

Summary
A number of chronic metabolic pathologies, including obesity, diabetes, cardiovascular disease, asthma, and cancer cluster together to present the greatest threat to human health. As research in this field has advanced, it has become clear that unresolved metabolic inflammation, organelle dysfunction, and other cellular and metabolic stresses underlie the development of these chronic metabolic diseases. However, the relationship between these systems and pathological mechanisms is poorly understood. Here, we will discuss the role of cellular Ca2+ homeostasis as a critical mechanism integrating the myriad of cellular and subcellular dysfunctional networks found in metabolic tissues such as liver and adipose tissue in the context of metabolic disease particularly in obesity and diabetes.
Introduction
Inappropriate consumption of nutrients and chronic metabolic diseases are the hallmarks of this century, presenting the greatest global public health problem and a major unmet need for novel therapies. One way to consider such high prevalence of these diseases among modern humans is that they reflect inherent limitations in the adaptive capacity of the organism to offset chronic metabolic challenges. During normal feeding cycles, the intake of calories of all forms (lipids, proteins, and carbohydrates) leads to acute fluctuations in energy and nutrient load, demanding an adaptive response from the metabolic networks that cells have developed to reestablish homeostasis. Even massive quantities of caloric intake can be managed in short periods of time, such as in the striking example of the Burmese python, which is well adapted to a lifestyle characterized by extremely large meals (ingestion of prey up to 1.6 times the snake's mass) spaced over long intervals (Riquelme et al., 2011; Secor and Diamond, 1998). Chronic exposure to excessive nutrients and energy, however, presents a continuous challenge for which there has not been an opportunity for selection and proper development of these adaptive pathways, triggering local and systemic stress responses such as metaflammation and organelle dysfunction that ultimately lead to the development of chronic and complex metabolic diseases (Egger, 2012; Fu et al., 2012b; Gregor and Hotamisligil, 2011). Several important mechanisms have been demonstrated to contribute to the development of cellular stress responses and systemic metaflammation, however, how these systems relate to each other and are integrated is not well understood (Glass and Olefsky, 2012; Gregor and Hotamisligil, 2011; Hotamisligil, 2006; Kahn et al., 2006; Lumeng and Saltiel, 2011; Qatanani and Lazar, 2007; Shulman, 2000). Here, we will discuss a potential role for cellular Ca2+ homeostasis as an important contributing factor sustaining organelle dysfunction and the systemic integration of critical stress responses in metabolic organs such as liver and adipose tissue. The main premise of this discussion is that cellular Ca2+ homeostasis also fails to maintain its adaptive integrity in the face of structural and signaling defects introduced by chronic metabolic challenges. For example, continuous substrate flow can drive synthetic pathways such as lipogenesis, which would impair ER membrane properties and adaptive capacity through impaired Ca2+ fluxes and homeostasis in the target cells, creating a feed forward pathological cycle including the engagement of stress and metaflammatory pathways and impaired hormone secretion and action. If such a Ca2+-related framework is critical in the integrated pathological sequences that characterize chronic metabolic diseases, it may then be possible to exploit several elements for therapeutic interventions.
Ca2+ is the most abundant ion in the body, primarily stored in bones in the form of CaPO3 (hydroxyapatite). In bone, Ca2+ plays a structural role and also can be dissolved, serving as a source of cations in the blood (Pozzan et al., 1994). Outside of the skeleton, Ca2+ is a ubiquitous and versatile signaling molecule controlling a wide variety of cellular processes including muscle contraction, neuronal transmission, hormone secretion, organelle communication, cellular motility, fertilization and cell growth (Berridge, 2012; Clapham, 2007; Pozzan et al., 1994). Given these critical and diverse functions, cellular Ca2+ concentration is tightly regulated, and dysfunction of cellular Ca2+ homeostasis is associated with several pathological conditions as well as aging (Guerrero-Hernandez and Verkhratsky, 2014; Luo and Anderson, 2013; Toescu and Verkhratsky, 2007).
Alterations in Ca2+ homeostasis in the context of metabolic stress, obesity, and diabetes strongly affect tissues including heart and skeletal muscle, where Ca2+ plays an essential role in muscle contraction, and pancreas, where Ca2+ is indispensable for insulin and glucagon secretion in response to fluctuations in blood glucose. The role of intracellular Ca2+ homeostasis and dysfunction in these tissues has been the focus of intense research and is reviewed elsewhere (Carvajal et al., 2014; Eizirik et al., 2008; Erickson, 2014; Eshima et al., 2014; Fernandez-Velasco et al., 2014; Gilon et al., 2014; Guerrero-Hernandez and Verkhratsky, 2014; Pereira et al., 2014; Ramadan et al., 2011), and thus we will not cover this aspect of the field in depth here. We will provide an overview of Ca2+ homeostasis in key organelles, such as endoplasmic reticulum (ER) and mitochondria in metabolic tissues such as liver and adipose tissue. We will focus on recent research that leads us to propose that disruption of Ca2+ signaling can be an important mechanism in the myriad of cellular and subcellular dysfunctional networks in the context of obesity and type 2 diabetes, with potential implications for other complex diseases of immunometabolic nature.
Intracellular Ca2+ dynamics in ER and mitochondrial function
Ca2+ signaling occurs in a wide dynamic range, and its outcome depends on the speed, amplitude and oscillation of Ca2+ release patterns in a temporally and spatially regulated manner (Dolmetsch et al., 1998; Hajnoczky et al., 1995). For example, a Ca2+ signal can be transduced within milliseconds during muscle contraction, or can take minutes or hours, such as the case in transcriptional regulation and cell proliferation. Spatially, the rise in cytosolic Ca2+ levels can be localized, as it is during neuronal synapsis and cell-cell communication, or can also diffuse across the cell as a wave and trigger an effect at a location distant from the origin of the stimulus (Dolmetsch et al., 1998; Hajnoczky et al., 1995). Cytosolic Ca2+ levels are maintained at low levels (10–100nM) against a huge (20,000-fold) Ca2+ concentration gradient in intracellular stores and in the extracellular milieu (Clapham, 2007). The regulation of cytosolic and organelle Ca2+ levels is achieved by the orchestrated action of an extensive signaling machinery that comprises proteins able to either pump Ca2+ out, from the cytosol to the extracellular space, or into intracellular stores and compartments such as the ER and mitochondria (Pozzan et al., 1994). The complex structural nature, dynamics, and trafficking of these transport proteins and the interactions between subcellular organelles through specialized domains, all require precise membrane properties and integrity for proper homeostasis, which are challenged during metabolic stress limiting proper Ca2+ homeostasis.
Ca2+ homeostasis in the endoplasmic reticulum
The ER is a multifunctional organelle that serves as the most important Ca2+ store in the cell. This organelle is able to accumulate Ca2+ at mM levels in both free and protein-buffered forms (Mekahli et al., 2011; Pozzan et al., 1994; Prins and Michalak, 2011). The Ca2+ stored in the ER lumen is essential for the regulation of protein posttranslational processing, folding and export, and can be rhythmically released to the cytosol, providing sustained and precise Ca2+-mediated responses.
