

Abstract
Although a large number of studies have well documented a key role of toll-like receptor (TLR)4 in atherosclerosis, it remains undetermined if TLR4 antagonist attenuates atherogenesis in mouse model for type 2 diabetes. In this study, we induced type 2 diabetes in low-density lipoprotein receptor-deficient (LDLR−/−) mice by high-fat diet (HFD). At 8 weeks old, 20 mice were fed HFD and 20 mice fed regular chow (RC) for 24 weeks. In the last 10 weeks, half HFD-fed mice and half RC-fed mice were treated with Rhodobacter sphaeroides lipopolysaccharide (Rs-LPS), an established TLR4 antagonist. After the treatment, atherosclerotic lesions in aortas were analyzed. Results showed that the HFD significantly increased bodyweight, glucose, lipids including total cholesterol, triglycerides and free fatty acids, and insulin resistance, indicating that the HFD induced type 2 diabetes in LDLR−/− mice. Results also showed that Rs-LPS had no effect on HFD-increased metabolic parameters in both nondiabetic and diabetic mice. Lipid staining of aortas and histological analysis of cross-sections of aortic roots showed that diabetes increased atherosclerotic lesions, but Rs-LPS attenuated atherogenesis in diabetic mice. Furthermore, immunohistochemical studies showed that Rs-LPS reduced infiltration of monocytes/macrophages and expression of interleukin (IL)-6 and matrix metalloproteinase-9 in atherosclerotic lesions of diabetic mice. Finally, the antagonistic effect of Rs-LPS on TLR4 was demonstrated by our in vitro studies showing that Rs-LPS inhibited IL-6 secretion from macrophages and endothelial cells stimulated by LPS or LPS plus saturated fatty acid palmitate. Taken together, our study demonstrated that TLR4 antagonist was capable of attenuating vascular inflammation and atherogenesis in mice with HFD-induced type 2 diabetes.
Keywords: Toll-like receptor 4, Atherosclerosis, Inflammation, Diabetes
Introduction
Toll-like receptors (TLRs) serve as key host sensors for pathogen-associated molecular patterns (PAMPs) and lipopolysaccharide (LPS) is an important PAMP molecule for TLR4 (Dalpke and Heeg, 2002). Upon binding TLR4, LPS triggers signaling cascades, most notably the nuclear factor kappa B (NFκB) and mitogen-activated protein kinase (MAPK) pathways, and upregulates pro-inflammatory genes such as cytokines and chemokines (Brown, et al., 2011) that contribute to atherosclerosis (Curtiss and Tobias, 2007, den Dekker, et al., 2010). The important role of TLR4 in atherosclerosis is strongly indicated by the studies showing that deficiency of TLR4 or MyD88, an adaptor protein for the TLR4 signaling pathway, in apolipoprotein E-deficient mice is associated with a significant reduction of atherosclerosis (Michelsen, et al., 2004, Bjorkbacka, et al., 2004, Higashimori, et al., 2011).
In addition to atherosclerosis, it has been reported that deficiency of TLR4 also attenuates pro-inflammatory state in mouse with diabetes (Grieco, et al., 2011, Zipris, 2010, Devaraj, et al., 2011). In consistent with this finding, it was reported that monocytes isolated from diabetic patients have increased TLR4 expression and that TLR4 expression is correlated with serum levels of glycosylated hemoglobin and NFκB (Devaraj, et al., 2008). Thus, TLR4 appears to be critically involved in both atherosclerosis and diabetes, which are strongly associated with inflammation (Dandona, et al., 2003, Ray, et al., 2009, Theuma and Fonseca, 2003).
Type 2 diabetes accounts for 90 to 95% of the incidence of diabetes (American Diabetes Association, (2014). Furthermore, the metabolic disorders contributing to atherosclerosis in type 2 diabetes are different from those in type 1 diabetes (Lee, 2014, Zeadin, et al., 2013). Type 1 diabetes is associated with hyperglycemia that plays a crucial role in promoting atherosclerosis while type 2 diabetes is associated with not only hyperglycemia, but also dyslipidemia (Zeadin, et al., 2013). In consistence with the features of human diabetes, the animal models for type 1 and type 2 diabetes also manifest different metabolic disorders. For examples, the mouse model for streptozotocin-induced type 1 diabetes had hyperglycemia, but no significant changes of plasma lipids when compared with the nondiabetic mice (Lu, et al., 2013). In contrast, mouse model with high-fat diet (HFD)-induced type 2 diabetes had not only hyperglycemia, but also dyslipidemia (Lyngdorf, et al., 2003). Therefore, it is important to determine if administration of TLR4 antagonist in animal models with type 2 diabetes is effective to reduce atherosclerosis.
In this study, we employed LDL receptor-deficient (LDLR−/−) mice as an animal model for atherosclerosis and induced type 2 diabetes with HFD. Using this mouse model, we determined the effect of Rhodobacter sphaeroides LPS (Rs-LPS), an established TLR4 antagonist (Aida, et al., 1995, Kirkland, et al., 1991, Hammad, et al., 2009, Hutchinson, et al., 2010, Baker, et al., 1990), on atherosclerosis. We found that Rs-LPS attenuated atherogenesis in mice with HFD-induced type 2 diabetes.
Materials and Methods
Animals, diet and treatments
Forty 8-week old male LDLR−/− mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed at the animal facility of VA Medical Center in Charleston, SC. The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC). All mice were maintained on a 12-hour light-dark cycle in a pathogen-free environment and had ad libitum access to water and food. Half of mice were fed regular chow (RC) (D12450J) and half of mice fed HDF (D12492, Research Diets, Inc. New Brunswick, NJ) to induce type 2 diabetes. The mice were randomly divided into 4 groups: 1. RC-fed mice without Rs-LPS treatment; 2. RC-fed mice with Rs-LPS treatment; 3. HFD-fed mice without Rs-LPS treatment; 4. HFD-fed mice with Rs-LPS treatment. Rs-LPS was purchased from InvivoGen (San Diego, CA). Mice were fed RC or HFD for 24 weeks. During the last 10 weeks of the feeding, the mice in Groups 2 and 4 were treated with Rs-LPS (1 μg/mouse in phosphate-buffered saline (PBS) twice a week, intraperitoneal injection) while the mice in Group 1 and 3 were given equivalent amount of PBS. This dose of Rs-LPS was reported to be effective in blocking TLR4 by the previous studies (Hammad, et al., 2009, Baker, et al., 1990).
