzed data, wrote the paper.
W.L. analyzed microarrays.
References
- 1.Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006;5:1026–33. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 2.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–13. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 3.Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–51. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 4.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2:51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell. 2004;6:229–40. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 7.Lee MJ, Hatton BA, Villavicencio EH, Khanna PC, Friedman SD, Ditzler S, et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc Natl Acad Sci U S A. 2012;109:7859–64. doi: 10.1073/pnas.1114718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metcalfe C, de Sauvage FJ. Hedgehog fights back: mechanisms of acquired resistance against Smoothened antagonists. Cancer Res. 2011;71:5057–61. doi: 10.1158/0008-5472.CAN-11-0923. [DOI] [PubMed] [Google Scholar]
- 9.Gajjar A, Stewart CF, Ellison DW, Kaste S, Kun LE, Packer RJ, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res. 2013;19:6305–12. doi: 10.1158/1078-0432.CCR-13-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bucheit AD, Davies MA. Emerging insights into resistance to BRAF inhibitors in melanoma. Biochemical pharmacology. 2014;87:381–9. doi: 10.1016/j.bcp.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 11.Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–4. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71:435–44. doi: 10.1158/0008-5472.CAN-10-2876. [DOI] [PubMed] [Google Scholar]
- 13.Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature. 2013;494:484–8. doi: 10.1038/nature11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, et al. Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res. 2006;66:4215–22. doi: 10.1158/0008-5472.CAN-05-4505. [DOI] [PubMed] [Google Scholar]
- 15.Borghesani PR, Peyrin JM, Klein R, Rubin J, Carter AR, Schwartz PM, et al. BDNF stimulates migration of cerebellar granule cells. Development. 2002;129:1435–42. doi: 10.1242/dev.129.6.1435. [DOI] [PubMed] [Google Scholar]
- 16.Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–34. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–45. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408–14. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poschl J, Stark S, Neumann P, Grobner S, Kawauchi D, Jones DT, et al. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol. 2014;128:123–36. doi: 10.1007/s00401-014-1297-8. [DOI] [PubMed] [Google Scholar]
- 20.Peukert S, He F, Dai M, Zhang R, Sun Y, Miller-Moslin K, et al. Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem. 2013;8:1261–5. doi: 10.1002/cmdc.201300217. [DOI] [PubMed] [Google Scholar]
- 21.Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31:2927–35. doi: 10.1200/JCO.2012.48.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006;20:3161–73. doi: 10.1101/gad.1470806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23:23–34. doi: 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–72. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–9. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacDonald TJ, Brown KM, LaFleur B, Peterson K, Lawlor C, Chen Y, et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet. 2001;29:143–52. doi: 10.1038/ng731. [DOI] [PubMed] [Google Scholar]
- 28.Gilbertson RJ, Langdon JA, Hollander A, Hernan R, Hogg TL, Gajjar A, et al. Mutational analysis of PDGFR-RAS/MAPK pathway activation in childhood medulloblastoma. Eur J Cancer. 2006;42:646–9. doi: 10.1016/j.ejca.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 29.Fogarty MP, Emmenegger BA, Grasfeder LL, Oliver TG, Wechsler-Reya RJ. Fibroblast growth factor blocks Sonic hedgehog signaling in neuronal precursors and tumor cells. Proc Natl Acad Sci U S A. 2007;104:2973–8. doi: 10.1073/pnas.0605770104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emmenegger BA, Hwang EI, Moore C, Markant SL, Brun SN, Dutton JW, et al. Distinct roles for fibroblast growth factor signaling in cerebellar development and medulloblastoma. Oncogene. 2012 doi: 10.1038/onc.2012.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced Basal cell carcinoma. Arch Dermatol. 2012;148:1324–5. doi: 10.1001/archdermatol.2012.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aasi S, Silkiss R, Tang JY, Wysong A, Liu A, Epstein E, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242–3. doi: 10.1001/jamadermatol.2013.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gill HS, Moscato EE, Chang AL, Soon S, Silkiss RZ. Vismodegib for periocular and orbital basal cell carcinoma. JAMA Ophthalmol. 2013;131:1591–4. doi: 10.1001/jamaophthalmol.2013.5018. [DOI] [PubMed] [Google Scholar]