Ca2+ distribution within the ER is not homogeneous (Petersen et al., 2001) and its regulation across the ER membrane is facilitated by three classes of well-studied proteins: (1) Ca2+ release channels – inositol-1,4,5-triphosphate (IP3) receptors (IP3Rs) and ryanodine receptors (RyRs), (2) Ca2+ pumps – sarco-endoplasmic reticulum Ca2+-ATPases (SERCAs) that uptake Ca2+ from the cytosol to the ER lumen, and (3) Ca2+ binding proteins (Clapham, 2007; Mekahli et al., 2011; Pozzan et al., 1994; Prins and Michalak, 2011), Figure 1 and Table 1.
Figure 1. Ca2+ homeostasis in the ER.
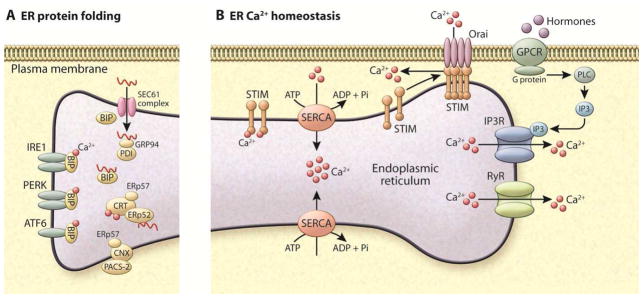
Right: tool-kit of Ca2+ handling proteins. Ligand binding of GPCRs leads to the biosynthesis of the intracellular messengers such as IP3, which diffuses into the cell and binds to IP3R, stimulating Ca2+ release from the ER. Ca2+ channels activated by IP3 release Ca2+ from ER to the cytosol. RyR also releases Ca2+ from ER to the cytosol. SERCA (Ca2+-ATPase) pumps Ca2+ from cytosol to the ER lumen at the expense of ATP hydrolysis. STIM is an ER membrane protein that possesses 2 EF hand domains. Under basal conditions, Ca2+ is bound to these domains and the protein remains in its monomeric form. After Ca2+ release from ER, Ca2+ dissociates from the of STIM proteins leading to oligomerization and trafficking of STIM through the ER membrane toward the plasma membrane contact sites. This localization, allows STIM to couple with the plasma membrane Ca2+ channel Orai and Ca2+ enters the cell from the extracellular space, a process known as capacitive or store operator Ca2+ entry (SOCE). SERCA colocalizes with STIM/Orai complex, and is thought to pump Ca2+ directly from cytosol to ER lumen. Left: ER protein folding machinery: During glycoprotein biosynthesis, the translation of nascent polypeptides is followed by their translocation of the peptides through the SEC61 pore. Sugar molecules are added to the polypeptides that are recognized by lectin chaperones such as calnexin (CNX) and calretculin (CRT), which function in complex with ERp57 and ERp72. Additional chaperone assistance is provided by the ATP-driven chaperone BiP (GRP78). Calnexin and calreticulin also bind Ca2+ with high capacity, functioning as the main Ca2+ buffers in the ER lumen. Ca2+ depletion leads to accumulation of misfolded proteins and the activation of the unfolded protein response (UPR), mediated by three canonical ER luminal sensors, IRE1, PERK, and ATF6, which are inactive and bound to BiP in the absence of stress. GPCR: G protein–coupled receptor. IP3: inositol triphosphate. RyR: Ryanodine receptor. STIM1: stroma interacting protein.
Table 1.
Proteins regulating Ca2+ homeostasis in the ER and mitochondria
| Name | Isoforms | Tissue Expression | Function | Location | Main Regulators | Knockout Phenotype | References |
|---|---|---|---|---|---|---|---|
| SERCA | SERCA 1a | Skeletal Muscle/brown adipose tissue | Ca2+ uptake from cytosol to ER lumen | ER membrane | Endogenous: Sarcolipin (skeletal muscle and heart) (reversible inhibitor) Phospholambam (skeletal muscle and heart) (reversible inhibitor) Pharmacological: Thapsigargin (inhibitor) Cyclopiazonic acid (reversible inhibitor) BHQ (inhibitor) |
Gasping respiration, cyanotic phenotype, and postnatal lethality | (de Meis and Vianna, 1979; Prasad et al., 2004; Toyoshima and Inesi, 2004; Wu et al., 1995) |
| SERCA 1b | Skeletal muscle | ||||||
| SERCA 2a | Skeletal muscle/heart | Embryonic lethality | |||||
| SERCA 2b | Ubiquitous including liver and adipose tissue | ||||||
| SERCA 3 | Non-muscle tissues including blood cells, epithelial cells and respiratory tract, pancreatic β cells | No gross phenotype | (Vandecaetsbeek et al., 2011) | ||||
| IP3R | IP3R1 | Ubiquitous, predominantly in brain | Ca2+ release from ER to cytosol | ER membrane | Endogenous: IP3 Low Ca2+ levels (activator) High Ca2+ levels (inhibitor) ATP Regulatory kinases: PKA,PKC,AKT,PKG,CaMkinase Protein interaction: Numerous proteins Pharmacological: Xestopongin C (inhibitor) |
Neurological defects and early lethality | (Choe and Ehrlich, 2006; Foskett et al., 2007) |
| IP3R2 | Ubiquitous. Represents the predominant isoform in liver and adipose tissue | IP3R2−/− exhibits some cardiac dysfunction. IP3R2/IP3R3 double deficiency leads to defective saliva and gastric juice secretion | |||||
| IP3R3 | Various tissues and culture cell | ||||||
| RyaR | RyR1 | Muscle (predominantly skeletal)/brain | Ca2+ release from ER to cytosol | ER membrane | Endogenous: Low Ca2+ levels (activator) High Ca2+ levels (inhibitor) Kinase proteins: PKA, PKG and CaMKinase High Mg+ (inhibitor) Pharmacological: Low Ryanodine levels (activator) High Ryanodine levels (inhibitor) Caffeine (activator) Rutheniun Red (inhibitor) |
Perinatal lethality with gross abnormalities of skeletal muscle | (Fill and Copello, 2002; Meissner, 1994; Van Petegem, 2012) |
| RyR2 | Muscle (predominantly heart)/brain | Embryonic lethality (~e10) with morphologic abnormalities in the heart tube | |||||
| RyR3 | Predominantly brain | Fertile and displays no gross abnormalities. Increased locomotor activity | |||||
| STIM | STIM1 | Ubiquitous, predominant isoform in most tissues | Ca2+ entry from extracellular space to cytosol | ER membrane | Endogenous:Ca2+ (depletion activates) ROS (activates) Temperature (high temperature activates) Hypoxia (activates) Regulatory Kinases: SGK1, AMPK Pharmacological: Low 2APB levels (activate) High 2APB levels (inhibit) |
Embryonic or perinatal lethality with respiratory failure | (Carrasco and Meyer, 2011; Soboloff et al., 2012) |
| STIM2 | Ubiquitous, prevailing isoform in brain and dendritic cells | Lethality at 4–5 weeks of age | |||||
| Orai | Orai1 | Ubiquitous, predominant skin and lymphocytes | Ca2+ entry from extracellular milieu to cytosol, coupled with STIM | Plasma membrane | Pharmacological: lanthanides Gd3+ and La3+ (inhibitors) |
Smaller size, eyelid irritation, and sporadic hair loss; immune system, lymphocyte development is normal but T- and B-cell function is impaired. | (Cao et al., 2015; Gwack et al., 2008; Hogan et al., 2010) |
| Orai2 | Ubiquitous | ----- | |||||
| Orai3 | Ubiquitous | ----- | |||||