Metabolic measurements
Blood samples were obtained under the fasted condition and glucose level was determined using a Precision QID glucometer (MediSense Inc., Bedford, MA). Serum cholesterol and triglycerides were assayed using Cholestech LDX Lipid monitoring System (Fisher Scientific, Pittsburgh PA). Serum fatty acids were determined using the EnzyChrom™ free fatty acid kit (BioAssay systems, Hayward, CA). Serum fasting insulin was assayed using the Ultra Sensitive Insulin ELISA Kit (Crystal Chem, Inc., Downers Grove, IL, USA). Fasting whole-body insulin sensitivity was estimated with the homeostasis model assessment of insulin resistance (HOMA-IR) according to the formula [fasting plasma glucose (mg/dL) × fasting plasma insulin (μU/mL)]/405.
En face analysis
Mice were euthanized and aortas from heart to the iliac arteries were dissected out, soaked for 24 h in 4% paraformaldehyde for fixation, excised longitudinally, and then stained with 0.5% of Sudan IV as described previously (Lloyd, et al., 2011). After staining, the aortas were laid onto the sponge block surface with the intimal surface up and pined down using the Minutien (Fine Science Tools, Inc., San Francisco, CA). The images of the aortas were taken using an EPSON Perfection 2450 photo scanner and analyzed with the Photoshop software as described (Schuyler, et al., 2011).
Histological analysis of atherosclerotic lesions
The tissues of aortic root were embedded in Tissue-Tek® OCT™ compound (EMS, Hatfied, PA), immediately frozen on dry ice and stored at −80°C. Starting from the aortic root, cryosetions with 6 μm thickness were cut and sections with a distance of 480 μm were collected and mounted on slides. Slides were fixed in 10% of formaline for 10 minutes, stained with Harris modified hematoxylin (Sigma-Aldrich, St. Louis, MO) for 10 minutes and then rinsed in deionized water. Staining of hematoxylin was then developed in tap water for 5 minutes and differentiated in 1% acid ethanol and bluing the staining for 50 seconds. Slides were stained with eosin Y solution (Sigma-Aldrich) for 2 minutes. Slides were placed in Coplin jars with 95% ethanol three times for 5 minutes each and the process was then repeated with 100% ethanol. Slides were further dehydrated in 99.5% xylene (Sigma Aldrich) and mounted in Permount mounting media (Fisher Scientific). Photomicrographs of tissue sections were taken using an Olympus BX53 digital microscope with Cellsens digital image software (Olympus American Inc., Center Valley, PA). The area with positive staining was quantified using a computer based morphometry software (Image-Pro Plus 6, Media Cybernetics, Bethesda, MD) as described previously (Patel, et al., 2010). The area of intima was expressed as the percentage of the total aortic area including intima, media, and lumen.
Immunohistochemical analysis of protein expression
Aortic roots were fixed in 4% paraformaldehyde for 10 min and frozen sections were made using a cryostate. Sections were rinsed in 0.01 M PBS, pH 7.4, and then incubated in 0.3% H2O2/methanol for 30 minutes to quench endogenous peroxidase. After being rinsed with 0.01 M PBS, sections were incubated with 3% normal serum in 0.01 M PBS for 1 hour to block nonspecific binding. Sections were then blocked with avidin-biotin solution from the ABC Elite kit (Vector Laboratories, Burlingame, CA) for 30 minutes and incubated with primary antibodies: Rabbit anti-IL-6 antibody (1:300) (Abcam, Cambridge, MA), goat anti-matrix metalloproteinase (MMP)-9 antibody (1:100) (R&D Systems, Minneapolis, MN) or rat anti-CD68 antibody (1:300) (AbD Serotec, Raleigh, NC) overnight at 4°C. Sections were incubated with secondary biotinylated-antibody (1:250) from the ABC Elite kit (Vector Laboratories) for 1 hour and then the ABC reagent (Vector Laboratories) for 30 minutes. Slides were rinsed in 0.01 M PBS and covered with diaminobenzidine peroxidase substrate solution from the Impack DAB kit (Vector Laboratories) for 2 minutes and then rinsed in water. Counterstaining was performed with hematoxylin solution, Gill No. 2 (Sigma-Aldrich). Slides were then dehydrated using increasing concentrations of ethanol and xylenes and mounted. Staining with normal IgG was used as a negative control. Images were taken and analyzed as described above for the histological analysis.
Collagen staining
Collagen staining using Sirius red was performed as described previously (He, et al., 2006). Briefly, after stained nuclei with hematoxylin, the tissue sections were incubated with 1.3 % Sirius red F3BA in saturated picric acid for 1 hour and then rinsed with 0.5% acetic acid solution for 1 minute twice. The sections were then dehydrated and mount in a mounting media. The area with positive staining in atherosclerotic lesions was quantified using software Image-Pro Plus 6 as described above.
Cell culture and treatment
Human monocytes were isolated as described previously from blood obtained from healthy donors (Seager Danciger, et al., 2004). The blood donation for monocyte isolation was approved by the Medical University of South Carolina Institutional Review Board. Human monocytes were differentiated into macrophages by incubation with RPMI 1640 medium (Life Technologies, Grand Island, NY) containing 10% human serum, 1% minimum Eagle’s medium nonessential amino acid solution, and 0.6 g/100 ml HEPES for 7 days. Human aortic endothelial cells (HAECs) (Life Technologies) were grown in Medium 200 supplemented with low serum growth supplement (LSGS) (Life Technologies) containing 2% fetal bovine serum, hydrocortisone, human epidermal growth factor, basic fibroblast growth factor and heparin. The HAEC cultures were 100% confluent before the treatments. For cell treatment, LPS from E. coli and palmitic acid (PA) (Sigma, St. Louis, MO) were used. The LPS was highly purified by phenol extraction and gel filtration chromatography and was cell culture tested. PA used in this study was bovine serum albumin-free as described previously (Schwartz, et al., 2010). To prepare PA, PA was dissolved in 0.1 N NaOH and 70% ethanol at 70°C to make 50 mM. The solution was kept at 55°C for 10 min, mixed, and brought to room temperature.