- 34.Iarrobino A, Messina JL, Kudchadkar R, Sondak VK. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33–4. doi: 10.1016/j.jaad.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 35.Zhu GA, Sundram U, Chang AL. Two Different Scenarios of Squamous Cell Carcinoma Within Advanced Basal Cell Carcinomas: Cases Illustrating the Importance of Serial Biopsy During Vismodegib Usage. JAMA Dermatol. 2014 doi: 10.1001/jamadermatol.2014.583. [DOI] [PubMed] [Google Scholar]
- 36.Saintes C, Saint-Jean M, Brocard A, Peuvrel L, Renaut JJ, Khammari A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with Vismodegib. Journal of the European Academy of Dermatology and Venereology : JEADV. 2014 doi: 10.1111/jdv.12526. [DOI] [PubMed] [Google Scholar]
- 37.Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene. 2004;23:7267–73. doi: 10.1038/sj.onc.1207946. [DOI] [PubMed] [Google Scholar]
- 38.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 39.Huang X, Ketova T, Litingtung Y, Chiang C. Isolation, enrichment, and maintenance of medulloblastoma stem cells. J Vis Exp. 2010 doi: 10.3791/2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duplan SM, Theoret Y, Kenigsberg RL. Antitumor activity of fibroblast growth factors (FGFs) for medulloblastoma may correlate with FGF receptor expression and tumor variant. Clin Cancer Res. 2002;8:246–57. [PubMed] [Google Scholar]
- 41.Emmenegger BA, Hwang EI, Moore C, Markant SL, Brun SN, Dutton JW, et al. Distinct roles for fibroblast growth factor signaling in cerebellar development and medulloblastoma. Oncogene. 2013 doi: 10.1038/onc.2012.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward RJ, Lee L, Graham K, Satkunendran T, Yoshikawa K, Ling E, et al. Multipotent CD15+ cancer stem cells in patched-1-deficient mouse medulloblastoma. Cancer Res. 2009;69:4682–90. doi: 10.1158/0008-5472.CAN-09-0342. [DOI] [PubMed] [Google Scholar]
- 43.Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31:306–10. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 44.Weiner HL, Bakst R, Hurlbert MS, Ruggiero J, Ahn E, Lee WS, et al. Induction of medulloblastomas in mice by sonic hedgehog, independent of Gli1. Cancer Res. 2002;62:6385–9. [PubMed] [Google Scholar]
- 45.Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–61. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- 46.Lusher ME, Lindsey JC, Latif F, Pearson AD, Ellison DW, Clifford SC. Biallelic epigenetic inactivation of the RASSF1A tumor suppressor gene in medulloblastoma development. Cancer Res. 2002;62:5906–11. [PubMed] [Google Scholar]
- 47.Lindsey JC, Lusher ME, Anderton JA, Bailey S, Gilbertson RJ, Pearson AD, et al. Identification of tumour-specific epigenetic events in medulloblastoma development by hypermethylation profiling. Carcinogenesis. 2004;25:661–8. doi: 10.1093/carcin/bgh055. [DOI] [PubMed] [Google Scholar]
- 48.Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008;18:73–9. doi: 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 49.Sebens S, Schafer H. The tumor stroma as mediator of drug resistance–a potential target to improve cancer therapy? Curr Pharm Biotechnol. 2012;13:2259–72. doi: 10.2174/138920112802501999. [DOI] [PubMed] [Google Scholar]
- 50.Snuderl M, Batista A, Kirkpatrick ND, Ruiz de Almodovar C, Riedemann L, Walsh EC, et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell. 2013;152:1065–76. doi: 10.1016/j.cell.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benazet JD, Zeller R. Vertebrate limb development: moving from classical morphogen gradients to an integrated 4-dimensional patterning system. Cold Spring Harb Perspect Biol. 2009;1:a001339. doi: 10.1101/cshperspect.a001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bertrand N, Dahmane N. Sonic hedgehog signaling in forebrain development and its interactions with pathways that modify its effects. Trends Cell Biol. 2006;16:597–605. doi: 10.1016/j.tcb.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 53.Lauth M, Bergstrom A, Shimokawa T, Tostar U, Jin Q, Fendrich V, et al. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat Struct Mol Biol. 2010;17:718–25. doi: 10.1038/nsmb.1833. [DOI] [PubMed] [Google Scholar]
- 54.Takahashi K, Mitsui K, Yamanaka S. Role of ERas in promoting tumour-like properties in mouse embryonic stem cells. Nature. 2003;423:541–5. doi: 10.1038/nature01646. [DOI] [PubMed] [Google Scholar]
- 55.Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012;482:529–33. doi: 10.1038/nature10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mumert M, Dubuc A, Wu X, Northcott PA, Chin SS, Pedone CA, et al. Functional genomics identifies drivers of medulloblastoma dissemination. Cancer Res. 2012;72:4944–53. doi: 10.1158/0008-5472.CAN-12-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ratushny V, Gober MD, Hick R, Ridky TW, Seykora JT. From keratinocyte to cancer: the pathogenesis and modeling of cutaneous squamous cell carcinoma. J Clin Invest. 2012;122:464–72. doi: 10.1172/JCI57415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.