| Calreticulin | Ubiquitous | Ca2+-binding Chaperone | ER lumen | Embryonic lethality due to impaired cardiac development. Fibroblast cells derived from calreticulin-deficient embryos show significantly reduced ER Ca2+ capacity. |
(Michalak et al., 2009; Prins and Michalak, 2011) | ||
| Calnexin | Ubiquitous | Ca2+-binding Chaperone | ER lumen | 50% lethality within the first 48h after birth, and the cause of the death remains to be determined. The survivors exhibit motor abnormalities. | (Caramelo and Parodi, 2008; Prins and Michalak, 2011) | ||
| VDAC | VDAC1 | Ubiquitous and the most abundant isoform | Forms a channel through the mitochondrial outer membrane and allows diffusion of small hydrophilic molecules such as Ca2+ | OMM | Endogenous: Interacting proteins: HK, Bcl2 ROS Phosphorylation: PKA PKCe Pharmacological: Ruthenium red (inhibitor) Pharmacological: Ru360 (inhibitor) |
Defects in skeletal muscle. Modifications of kinetic parameters of some mitochondrial enzymes and mitochondrial ultrastructure abnormalities. No major functional deficits |
(Colombini, 2012; Messina et al., 2012; Shoshan-Barmatz and Ben-Hail, 2012) |
| VDAC2 | Ubiquitous | ------ | |||||
| VDAC3 | Ubiquitous | Male infertility and altered mitochondrial ultrastructure. | |||||
| MCU | Ubiquitous | Ca2+uptake into the mitochondria | IMM | Endogenous: Interacting proteins: MICU1/MICU2/MICUR1/EMRE Pharmacological: Ru360 (inhibitor) |
Dependent on the genetic background. MCU knockout mice on a mixed background are viable , mitochondria Ca2+ uptake by isolated mitochondria and cells of this animals are blunted but their respiratory capacity and morphology are normal. Also the animals don’t show any gross phenotype except impaired skeletal muscle performance under situations requiring high-energy expenditure. | (Chaudhuri et al., 2013; De Stefani et al., 2011; De Stefani and Rizzuto, 2014; Foskett and Philipson, 2015) | |
| Na+/Ca2+ channel, NCLX | NCLX | Ubiquitous | Ca2+ efflux from the mitochondria to the cytosol | Mitochondrial inner membrane | Endogenous: K+ (activator) Ni2+, Mg2+, Ba2+ and La3+ (inhibitor) Kinase proteins: PKC, PINK1 |
(Palty et al., 2012) |
Ca2+ uptake into the ER from the cytoplasm by the SERCA enzymes occurs against its chemical gradient and requires energy released from ATP hydrolysis (de Meis and Vianna, 1979; Toyoshima and Inesi, 2004). Ca2+ stored in the ER is then released to the cytosol through the IP3R and RyR Ca2+ channels. The release of Ca2+ from ER during the generation of cytosolic Ca2+ signals leads to depletion of ER luminal Ca2+. Two homologous ER proteins called STIM1 and STIM2 can sense this decrease in Ca2+ through EF hand domains and respond by activating the entry of Ca2+ from the extracellular environment into the cell, a process known as capacitive or store operated calcium entry (SOCE). Ca2+ dissociation from the EF hands domains of STIM proteins leads to a rapid oligomerization and trafficking of STIM in the ER membrane toward the plasma membrane. At this site, STIM couples with the plasma membrane channel Orai, allowing Ca2+ to enter the cell from the extracellular milieu. Upon the entrance of Ca2+ to the cytosol, SERCA pumps Ca2+ into the ER to replenish the deficit.
Inside the ER, high levels of Ca2+ are essential for protein folding and processing, in part due to the allosteric regulation of many intraluminal proteins. These include buffering proteins, such as calsequestrin and calreticulin, and chaperones, such as GRP78 (BiP, immunoglobulin binding protein), GRP94 (endoplasmin), calnexin, GRP170/ORP150 and ERp57. A third class of Ca2+-regulated proteins includes the oxidoreductases PDI, ERp57 and ERO1L, which provide an electron transport pathway from thiol residues to molecular oxygen during disulfide bond formation (Gorlach et al., 2006). ER Ca2+ depletion impairs the function of all these proteins, and if persistent, results in the accumulation of unfolded proteins. This situation triggers a well-known adaptive response called the unfolded protein response (UPR), with the goal of decreasing protein translation while selectively increasing chaperone expression and increasing protein degradation. When unsuccessful, this response can also trigger cell death. ER dysfunction is a well-established component of metabolic pathologies and homeostasis and the canonical UPR pathways as well as their metabolic relevance are reviewed in detail elsewhere and will not be covered here (Hotamisligil, 2010b; Marciniak and Ron, 2006; Mori, 2000; Zhao and Ackerman, 2006).
Ca2+ homeostasis in mitochondria
Ca2+ uptake in isolated mitochondria was directly measured for the first time more than 50 years ago (Deluca and Engstrom, 1961), and it is now well accepted that Ca2+ uptake in the mitochondria is essential for its function and also contributes to the regulation of cytosolic Ca2+ concentrations.
The main force driving mitochondrial Ca2+ uptake is the electrochemical gradient formed across the mitochondrial inner and outer membranes (Rizzuto et al., 1987). To get into the mitochondrial matrix, Ca2+ first crosses the outer mitochondrial membrane (OMM), which is permeable to Ca2+ through voltage-dependent anion channels (VDACs). VDACs act as general diffusion pores for small hydrophilic molecules and connects with IP3R in the ER through linkage with the chaperone GRP75. This structure allows for Ca2+ transfer between the ER and the mitochondria, as will be described in further detail below. The inner mitochondrial membrane (IMM) is impermeable to ions and the transport of Ca2+ occurs through the mitochondrial Ca2+ uniporter (MCU), a low affinity (Kd of 20–30μM under physiological conditions), low conductance channel, which allows rapid accumulation of Ca2+ depending on the electrochemical gradient (Chaudhuri et al., 2013; Kirichok et al., 2004). MCU is regulated by various interacting proteins forming a large complex in the IMM (Figure 2 and Table 1). Rapid Ca2+ uptake into mitochondria is counteracted by an extrusion mechanism mediated by the mitochondrial antiporter exchanging Na+/Ca2+ (in excitable tissues such as neurons and skeletal muscle) or H+ (in the liver and other tissues).