Enzyme-linked immunosorbent assay
IL-6 in medium was quantified using sandwich enzyme-linked immunosorbent assay (ELISA) kits according to the protocol provided by the manufacturer (Biolegend, San Diego, CA). Mouse serum IL-1β and TNFα were quantified using ELISA kits manufactured by R&D System (Minneapolis, MN).
Statistic analysis
Data were presented as mean ± SD. Student t tests were performed to determine the statistical significance of differences of intimal lesion size and protein expression among different experimental groups. A value of P< 0.05 was considered significant.
Results
Rs-LPS has no effect on glucose and lipid changes induced by high-fat diet
The HFD markedly increased bodyweight, fasting blood glucose, plasma lipids including cholesterol, triglycerides, free fatty acids, insulin and HOMA-IR as compared with feeding with the RC (Fig. 1 and Table 1), indicating that the HFD induced type 2 diabetes in LDLR−/− mice. Furthermore, the metabolic data showed that Rs-LPS had no significant effect on the metabolic parameters in mice fed the HFD or RC (Fig. 1 and Table 1).
Figure 1.
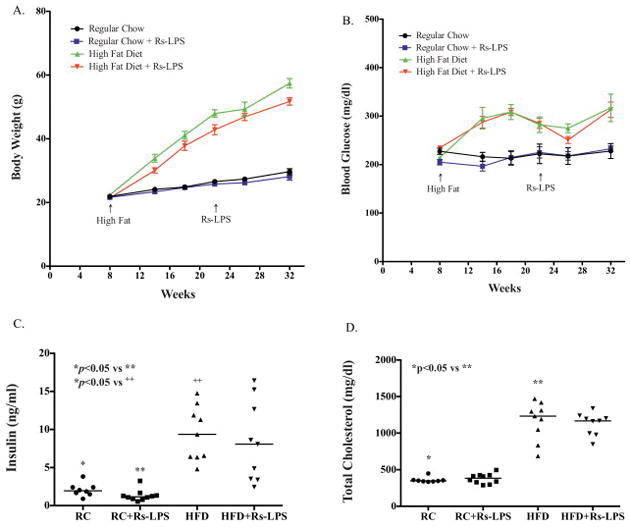
The effects of regular chow (RC) or high-fat diet (HFD) and Rs-LPS on bodyweight, glucose, insulin and cholesterol. A and B: The changes of bodyweight and fasting blood glucose during 24 weeks. C and D: Fasting plasma insulin and total cholesterol levels at the end of the study. The lines reflect the means of all mice in each group.
HOMA-IR: homeostasis model assessment of insulin resistance (see calculation formula in Materials and Methods). Data are mean +/− SD (n=10). **p<0.05 vs *.
Table 1.
The effect of high-fat diet and Rs-LPS on metabolic parameters in LDLR−/− mice
| Nondiabetic mice | Diabetic mice | |||
|---|---|---|---|---|
| Without Rs-LPS | With Rs-LPS | Without Rs-LPS | With Rs-LPS | |
| Weight (g) | 30 ± 3* | 28 ± 3 | 57 ± 4** | 52 ± 4 |
| Glucose (mg/dl) | 228 ± 46* | 233 ± 19 | 317 ± 85** | 313 ± 50 |
| Cholesterol (mg/dl) | 359 ± 37* | 378 ± 67 | 1165 ± 263** | 1125 ± 153 |
| LDL (mg/dl) | 242 ± 44* | 284 ± 84 | 955 ± 204** | 953 ± 150 |
| Triglycerides (mg/dl) | 151 ± 29* | 165 ± 65 | 503 ± 365** | 407 ± 205 |
| Free fatty acids (mg/dl) | 503 ± 98* | 560 ± 142 | 833 ± 372** | 832 ± 367 |
| Insulin (ng/ml) | 2.07 ± 0.86* | 1.32 ± 0.75 | 9.43 ± 3.57** | 8.35 ± 5.32 |
| HOMA-IR | 29.13 ± 2.4* | 18.99 ± 0.9 | 184.53 ± 18.7** | 161.33 ±16.4 |
Rs-LPS inhibits atherosclerosis in diabetic LDLR−/− mice
To determine the effect of Rs-LPS treatment on atherosclerosis, we performed both lipid staining on the en face aortas and HE staining on the cross-sections of aortic roots. As shown in Fig. 2A and 2B, the lipid staining showed that the surface area with positive staining on the en face aortas of diabetic LDLR−/− mice was significantly increased when compared with that of nondiabetic LDLR−/− mice, and the treatment with Rs-LPS significantly reduced the en face lesions in both nondiabetic and diabetic mice. Furthermore, the HE staining showed that the intimal size of atherosclerotic lesions in the cross-sections of aortic roots from diabetic mice was larger than that from nondiabetic mice and Rs-LPS reduced the intimal size in diabetic mice (Fig. 2C and 2D).
Figure 2.
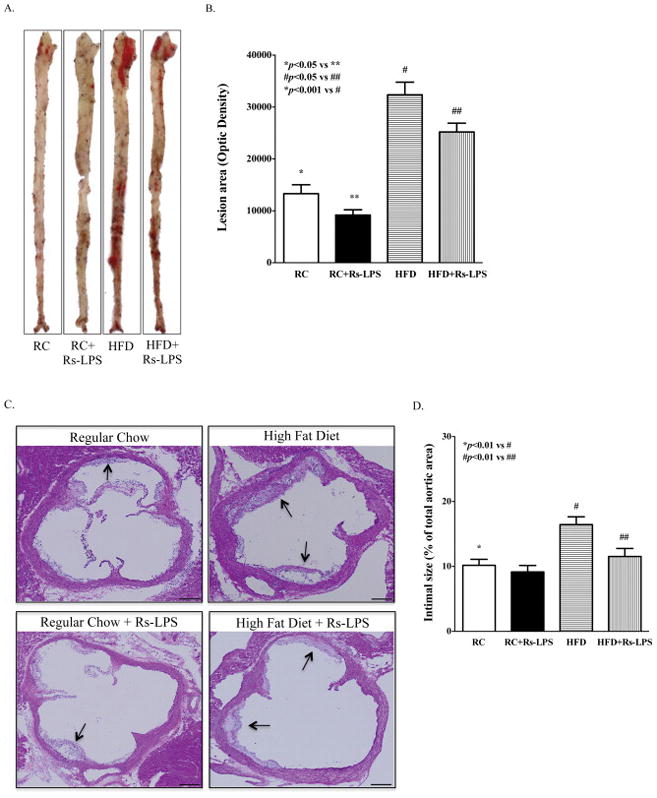
Reduction of atherosclerotic lesions in LDLR−/− mice by Rs-LPS. A: Sudan IV staining of en face aortas. B: Quantification of atherosclerotic lesion area. Data show mean ± SD (n=10). C: Representative images from histological analysis of atherosclerotic lesions in aortic roots. The arrows point to the intimal lesions. D: Quantification of intimal lesion area of atherosclerotic plaques. Data show mean ± SD (n=10). RC: regular chow. HFD: high-fat diet. Bar = 500 μm.
Rs-LPS reduces monocytes and macrophages in atherosclerotic lesions in diabetic LDLR−/− mice
Monocytes and macrophages in atherosclerotic plaques were detected by immunohistochemical staining of CD68, a marker for cells in the macrophage lineage, including monocytes, dendritic cells and histiocytes (Holness and Simmons, 1993). Results showed that diabetic mice had significantly increased content of monocytes and macrophages as compared to nondiabetic mice, but Rs-LPS treatment reduced the content in both nondiabetic and diabetic mice (Fig. 3A and 3B).
Figure 3.
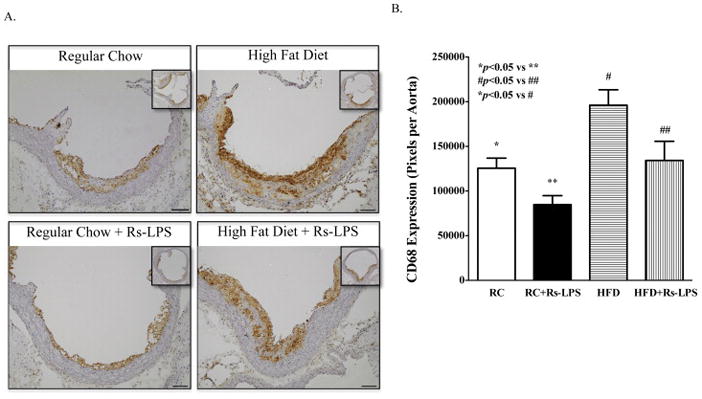
Inhibition by Rs-LPS of monocyte and macrophage accumulation in atherosclerotic lesions of LDLR−/− mice. A: Immunohistochemical staining of CD68. B: Quantification of lesion area with positive CD68 immunohistochemical staining. Data show mean ± SD (n=10). RC: regular chow. HFD: high-fat diet. Bar = 200 μm.
Rs-LPS inhibits IL-6 and MMP-9 expression in atherosclerotic plaques in diabetic LDLR−/− mice
To determine the effect of Rs-LPS on vascular inflammation, we focused on IL-6 and MMP-9 expression since both of them have been shown to play an important role in atherosclerosis (Hamirani, et al., 2008). Results showed that diabetic LDLR−/− mice had significantly increased expression of IL-6 (Fig. 4A and B) and MMP-9 (Fig. 4C and D) in atherosclerotic plaques as compared to nondiabetic mice and Rs-LPS treatment reduced the expression of IL-6 and MMP-9 in both nondiabetic and diabetic LDLR−/− mice.
Figure 4.
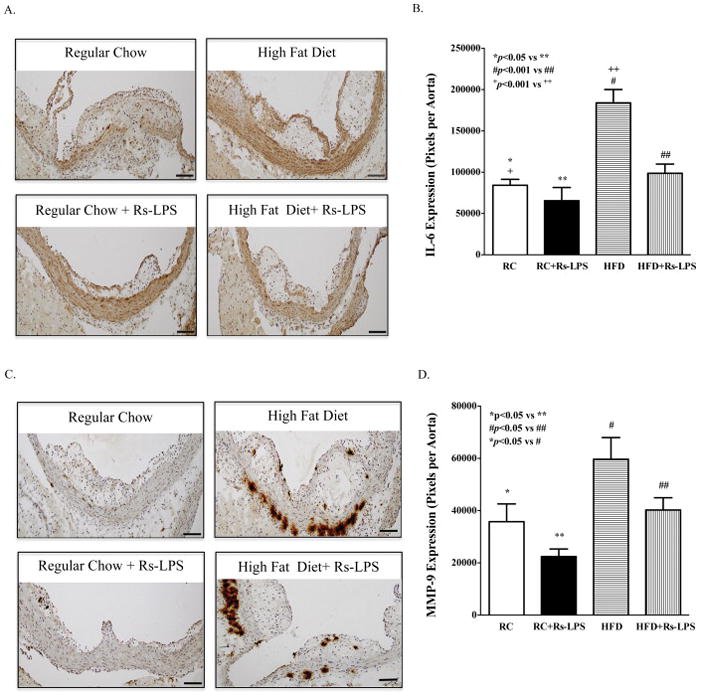
Inhibition of IL-6 and MMP-9 expression in atherosclerotic lesions of LDLR−/− mice by Rs-LPS. A and C: Immunohistochemical staining of IL-6 (A) and MMP-9 (C). B and D: Quantification of IL-6 (B) and MMP-9 protein (D). Data show mean ± SD (n=10). RC: regular chow. HFD: high-fat diet. Bar = 200 μm.
Rs-LPS reduces collagen content in diabetic mice
Smooth muscle cell (SMC) proliferation and SMC-derived collagen accumulation in the intima are important early changes in atherosclerosis (Kunz, 2007). Therefore, we determined the effect of Rs-LPS on collagen content in the intima of atherosclerosis. Results showed that collagen content was markedly increased in atherosclerotic lesions in diabetic mice, but Rs-LPS abolished the stimulatory effect of diabetes on the collagen content (Fig. 5).
Figure 5.