Figure 2. Components of the mitochondria associated ER membranes (MAMs) and Ca2+ homeostasis in the mitochondria (A).

Electron microscopic images exemplifying the close contacts between smooth ER and mitochondria in mouse liver cells. Dense areas show the proteinaceous bridges formed between the two membranes. (B) The MAM connection is mediated by protein tethers such as mitofusin2 (MFN2) and stabilized by proteins such as PACS-2 bound to ER-resident calnexin (CNX). The MAMs are enriched in proteins that regulate Ca2+ transport from ER to the mitochondria such as IP3R, connected to the mitochondria anion transporter (VDAC) by the chaperone GRP75. Through these structures Ca2+ leaves the ER and enters the mitochondria matrix via the mitochondria Ca2+ uniporter (MCU) located in the IMM. MCU is regulated by a series of binding partners such as MICU1 and MICU2, MICUR1 and EMRE. MICU1 functions as a gatekeeper for MCU-mediated Ca2+ uptake. MICU2 seems to inhibit MCU function. EMRE is essential for the interaction between MCU and the MICU proteins and is indispensable for MCU-mediated Ca2+ transport. The exit pathway for mitochondrial Ca2+ is mediated by the NCXL antiporter that exchanges Na+/Ca2+. MAMs are also enriched in key enzymes of lipid biosynthesis to facilitate the conversion of PA to PE and PC, as described in the text. (C) Within the mitochondria, Ca2+ regulates the activity of matrix and citric acid cycle enzymes such as pyruvate dehydrogenase phosphatase (PDP1), α-ketoglutarate dehydrogenase (aKGDH), and isocitrate dehydrogenase (IDH). This culminates in increased NADH production that feeds the respiratory chain to produce ATP. (D) Mitochondrial Ca2+ overload leads to increased mitochondrial ROS production and the opening the mitochondrial permeability transition pore (PTP). The opening of the PTP leads to a collapse of membrane potential and mitochondrial swelling, with consequent loss of nucleotides and cytochrome C (CytoC), and is directly linked with apoptotic cell death. Apoptosis is regulated by the BCL2 class of proteins such as BID, BAX and BAD. Under normal conditions, the proapoptotic protein BAX is in the cytosol. However, under apoptotic condition this protein interacts with BAD or BID located in the OMM, stimulating the permeabilization of the OMM.
Within the mitochondria, Ca2+ regulates intrinsic functions of the organelle, such as the activity of the citric acid cycle and NADH and ATP synthesis. Three matrix dehydrogenases are activated by Ca2+: pyruvate dehydrogenase phosphatase, α-ketoglutarate dehydrogenase, and isocitrate dehydrogenase, and their stimulation by Ca2+ increases NADH availability. The increased level of NADH drives an increase in the flow of electrons through the respiratory chain, thus enhancing ATP synthesis to meet the needs of the cell. Therefore, mitochondrial Ca2+ flux is a critical regulator of aerobic metabolism (Rizzuto et al., 2012). On the other hand, excessive Ca2+ accumulation is associated with mitochondrial dysfunction. For example, Ca2+ overload leads to increased mitochondrial ROS production by activating ROS-generating enzymes such as glycerol phosphate and α-ketoglutarate dehydrogenase, by enhancing nitric oxide generation and consequent respiratory inhibition, and by stimulating the opening the mitochondrial permeability transition pore (PTP) (Kowaltowski et al., 2009; Peng and Jou, 2010). Opening of the PTP leads to a collapse of membrane potential and mitochondrial swelling, with consequent loss of nucleotides and cytochrome C, and is directly linked with apoptotic cell death (Baines et al., 2005; Kowaltowski et al., 2009). Recently, mitochondrial Ca2+ overload has also been implicated in mitophagy, the selective degradation of damaged mitochondria through autophagy (Rimessi et al., 2013) Thus, the precise regulation of mitochondrial Ca2+ levels is crucial for the maintenance of balanced cell function, and the fine-tuned control of mitochondrial Ca2+ uptake is a critical determinant of cellular function and survival.
The recent identification of the proteins involved in regulating mitochondrial Ca2+ uptake and efflux has led to a flurry of publications and renewed interest in this field, highlighting the gaps that remain to be filled in terms of our understanding of mitochondrial Ca2+ flux in cellular function and metabolism (Reviewed by: Chaudhuri et al., 2013; De Stefani et al., 2011; Foskett and Philipson, 2015).
Ca2+ homeostasis and ER-mitochondria interaction
Although ER and mitochondria play distinct cellular roles, these organelles are also functionally coupled and form physical interactions at sites defined as mitochondria associated ER-membranes (MAMs). The association between ER and mitochondria was first described by Copeland and Dalton over 50 years ago in pseudo branch gland cells, and their function was originally characterized biochemically (Copeland and Dalton, 1959). Recently, advances in molecular biology and imaging technologies allowed cell biologists to better understand the specificity and dynamic nature of these contacts and to unveil their physiological functions (reviewed in Giorgi et al., 2009; Rowland and Voeltz, 2012). MAMs comprise ~5–20% of the total mitochondrial surface and function as a lipid raft (Hayashi and Su, 2003; Vance, 1990) (Figure 2). MAM connections are tight and strong, maintained in part by proteins such as phosphofurin acidic cluster sorting protein 2 (PACS-2), which has been shown to be necessary to stabilize MAM connections. PACS-2 is enriched in MAMs through its direct interaction with the ER protein calnexin (Myhill et al., 2008) and experimental suppression of PACS-2 can reduce MAM stability (Simmen et al., 2005). MAMs are also thought to be secured by protein tethers such as mitofusin-2, ablation of which leads to ER-mitochondrial disconnection (de Brito and Scorrano, 2008). Interestingly, a recent report calls this concept into question by demonstrating that deletion of mitofusin-2 unexpectedly and in contrast to the predictions, led to increased mitochondria ER interactions (Filadi et al., 2015). Additional studies will be required to fully understand the details of the structural components of these important organelle connections and their physiological and pathological regulation.
MAMs are also enriched in proteins that regulate Ca2+ transport from ER to the mitochondria such as IP3R, which appears to be connected to VDACs on the mitochondrial side by chaperones such as GRP75. Indeed, suppression of GRP75 decreases MAM coupling and Ca2+ uptake into mitochondria (Szabadkai et al., 2006). Furthermore, the presence of MAMs allows the formation of microdomains of high Ca2 + concentrations (~10μM) that exceed those measured in the cytosol under healthy conditions. The existence of this structure explains how low-affinity MCU is able to take up Ca2+ under physiological conditions, solving what had been a major puzzle in the field (Csordas et al., 1999; Rizzuto et al., 1998; Rizzuto et al., 1992; Szalai et al., 2000).