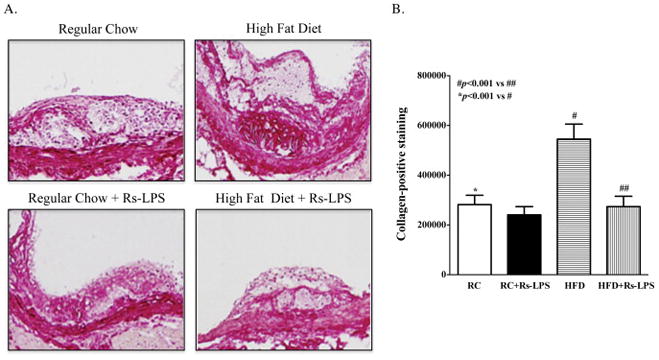
Inhibition of collagen accumulation in atherosclerotic lesions of diabetic LDLR−/− mice by Rs-LPS. A: Sirius red staining of collagen in atherosclerotic lesions. B: Quantification of collagen (pixels per aorta). Data show mean ± SD (n=10). RC: regular chow. HFD: high-fat diet.
Diabetes increases serum inflammatory cytokine levels, but Rs-LPS has no effect
To determine if Rs-LPS has effect on systemic inflammation, we quantified the serum levels of inflammatory cytokines IL-1β and TNFα. Results showed that serum levels of IL-1β (Fig. 6A) and TNFα (Fig. 6B) increased significantly in diabetic mice when compared to those in nondiabetic mice, but Rs-LPS had no effect on the serum levels of IL-1β and TNFα, suggesting that Rs-LPS is effective in controlling inflammation in inflamed tissues such as atherosclerotic plaques, but has no effect on the serum cytokine levels.
Figure 6.
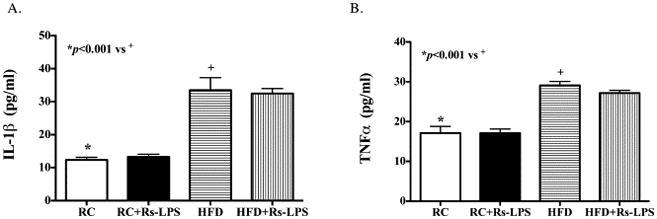
Diabetes, but not Rs-LPS, changes serum inflammatory cytokine levels. The serum levels of IL-1β (A) and TNFα (B) in mice of 4 groups were quantified using ELISA. RC: regular chow. HFD: high-fat diet.
Rs-LPS inhibits IL-6 secretion by macrophages and vascular endothelial cells
The above findings indicate that Rs-LPS inhibited atherosclerosis by suppressing vascular inflammation in mouse with type 2 diabetes. To further validate the antagonistic effect of Rs-LPS on TLR4-mediated proinflammatory cytokine expression, we employed human monocyte-derived macrophages and vascular endothelial cells. Since we have shown previously that LPS and PA, the most abundant saturated fatty acid, synergistically stimulate IL-6 expression in macrophages (Jin, et al., 2013), we treated macrophages with LPS, PA, or LPS plus PA in the absence or presence of Rs-LPS. Results (Fig. 7A) showed that while PA had no effect on IL-6 secretion from macrophages, LPS strongly induced it and the combination of LPS and PA further stimulated it. Interestingly, Rs-LPS attenuated the stimulatory effect of LPS or LPS plus PA on IL-6 secretion. We also assessed the effect of Rs-LPS on IL-6 secretion from vascular endothelial cells. Results (Fig. 7B) showed that either LPS or PA stimulated IL-6 secretion and the combination of LPS and PA synergistically increased IL-6 secretion. Similar as the finding in macrophages, Rs-LPS inhibited IL-6 secretion from vascular endothelial cells in response to LPS or LPS plus PA.
Figure 7.
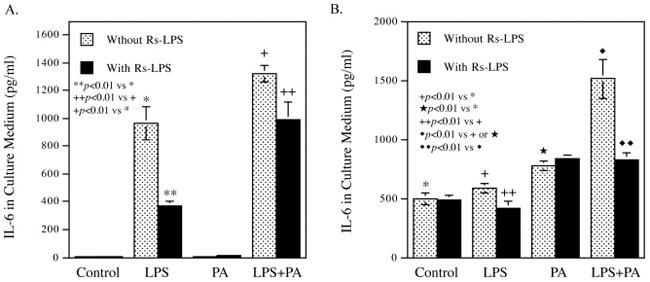
Inhibition of IL-6 secretion from macrophages (A) and vascular endothelial cells (B) by Rs-LPS. Human macrophages and aortic endothelial cells were treated with 100 ng/ml of LPS, 100 μM of palmitic acid (PA) or LPS plus PA in the absence or presence of 100 ng/ml of Rs-LPS for 24 h. After the treatment, IL-6 in culture medium was quantified using ELISA.
Discussion
In the present study, Rs-LPS was used as TLR4 antagonist since it competes with LPS for the same binding site on TLR4 (Kaltashov, et al., 1997, Visintin, et al., 2005). Previous studies have well demonstrated that Rs-LPS effectively and specifically blocks a number of TLR4-mediated pathological effects (Aida, et al., 1995, Kirkland, et al., 1991, Hammad, et al., 2009, Hutchinson, et al., 2010, Baker, et al., 1990). To determine the effect of Rs-LPS on type 2 diabetes-related atherosclerosis, we utilized LDLR−/− mice with HFD-induced type 2 diabetes that have been used by many studies to investigate the pathogenesis of atherosclerosis associated with type 2 diabetes (Wu, et al., 2006, Nguyen, et al., 2013, Radonjic, et al., 2013). Our findings from the control mice clearly showed that HFD-induced type 2 diabetes was associated with accelerated atherosclerosis in aortas. By quantifying both en face atherosclerotic lesions in whole aortas and intimal lesions in cross-sections of the aortic roots, our findings showed that Rs-LPS inhibited atherosclerosis in mice with type 2 diabetes. Since TLR4 antagonist Rs-LPS was given to mice 14 weeks after HFD feeding, this study indicates that Rs-LPS is capable of attenuating the early stage of atherosclerosis development.
Our metabolic studies clearly showed that the HFD induced type 2 diabetes by increasing bodyweight, glucose, lipids and insulin resistance. Our finding on insulin resistance in mice fed HFD based on the HOMA-IR data is in line with the study by Wu et al., in which glucose tolerance and insulin secretion in response to an intraperitoneal glucose load were determined (Wu, et al., 2006). The results showed that, as compared with low-fat diet-fed mice, HFD-fed LDLR−/− mice had impaired glucose tolerance and elevated insulin secretion. Interestingly, Rs-LPS had no effect on HFD-altered metabolic parameters, suggesting that Rs-LPS inhibited atherosclerosis in diabetic LDLR−/− mice through mechanisms independent of metabolic control. Given that inflammation plays a critical role in the initial development of atherosclerosis (Lamon and Hajjar, 2008, Lyon, et al., 2003, Munro and Cotran, 1988), we focused on the effect of Rs-LPS on vascular inflammation to elucidate the potential mechanisms by which Rs-LPS reduced atherosclerosis in diabetic LDLR−/− mice. Our study showed that diabetic LDLR−/− mice had increased content of infiltrated monocytes and macrophages, and expression of IL-6 and MMP-9 in atherosclerotic lesions when compared with nondiabetic mice, but Rs-LPS significantly attenuated the increase. Since IL-6 and MMP-9 play crucial roles in atherosclerosis (Moore, et al., 2013, Haddy, et al., 2003, Kim and Ko, 2014), these findings underscored the role of TLR4-mediated inflammation in diabetes-associated atherosclerosis.
In addition to quantification of IL-6 and MMP-9 expression in atherosclerotic lesions, we also quantified collagen content using Sirius red staining. Interestingly, results showed that diabetes increased MMP-9 and collagen expression simultaneously, suggesting that the two processes under diabetes are inter-related for the intimal remodeling that contributes to the development of atherosclerotic plaques. The similar findings in LDLR−/− mice fed HFD were reported previously (Hu, et al., 2008). In contrast to the stimulatory effect of diabetes, Rs-LPS inhibited both MMP-9 expression and collagen accumulation, which is likely to disrupt the intimal remodeling process and thus reduce atherosclerotic lesions.
To validate the antagonistic effect of Rs-LPS on inflammatory response of vascular cells, we demonstrated that Rs-LPS inhibited IL-6 secretion by cultured macrophages and endothelial cells in response to LPS or LPS plus PA. We have reported previously that PA enhanced LPS-triggered expression of IL-6 expression in macrophages (Jin, et al., 2013). Given that the serum level of PA as the most abundant saturated fatty acid is increased in patients with type 2 diabetes (Stahlman, et al., 2012), this observation may elucidate a mechanism whereby type 2 diabetes is associated with increased risk of atherosclerosis. Indeed, our current study showed that mice with type 2 diabetes had increased free fatty acids (833 vs 503 mg/dl) and it is likely that PA as a major free fatty acid was also increased in the diabetic mice in this study. Since PA and LPS exert a synergy on IL-6 upregulation and Rs-LPS targets the action of LPS, it is expect that Rs-LPS is capable of attenuating the synergistic effect of LPS and PA on IL-6 secretion.
In our present study, the mice were not treated with LPS or Gram-negative bacteria. Thus, it is likely that LPS and other TLR4 ligands were present in the circulation of the mice. Indeed, studies have shown that LPS enters blood circulation via intestines and a 4-week HFD chronically increased plasma LPS concentration two to three times, leading to a metabolic endotoxemia (Cani, et al., 2007, Cani, et al., 2008). Studies have also shown that metabolic endotoxemia contributes to HFD-induced obesity and diabetes (Cani, et al., 2007). In addition to the gut-derived LPS as TLR4 ligand, it has been reported that minimally modified LDL (mmLDL) was also capable of engaging TLR4 as an endogenous ligand (Miller, et al., 2003, Miller, et al., 2005). Therefore, it is likely that diabetes-associated metabolic endotoxemia and some bioactive lipids contribute to TLR4-mediated vascular inflammation and atherosclerosis.
In conclusion, this study has shown that antagonizing TLR4 is effective to reduce vascular inflammation and the early-stage atherosclerosis in mice with type 2 diabetes. The findings from this study suggest that TLR4 is a potential target for anti-inflammatory therapy to reduce cardiovascular complications in patients with type 2 diabetes.
Acknowledgments
This work was supported by a Merit Review grant from the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs and NIH grant R01 DE016353 (to Y.H.).