In recent years the recognized repertoire of proteins that are enriched at MAMs has grown, and now includes the sigma 1 receptor (Hayashi and Su, 2007), a chaperone that regulates IP3R activity, PERK, a key component of canonical UPR (Verfaillie et al., 2012), mTOR, a critical nutrient sensing molecule, and AKT, a key signaling molecule for hormones, including insulin (Betz et al., 2013), among many others. In addition, key enzymes of lipid biosynthesis such as PEMT2 (Cui et al., 1993), DGAT2 (Stone et al., 2009), and phosphatidyl serine synthase (PSS) (Vance, 1990) are all present in high concentrations at MAMs, indicating a role for this structure in lipid biosynthesis and trafficking (Figure 2). Indeed, phosphatidylserine (PS) is produced in the ER but needs to be transferred to the mitochondria to be converted into phosphatidylethanolamine (PE), and into phosphatidylcholine (PC). The PE precursor is translocated back to ER. Hence the MAM is a strategic location for the lipid synthesis machinery, facilitating lipid trafficking.
In addition, MAMs seem to be important for autophagosome formation; it was reported that upon suppression of mitofusin-2 and PACS-2, ER-mitochondria contacts are disrupted and there is a decrease in autophagosome biogenesis (Hamasaki et al., 2013). Another function of the MAM is to regulate mitochondrial dynamics. Taking advantage of high-resolution EM tomography and live-confocal fluorescence microscopy, the Voeltz group has shown that ER marks the sites for mitochondrial fission and is involved in the recruitment of proteins involved in mitochondrial division such DRP1 (Friedman et al., 2011). The impact of this structure on physiology and pathophysiology has been a topic of intense research in recent years, and these findings suggest that in addition to modulating ER-mitochondrial Ca2 + homeostasis, MAMs act as a signaling platform for the dynamic modulation of multiple metabolic pathways (Figure 2). Taken together, it is clear that many components of MAMs are linked to key metabolic pathways.
Cytosolic Ca2+ signaling
Cytosolic Ca2+ signaling is initiated by a stimulus that generates a rise in intracellular Ca2+, which in turn modulates downstream signaling pathways. In non-excitable cells this cascade is most commonly initiated by a hormone or other agonist binding to a G protein-coupled receptor (GPCR) and the activation of canonical signaling pathways. For example, glucagon (through the glucagon receptor) and catecholamines (through the β adrenergic receptors) activate the Gαs protein leading to stimulation of adenylate cyclase activity and the production of cAMP. These signals converge on protein kinase A (PKA), which is able to phosphorylate and activate IP3R leading to Ca2+ release from the ER (Bugrim, 1999). Alternatively, catecholamine signaling through the α-adrenergic receptor activates phospholipase C (PLC), which catalyzes the hydrolysis of phosphatidylinositol 4,5 bisphosphate (PIP2) to produce the intracellular messengers IP3 and DAG (Foskett et al., 2007). IP3 diffuses into the cell and binds to IP3R, stimulating Ca2+ release from the ER. DAG stimulates PKC, which regulates an array of signaling pathways including modulations of Ca2+ signaling (Clapham, D.E. (2007).
These signal-induced rises in cytosolic Ca2+ are transient, follow an oscillatory pattern, and can be sensed by different proteins. A central Ca2+ responsive protein is calmodulin (CaM), a protein which undergoes conformational change following Ca2+ binding to its four EF hand domains (Zhang et al., 1995). CaM binds to the regulatory regions of CaM-dependent kinases (CaMKs) to relieve autoinhibition and allow the phosphorylation of exogenous substrates. Among other substrates, CaMKs have been associated with direct phosphorylation of several transcription factors critical for metabolism, inflammation, and other related cellular processes including FOXO (Ozcan et al., 2012), CREB (Sheng et al., 1991), and CBP (Impey et al., 2002), with the nuclear translocation of NF-κB (Ishiguro et al., 2006), and activation of JNK (Timmins et al., 2009).
Another important Ca2+-dependent signaling molecule is Calcineurin, also known as protein phosphatase-3, a serine/threonine phosphatase that is activated by sustained high Ca2+ levels. Calcineurin transduces cytosolic Ca2+ signals to the nucleus, regulating the activity of at least three important transcription pathways involving Nuclear factor of activated T-cells (NFAT) (Choi et al., 1994), CREB-regulated transcription coactivator 2 (CRTC2) (Screaton et al., 2004), and myocyte enhancer factor-2 (MEF-2) (Mao and Wiedmann, 1999), and thus controls responses important in the immune, nervous, metabolic, and cardiovascular systems (Figure 3).
Figure 3. Intracellular Ca2+ signaling.

GPCR activation by hormones can induce Ca2+ release by activating phospholipase C (PLC) leading to the hydrolysis of phosphatidylinositol 4,5 bisphosphate (PIP2) to produce the intracellular messengers IP3 and DAG. IP3 diffuses in to the cell and binds to IP3R, stimulating Ca2+ release from the ER. Alternatively, GPCR activation leads to the production of cAMP, which activates the protein kinase A (PKA), enabling it to phosphorylate IP3R and induce its activity. In the cytosol the two main Ca2+ sensors involved in signal transduction are CaM kinase (CamK) and Calcineurin (CLN). CaMKs have been shown to directly phosphorylate many targets including the inflammatory proteins JNK and p38 and transcription factors including FOXO and CREB. CLN is activated by sustained high Ca2+ levels and regulates the activity of at least three important transcription pathways involving Nuclear factor of activated T-cells (NFAT) and CREB-regulated transcription coactivator 2 (CRTC2).
Alterations in Ca2+ homeostasis causing organelle stress and metabolic disease
In this section, we will primarily focus on studies in the liver and the emerging work applicable to adipose tissue and macrophages to illustrate the role of Ca2+ homeostasis in organelle stress and metabolic deterioration. However, these concepts are applicable to any metabolic or immunological target cell in influencing systemic metabolic homeostasis.
Liver metabolism, function, and Ca2+ homeostasis
The liver represents a major metabolic organ in its ability to control gluconeogenesis, glycogen storage, lipogenesis, and, cholesterol and bile acid metabolism. During fasting, the liver initially uses glycogen to mobilize glucose, and, when glycogen is depleted, de novo synthesis of glucose from non-carbohydrate precursors becomes the main determinant of the body’s ability to maintain normoglycemia. The liver is also a major site for de novo synthesis of fatty acids and triacylglycerol, and lipid excretion.