Abbreviations
- TLR
Toll-like receptor
- LDLR−/− mice
Low-density lipoprotein receptor-deficient mice
- RC
Regular chow
- HFD
High-fat diet
- LPS
Lipopolysaccharide
- Rs-LPS
Rhodobacter sphaeroides LPS
- NFκB
Nuclear factor kappa B
- MAPK
Mitogen-activated protein kinase
- PAMP
Pathogen-associated molecular pattern
- PBS
Phosphate-buffered saline
- HOMA-IR
Homeostasis model assessment of insulin resistance
- MMP
Matrix metalloproteinase
- IL-6
Interleukin-6
Footnotes
Conflict of Interests
There is no conflict of interests for all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dalpke A, Heeg K. Signal integration following Toll-like receptor triggering. Crit Rev Immunol. 2002;22:217–250. [PubMed] [Google Scholar]
- 2.Brown J, Wang H, Hajishengallis GN, Martin M. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res. 2011;90:417–427. doi: 10.1177/0022034510381264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curtiss LK, Tobias PS. The toll of Toll-like receptors, especially toll-like receptor 2, on murine atherosclerosis. Curr Drug Targets. 2007;8:1230–1238. doi: 10.2174/138945007783220605. [DOI] [PubMed] [Google Scholar]
- 4.den Dekker WK, Cheng C, Pasterkamp G, Duckers HJ. Toll like receptor 4 in atherosclerosis and plaque destabilization. Atherosclerosis. 2010;209:314–320. doi: 10.1016/j.atherosclerosis.2009.09.075. [DOI] [PubMed] [Google Scholar]
- 5.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bjorkbacka H, Kunjathoor VV, Moore KJ, Koehn S, Ordija CM, Lee MA, Means T, Halmen K, Luster AD, Golenbock DT, Freeman MW. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med. 2004;10:416–421. doi: 10.1038/nm1008. [DOI] [PubMed] [Google Scholar]
- 7.Higashimori M, Tatro JB, Moore KJ, Mendelsohn ME, Galper JB, Beasley D. Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:50–57. doi: 10.1161/ATVBAHA.110.210971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grieco FA, Vendrame F, Spagnuolo I, Dotta F. Innate immunity and the pathogenesis of type 1 diabetes. Semin Immunopathol. 2011;33:57–66. doi: 10.1007/s00281-010-0206-z. [DOI] [PubMed] [Google Scholar]
- 9.Zipris D. Toll-like receptors and type 1 diabetes. Adv Exp Med Biol. 2010;654:585–610. doi: 10.1007/978-90-481-3271-3_25. [DOI] [PubMed] [Google Scholar]
- 10.Devaraj S, Tobias P, Jialal I. Knockout of toll-like receptor-4 attenuates the pro-inflammatory state of diabetes. Cytokine. 2011;55:441–445. doi: 10.1016/j.cyto.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 11.Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab. 2008;93:578–583. doi: 10.1210/jc.2007-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dandona P, Aljada A, Chaudhuri A, Bandyopadhyay A. The potential influence of inflammation and insulin resistance on the pathogenesis and treatment of atherosclerosis-related complications in type 2 diabetes. J Clin Endocrinol Metab. 2003;88:2422–2429. doi: 10.1210/jc.2003-030178. [DOI] [PubMed] [Google Scholar]
- 13.Ray A, Huisman MV, Tamsma JT, van Asten J, Bingen BO, Broeders EA, Hoogeveen ES, van Hout F, Kwee VA, Laman B, Malgo F, Mohammadi M, Nijenhuis M, Rijkee M, van Tellingen MM, Tromp M, Tummers Q, de Vries L. The role of inflammation on atherosclerosis, intermediate and clinical cardiovascular endpoints in type 2 diabetes mellitus. Eur J Intern Med. 2009;20:253–260. doi: 10.1016/j.ejim.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Theuma P, Fonseca VA. Inflammation and emerging risk factors in diabetes mellitus and atherosclerosis. Curr Diab Rep. 2003;3:248–254. doi: 10.1007/s11892-003-0072-3. [DOI] [PubMed] [Google Scholar]
- 15.Diagnosis and classification of diabetes mellitus. Diabetes Care. 37(Suppl 1):S81–90. doi: 10.2337/dc14-S081. [DOI] [PubMed] [Google Scholar]
- 16.Lee MS. Role of Innate Immunity in the Pathogenesis of Type 1 and Type 2 Diabetes. J Korean Med Sci. 2014;29:1038–1041. doi: 10.3346/jkms.2014.29.8.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeadin MG, Petlura CI, Werstuck GH. Molecular mechanisms linking diabetes to the accelerated development of atherosclerosis. Can J Diabetes. 2013;37:345–350. doi: 10.1016/j.jcjd.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Lu Z, Zhang X, Li Y, Jin J, Huang Y. TLR4 antagonist reduces early-stage atherosclerosis in diabetic apolipoprotein E-deficient mice. J Endocrinol. 2013;216:61–71. doi: 10.1530/JOE-12-0338. [DOI] [PubMed] [Google Scholar]
- 19.Lyngdorf LG, Gregersen S, Daugherty A, Falk E. Paradoxical reduction of atherosclerosis in apoE-deficient mice with obesity-related type 2 diabetes. Cardiovasc Res. 2003;59:854–862. doi: 10.1016/s0008-6363(03)00506-6. [DOI] [PubMed] [Google Scholar]
- 20.Aida Y, Kusumoto K, Nakatomi K, Takada H, Pabst MJ, Maeda K. An analogue of lipid A and LPS from Rhodobacter sphaeroides inhibits neutrophil responses to LPS by blocking receptor recognition of LPS and by depleting LPS-binding protein in plasma. J Leukoc Biol. 1995;58:675–682. doi: 10.1002/jlb.58.6.675. [DOI] [PubMed] [Google Scholar]
- 21.Kirkland TN, Qureshi N, Takayama K. Diphosphoryl lipid A derived from lipopolysaccharide (LPS) of Rhodopseudomonas sphaeroides inhibits activation of 70Z/3 cells by LPS. Infect Immun. 1991;59:131–136. doi: 10.1128/iai.59.1.131-136.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15:410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, Watkins LR. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun. 2010;24:83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker PJ, Taylor CE, Stashak PW, Fauntleroy MB, Haslov K, Qureshi N, Takayama K. Inactivation of suppressor T cell activity by the nontoxic lipopolysaccharide of Rhodopseudomonas sphaeroides. Infect Immun. 1990;58:2862–2868. doi: 10.1128/iai.58.9.2862-2868.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lloyd DJ, Helmering J, Kaufman SA, Turk J, Silva M, Vasquez S, Weinstein D, Johnston B, Hale C, Veniant MM. A volumetric method for quantifying atherosclerosis in mice by using microCT: comparison to en face. PLoS One. 2011;6:e18800. doi: 10.1371/journal.pone.0018800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuyler CA, Ta NN, Li Y, Lopes-Virella MF, Huang Y. Insulin treatment attenuates diabetes-increased atherosclerotic intimal lesions and matrix metalloproteinase 9 expression in apolipoprotein E-deficient mice. J Endocrinol. 2011;210:37–46. doi: 10.1530/JOE-10-0420. [DOI] [PubMed] [Google Scholar]
- 27.Patel S, Chung SH, White G, Bao S, Celermajer DS. The “atheroprotective” mediators apolipoprotein A-I and Foxp3 are over-abundant in unstable carotid plaques. Int J Cardiol. 2010;145:183–187. doi: 10.1016/j.ijcard.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 28.He L, Game BA, Nareika A, Garvey WT, Huang Y. Administration of pioglitazone in low-density lipoprotein receptor-deficient mice inhibits lesion progression and matrix metalloproteinase expression in advanced atherosclerotic plaques. J Cardiovasc Pharmacol. 2006;48:212–222. doi: 10.1097/01.fjc.0000248831.21973.c4. [DOI] [PubMed] [Google Scholar]
- 29.Seager Danciger J, Lutz M, Hama S, Cruz D, Castrillo A, Lazaro J, Phillips R, Premack B, Berliner J. Method for large scale isolation, culture and cryopreservation of human monocytes suitable for chemotaxis, cellular adhesion assays, macrophage and dendritic cell differentiation. J Immunol Methods. 2004;288:123–134. doi: 10.1016/j.jim.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz EA, Zhang WY, Karnik SK, Borwege S, Anand VR, Laine PS, Su Y, Reaven PD. Nutrient modification of the innate immune response: a novel mechanism by which saturated fatty acids greatly amplify monocyte inflammation. Arterioscler Thromb Vasc Biol. 2010;30:802–808. doi: 10.1161/ATVBAHA.109.201681. [DOI] [PubMed] [Google Scholar]
- 31.Holness CL, Simmons DL. Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood. 1993;81:1607–1613. [PubMed] [Google Scholar]
- 32.Hamirani YS, Pandey S, Rivera JJ, Ndumele C, Budoff MJ, Blumenthal RS, Nasir K. Markers of inflammation and coronary artery calcification: a systematic review. Atherosclerosis. 2008;201:1–7. doi: 10.1016/j.atherosclerosis.2008.04.045. [DOI] [PubMed] [Google Scholar]
- 33.Kunz J. Matrix metalloproteinases and atherogenesis in dependence of age. Gerontology. 2007;53:63–73. doi: 10.1159/000096351. [DOI] [PubMed] [Google Scholar]
- 34.Jin J, Zhang X, Lu Z, Perry DM, Li Y, Russo SB, Cowart LA, Hannun YA, Huang Y. Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentrations in macrophages. Am J Physiol Endocrinol Metab. 2013;305:E853–867. doi: 10.1152/ajpendo.00251.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaltashov IA, Doroshenko V, Cotter RJ, Takayama K, Qureshi N. Confirmation of the structure of lipid A derived from the lipopolysaccharide of Rhodobacter sphaeroides by a combination of MALDI, LSIMS, and tandem mass spectrometry. Anal Chem. 1997;69:2317–2322. doi: 10.1021/ac9612943. [DOI] [PubMed] [Google Scholar]
- 36.Visintin A, Halmen KA, Latz E, Monks BG, Golenbock DT. Pharmacological inhibition of endotoxin responses is achieved by targeting the TLR4 coreceptor, MD-2. J Immunol. 2005;175:6465–6472. doi: 10.4049/jimmunol.175.10.6465. [DOI] [PubMed] [Google Scholar]
- 37.Wu L, Vikramadithyan R, Yu S, Pau C, Hu Y, Goldberg IJ, Dansky HM. Addition of dietary fat to cholesterol in the diets of LDL receptor knockout mice: effects on plasma insulin, lipoproteins, and atherosclerosis. J Lipid Res. 2006;47:2215–2222. doi: 10.1194/jlr.M600146-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen N, Naik V, Speer MY. Diabetes mellitus accelerates cartilaginous metaplasia and calcification in atherosclerotic vessels of LDLr mutant mice. Cardiovasc Pathol. 2013;22:167–175. doi: 10.1016/j.carpath.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radonjic M, Wielinga PY, Wopereis S, Kelder T, Goelela VS, Verschuren L, Toet K, van Duyvenvoorde W, van der Werff van der Vat B, Stroeve JH, Cnubben N, Kooistra T, van Ommen B, Kleemann R. Differential effects of drug interventions and dietary lifestyle in developing type 2 diabetes and complications: a systems biology analysis in LDLr−/− mice. PLoS One. 2013;8:e56122. doi: 10.1371/journal.pone.0056122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamon BD, Hajjar DP. Inflammation at the molecular interface of atherogenesis: an anthropological journey. Am J Pathol. 2008;173:1253–1264. doi: 10.2353/ajpath.2008.080442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyon CJ, Law RE, Hsueh WA. Minireview: adiposity, inflammation, and atherogenesis. Endocrinology. 2003;144:2195–2200. doi: 10.1210/en.2003-0285. [DOI] [PubMed] [Google Scholar]
- 42.Munro JM, Cotran RS. The pathogenesis of atherosclerosis: atherogenesis and inflammation. Lab Invest. 1988;58:249–261. [PubMed] [Google Scholar]
- 43.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haddy N, Sass C, Droesch S, Zaiou M, Siest G, Ponthieux A, Lambert D, Visvikis S. IL-6, TNF-alpha and atherosclerosis risk indicators in a healthy family population: the STANISLAS cohort. Atherosclerosis. 2003;170:277–283. doi: 10.1016/s0021-9150(03)00287-9. [DOI] [PubMed] [Google Scholar]
- 45.Kim J, Ko J. Human sLZIP promotes atherosclerosis via MMP-9 transcription and vascular smooth muscle cell migration. FASEB J. 2014;28:5010–5021. doi: 10.1096/fj.14-259218. [DOI] [PubMed] [Google Scholar]
- 46.Hu C, Dandapat A, Sun L, Chen J, Marwali MR, Romeo F, Sawamura T, Mehta JL. LOX-1 deletion decreases collagen accumulation in atherosclerotic plaque in low-density lipoprotein receptor knockout mice fed a high-cholesterol diet. Cardiovasc Res. 2008;79:287–293. doi: 10.1093/cvr/cvn110. [DOI] [PubMed] [Google Scholar]
- 47.Stahlman M, Pham HT, Adiels M, Mitchell TW, Blanksby SJ, Fagerberg B, Ekroos K, Boren J. Clinical dyslipidaemia is associated with changes in the lipid composition and inflammatory properties of apolipoprotein-B-containing lipoproteins from women with type 2 diabetes. Diabetologia. 2012;55:1156–1166. doi: 10.1007/s00125-011-2444-6. [DOI] [PubMed] [Google Scholar]
- 48.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 49.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 50.Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J Biol Chem. 2003;278:1561–1568. doi: 10.1074/jbc.M209634200. [DOI] [PubMed] [Google Scholar]
- 51.Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25:1213–1219. doi: 10.1161/01.ATV.0000159891.73193.31. [DOI] [PubMed] [Google Scholar]