Fluctuations in cytosolic and organelle Ca2+ following glucagon or epinephrine stimulation play a major role in hepatic control of glucose production, bile fluid movement and excretion, fatty acid, amino acid and xenobiotic metabolism, protein synthesis and secretion, cell cycle and cell proliferation, among other functions (Amaya and Nathanson, 2013; Bollen et al., 1987; Nathanson et al., 1992). The regulation of glucose metabolism by cytosolic Ca2+ results from its ability to directly modulate the activity of key enzymes in the gluconeogenesis pathway such as pyruvate carboxylase, an enzyme that mediates the synthesis of oxaloacetate from pyruvate, and phosphoenolpyruvate carboxykinase, which converts oxaloacetate into phosphoenol-pyruvate. Also, a rise in cytosolic Ca2+ levels leads to activation of the CaMKinase-FOXO pathway, which regulates the expression of key gluconeogenesis genes (discussed in more detail below). In addition to gluconeogenesis, Ca2+ regulates glycogenolysis through the stimulation of the phosphorylase kinase and activation of glycogen phosphorylase, the enzyme responsible for the catalysis of glucagon to glucose-1-phosphate (Amaya and Nathanson, 2013; Exton, 1987). Opposing the effects of glucagon and epinephrine is insulin, the primary hormone stimulating glucose storage. Insulin influences cytosolic Ca2+ levels through the stimulation of AKT. It has been shown that AKT is able to directly phosphorylate IP3R, inhibiting its function, attenuating cytosolic Ca2+ signaling and leading to increased ER Ca2+ concentrations. IP3R phosphorylation by AKT also has a strong antiapoptotic effect (Khan et al., 2006; Marchi et al., 2012).
In the setting of obesity, the reduction in insulin sensitivity and the continuous breakdown of lipids in adipose tissue and the associated hormonal signals lead to over-activation of gluconeogenesis, contributing to hyperglycemia. Reduction in insulin sensitivity also contributes to decreased fatty acid oxidation and increased de novo lipid synthesis in the liver (Fu et al.,2012b). Dysfunction of cellular organelles including the ER and mitochondria are key mechanisms in the development of abnormal insulin action and metabolic alterations in the liver (Fu et al., 2012b; Mantena et al., 2008; Ozcan et al., 2004), and emerging evidence suggests that disruption of Ca2+ homeostasis in these organelles plays a central role.
In searching for the mechanisms underlying the development of ER dysfunction and stress in metabolic disease, our group identified obesity-induced dysfunctional ER Ca2+ transport as an important cause of ER stress in the liver (Fu et al., 2011). Excessive energy intake in obesity stimulates de novo lipogenesis in the liver leading to accumulation of lipid droplets and also to an imbalance in the synthesis of PC and PE, the main phospholipids comprising the organelle membranes. Increased PC/PE ratio of the ER membrane leads to altered membrane fluidity which reduces SERCA2b function, resulting in decreased Ca2+ accumulation in the ER lumen and impaired protein folding capacity (Fu et al., 2011). Similarly, two independent groups have recently shown that the incorporation of saturated fatty acids into the ER also reduces membrane fluidity and SERCA function, decreasing luminal ER Ca2+ (Egnatchik et al., 2014; Zhang et al., 2014b). It has also been shown that modulation of LXRs regulates ER function through changes in membrane phospholipid composition and lipogenic and inflammatory responses (Rong et al., 2013). The importance of this mechanism for the development of fatty liver and diabetes is evidenced by the fact that overexpression of SERCA improves ER folding capacity and results in better glycemic control in obese leptin-deficient mice (Fu et al., 2011; Park et al., 2010).
Based on high-throughput functional ER screening systems, our group recently identified a new compound able to increase SERCA2b expression resulting in increased ER Ca2+ accumulation. Interestingly, this compound shows strong anti-diabetic activities in mouse models (Fu, 2015). Similarly, rosiglitazone, an insulin-sensitizing agent in clinical use has also been shown to induce SERCA expression (Shah et al., 2005). In a genome wide association meta analysis study, a SNP at the SERCA1 (ATP2A1) gene locus was found to be associated with BMI (Locke et al., 2015), and SNPs in the SERCA3 locus have independently been linked to diabetes susceptibility (Varadi et al., 1999). Taken together, these studies strongly support an important role for SERCA function in metabolic homeostasis, and suggest that targeting SERCA may be a promising potential therapeutic avenue. Indeed small molecule SERCA activators have been identified (Cornea et al., 2013; Khan et al., 2009) but their potential effects on metabolic disease have not yet been tested.
Alterations in Ca2+ fluxes also contribute to hepatic mitochondrial dysfunction in obesity. We recently showed that in the context of overnutrition and obesity, the total ER membrane content in liver cells is diminished and reorganized, with significantly increased ER-mitochondrial contact (MAMs) (Arruda et al., 2014). The ER structure comprises two major morphologies that include the nuclear envelope (NE) and the peripheral ER that is subdivided in cisternae (sheets) and a tubular network (Westrate et al., 2015). The cisternae are densely studded with ribosomes and are preferentially involved in protein synthesis. Since a generalized decrease in protein synthesis is evident in liver tissue in obesity (Fu et al., 2012a), it is likely that the reduced ER content in hepatocytes seen in this condition (Arruda et al., 2014) is primarily related to the cisternae. It will be interesting to carefully characterize and quantitate the relative alterations in different ER domains in the future.
In addition to a greater degree of ER-mitochondrial connectivity, the expression of the Ca2+ channel IP3R and tethering proteins such as PACS-2 were significantly increased in the MAMs isolated from liver cells in two different models of obesity. This resulted in increased Ca2+ transfer from the ER to mitochondria, leading to enhanced oxidative stress and compromised oxidative phosphorylation. Remarkably, experimental suppression of IP3R1 or PACS-2 improved mitochondrial function and resulted in significantly improved glucose tolerance in obese mice (Arruda et al., 2014).
Increased ER-mitochondria interaction and Ca2+ transport has also been shown in conditions of ER stress, such as upon tunicamycin treatment, both in vivo (Arruda et al., 2014) and in vitro (Bravo et al., 2011). Treatment of primary hepatocytes and immortalized H4IIEC3 hepatic cells with palmitate decreased ER Ca2+ levels, elevated mitochondrial Ca2+ content, reduced mitochondrial membrane potential, and elevated ROS; importantly, treatment of these cells with the Ca2+ chelator BAPTA abrogated the lipotoxic effects of palmitate (Egnatchik et al., 2014). It has also been shown that ERO1α, an oxidoreductase involved in protein folding, is increased during ER stress and can bind to IP3R1 activating Ca2+ release from the ER directly modulating mitochondria Ca2+ levels (Anelli et al., 2012). Furthermore, it was recently shown that in the postprandial state (5h after feeding) the area of mitochondria covered with ER doubles, and this is correlated with 20% decrease in mitochondrial oxidative capacity (Sood et al., 2014). Taken together, these findings suggest that Ca2+ is a critical link between ER and mitochondrial stress during nutrient fluctuations and overload. Another group recently reported however, that MAM content might be decreased in the liver of obese mice (Tubbs et al., 2014) although an indirect protein interaction based secondary assay system was employed to evaluate MAM formation. As these inter-organelle interactions, as well as ER and mitochondria abundance is dynamically regulated during metabolic exposures and stage of the pathologies, more research will be required to understand the dynamic regulation and modulation of these connections during metabolic disease and contribution to the pathogenesis.
The role of Ca2+ flux through IP3R in human metabolic disease has been suggested by a recent GWAS meta analysis which identified a SNP in the IP3R2 locus associated with alterations in waist-to-hip ratio adjusted for BMI (Shungin et al., 2015). In addition, a role in hepatic metabolic dysfunction is supported by two other important studies. First, work from the Montminy laboratory demonstrated that protein expression as well as phosphorylation status of IP3R are increased in a genetic model of obesity (db/db mice), resulting in higher cytosolic Ca2+. Upon fasting, high levels of glucagon signaling leads to increased cAMP and activation of PKA, which in turn leads to phosphorylation and activation of IP3R (Wang et al., 2012). Higher cytosolic Ca2+ culminates in the activation of Calcineurin, which promotes CRTC2-mediated induction of the FOXO1 transcriptional partner, PGC1α, to regulate gluconeogenesis, an important contributor to hyperglycemia in obesity. Furthermore, suppression of IP3R1 in the liver enhances glucose metabolism in obese mice (Wang et al., 2012).
Secondly, increased hepatic glucose production mediated by the rise in cytosolic Ca2+ also seems to involve the Ca2+-sensing enzyme CaMKII. Ira Tabas’s group has shown that under fasting conditions, glucagon stimulates Ca2+ release from the ER, which in turn leads to activation of CaMKII. CaMKII phosphorylates p38 and FOXO, leading to FOXO translocation to the nucleus and regulation of gluconeogenic genes. In agreement with the finding of higher cytosolic Ca2+ in obesity, CaMKII is hyperphosphorylated in hepatocytes of obese mice, which contributes to JNK activation, inflammatory signaling, and excessive glucose production (Ozcan et al., 2012). JNK has previously been established as a key inflammatory kinase causing impaired metabolic homeostasis in obesity (Hirosumi et al., 2002; Manning and Davis, 2003; Vallerie and Hotamisligil, 2010), and activation of this pathway is reverted by blocking IP3R1 both pharmacologically and genetically (Ozcan et al., 2012). The CaMKII-p38α pathway also negatively impacts insulin signaling by modulating PERK, a component of the UPR. As a consequence, levels of ATF4, a PERK-regulated transcription factor target, increase, inducing the expression of TRB3, which directly inhibits insulin-mediated AKT phosphorylation. Inhibition of the CaMKII–p38α pathway in hepatocytes makes the cells resistant to UPR activation, and in obese mice, genetic or pharmacological inhibition of CaMKII results in a marked improvement in metabolism, including lowering of blood glucose and insulin (Ozcan et al., 2013). In addition, CaMKII-deficient obese mice have higher nuclear ATF6 levels, which seem to have beneficial effects in obesity and type 2 diabetes. It has been reported that ATF6 suppresses hepatic glucose production through disruption of CREB-CRTC2 interaction, a functionality that is diminished in obesity in liver (Wang et al., 2009) and pancreas (Engin et al., 2014; Engin et al., 2013). Taken together, these findings directly link defective Ca2+ homeostasis with ER stress, inflammatory signaling, and deregulation of gluconeogenesis. In line with this evidence, mice deficient for CaMKK2, an enzyme that activates and phosphorylates CaMKs, are protected from diet induced obesity, hyperglycemia and glucose intolerance, and deletion of this enzyme specifically in the liver improves glucose tolerance (Anderson et al., 2012).
Increased cytosolic Ca2+ in the liver in the context of obesity seems also to have an implication for the control of autophagy, the main pathway through which organelles and single proteins are recycled. It has been shown that in late-stage obesity, autophagic flux is decreased in the liver, and the expression levels of the autophagy machinery proteins, such as ATG7, are markedly suppressed (Yang et al., 2010). Importantly, this seems to be connected with the overexpression of calpain, a Ca2+-dependent protease that cleaves ATG7. Inhibition of calpain activity in vivo by two independent calpain inhibitors resulted in enhanced ATG7 protein expression level and increased autophagy (Yang et al., 2010). More recently, it was shown that lipotoxicity-induced elevation of cytosolic Ca2+ leads to inhibition of fusion between autophagosomes and lysosomes, thereby attenuating the autophagic flux (Park and Lee, 2014). As a result, there is an accumulation of undegraded autophagic substrates such as ubquitinated proteins and lipid droplets. Importantly, treating obese animals with a Ca2+ channel blocker, verapamil, improves ER stress, general autophagic flux, and glucose metabolism in obese animals (Park et al., 2014). These results support a model where ER-mitochondria interactions and Ca2+ fluxes may play an important role in regulation of autophagy, especially in the context of metabolic disease (Figure 4A).
Figure 4. Dysfunction of Ca2+ homeostasis in metabolic disease.

(A) In the liver of obese humans and animal models, the UPR is activated and the phosphorylation of IRE1 (inositol required 1), PERK (PKR-like endoplasmic reticulum localized kinase), and eIF2α are increased. However, the levels of sXBP1 and ATF6 (activating transcription factor 6) activity are decreased in obesity in the liver (similar to beta cells). Due to changes in ER membrane composition (increased ratio of PC/PE) the transport of Ca2+ from cytosol to ER lumen by SERCA is impaired, leading to decreased Ca2+ levels in ER. Also, in the liver of obese animals IP3R1 expression and activity are increased, which contributes both to decreased levels of ER Ca2+ and increased cytosolic and mitochondrial Ca2+. In the mitochondria, Ca2+ overload correlates with increased oxidative stress (ROS) and decreased oxidative function. Increased cytosolic Ca2+ also affects several cellular pathways such as autophagosome accumulation, and activates proteases such as calpain that cleaves and degrades ATG7. Both phenomena culminate in decreased autophagy, as seen in liver in obesity. Increased cytosolic Ca2+ also activates calcineurin leading to dephosphorylation of CRTC2 and its nuclear translocation, and activates CaMK to phosphorylate FOXO, with a direct impact on gluconeogenesis. CaMK also phosphorylates p38 and JNK leading to increased inflammation.
In both adipocytes (B) and macrophages (C) of obese animal models, the UPR is activated and the phosphorylation of IRE1, PERK, ATF6 and eIF2α are increased. (B) In hypertrophic adipocytes there is increased deposition of Ca2+ surrounding the lipid droplets and increased cytosolic Ca2+ levels. NFAT transcription factors isoforms 2 and 4 are induced in obese adipose tissue driving expression of inflammatory genes. TRPV4 (Transient receptor potential cation channel subfamily V member 4) is a Ca2+-permeable ion channel that was first identified as an osmolality sensor. In adipocytes, it acts as a negative regulator of PGC1α and a positive regulator of inflammatory genes and proteins and JNK activity. These effects are related to the activation of ERK1/2. (C) Macrophages derived from obese animals show increased IP3R1 and ERO1α activity, which leads to higher Ca2+ release from ER to cytosol. In addition reduced levels of SERCA2b mRNA and protein expression is observed in this condition. Rise in cytosolic Ca2+ and mitochondrial oxidative stress have been shown to engage the NLRP3 inflammasome pathway. In addition, the alterations in macrophage activity in obesity have been shown to activate CaMKK (Calcium/calmodulin-dependent protein kinase kinase 2), the deletion of which leads to impaired cytokine secretion and phagocytosis and induces morphological changes.
Adipose tissue function and Ca2+ homeostasis
Adipose tissue is a critical regulator of energy metabolism, as it is able to store energy as lipids in triglycerides (TG) under circumstances where excess nutrients are available and to hydrolyze TG, releasing fatty acids in the setting of insufficient caloric intake. Adipose tissue also supports lactational metabolism, and this function requires robust ER function (Gregor et al., 2013). In the setting of obesity or chronic nutrient excess however, perturbations in adipose lipid metabolism occur in response to the integration of various mechanisms, such as lipid accumulation, inflammation, hypoxia and oxidative stress, with pathogenic consequences (Gregor and Hotamisligil, 2007). Organelle stress such as increased mitochondrial lipid catabolism, production of ROS and UPR activation are key mechanisms strongly contributing to adipose tissue inflammation including JNK activation, decline in metabolic function, decreased survival, and the efflux of free fatty acids and other mediators from adipocytes in to the circulation (Cao et al., 2008; Eissing et al., 2013; Gregor and Hotamisligil, 2007; Guilherme et al., 2008; Herman et al., 2012; Kershaw and Flier, 2004; Ozcan et al., 2004; Rajala and Scherer, 2003; Sun et al., 2011). However, adipocytes present unique challenges to study these processes and, in general, there are important gaps in studies exploring the ER or mitochondrial structure and function in adipocytes.
In mammals, investigation of the role of Ca2+ homeostasis in adipose tissue biology and pathophysiology is a new area of activity, however the accumulating evidence so far suggests that Ca2+ imbalance may have important consequences for adipocyte function. For example, a rise in cytosolic Ca2+ has been shown to promote triglyceride accumulation and lipid storage by controlling de novo lipogenesis through the regulation of FAS expression (Jones et al., 1996; Zemel et al., 1995; Zemel et al., 2000). It is important to note that an acute rise in cytosolic Ca2+ can transduce a lipolytic signal, while sustained high levels of Ca2+ inhibit lipolysis (Zemel et al., 2000). Modulation of cytosolic Ca2+ has also been shown to regulate adipogenesis: increasing cytosolic Ca2+ by either inhibiting Ca2+-ATPase or stimulating Ca2+ influx inhibits the early stages of murine adipocyte differentiation in mouse models and human cell lines (Shi et al., 2000). Furthermore, increasing cytosolic Ca2+ induces NFAT transcription factors, which in addition to promoting the production of inflammatory cytokines and other immune mediators also play a role in adipocyte differentiation and function by interacting with C/EBP (Ho et al., 1998). It is likely that other transcriptional and hormonal programs are also responsive to changes in Ca2+ fluxes and impact adipocyte differentiation, function, and survival. For example, increasing Ca2+ levels in adipocytes also induces the secretion of hormones and cytokines such as aP2 or FABP4 (Ertunc et al., 2015; Fu et al., 2012b; Schlottmann et al., 2014), and Ca2+ also seems to regulate the thermogenic capacity of brown adipocytes (Leaver and Pappone, 2002) (Figure 4B).
Given these findings, it follows that alterations in Ca2+ signaling may play a role in adipose tissue dysfunction in metabolic disease. For example, within hypertrophic adipocytes it has been demonstrated that there is deposition of Ca2+ surrounding the lipid droplets, possibly indicating increased cytosolic Ca2+ in this condition (Giordano et al., 2013). Also, adipocytes isolated from obese elderly humans display elevated cytosolic Ca2+ levels (Byyny et al., 1992). In agreement with the finding that obese adipocytes contain elevated cytosolic Ca2+, it has been shown that NFAT transcription factor isoforms 2 (NFATc2) and 4 (NFATc4) are induced in obesity and also during adipogenesis (Yang et al., 2006). Interestingly, mice deficient for both NFATc2 and NFATc4 have decreased production of resistin in adipose tissue, are insulin sensitive, and resistant to diet-induced obesity (Yang et al., 2006). In obesity, both in experimental models and in humans, there is significantly increased JNK activity in adipose tissue, which has significant metabolic consequences (Boden et al., 2008; Gregor et al., 2009; Tobar et al., 2011; Tsukumo et al., 2007). Since this pathway is activated by CaMK in liver, a similar regulatory link can also be envisioned for adipose tissue supporting a role for Ca2+ fluxes in activation of inflammatory and stress signaling pathways.
Another piece of evidence of the connection between Ca2+ signaling and adipose tissue dysfunction was recently reported during the search for small molecules that stimulate mitochondrial function and proliferation based on PGC1α activation. Using this approach, a plasma membrane Ca2+ channel, TRPV4, was identified as a negative regulator of PGC1α and a positive regulator of inflammatory genes. TRPV4 is highly expressed in adipose tissue, and deletion or pharmacological inhibition in vivo leads to activation of energy expenditure, activation of brown fat function, and decreased inflammation, and protects mice from diet-induced obesity (Ye et al., 2012). Although not directly measured, this effect may also involve fluctuations in Ca2+ import to the cell.
The most compelling evidence implicating dysfunction in Ca2+ metabolism and its implication to lipid metabolism comes from studies performed in the model organism Drosophila melanogaster. In Drosophila, systemic metabolism is governed largely by the fat body, an important immunometabolic organ which may be considered the equivalent of the mammalian liver, adipose tissue, and hematopoietic and immune systems (Hotamisligil, 2006). Thus the fly’s fat body serves a central role in sensing energy and nutrient availability and coordinating the appropriate metabolic, immune, and survival responses. Thus, mechanisms studied therein may serve as models for evolutionarily conserved pathways in multiple mammalian tissues (Tong et al., 2000). In Drosophila it has been recently shown that STIM acts as a potential regulator of adiposity. Genetic downregulation of STIM1 leads to decreased intracellular Ca2+, which correlates with an increase in body fat accumulation in the flies. The mechanisms through which STIM-deficiency directly regulates adipose tissue content are not known but seem to involve a fat body-brain signaling axis (Baumbach et al., 2014). In addition, Drosophila bearing mutations in IP3R exhibit obesity (Subramanian et al., 2013), indicating that changes in Ca2+ homeostasis directly impact fat deposition in this organism.
Alterations in SERCA function and ER Ca2+ uptake also seem to connect Ca2+ homeostasis with adiposity in Drosophila, as lipodystrophy mediated by disruption of Seipin, a protein involved in adipocyte differentiation and lipid droplet formation, can be explained by decreased ER Ca2+ transport through SERCA. Expression of mutant seipin in Drosophila fat bodies results in defective SERCA activity, lower ER content and elevated cytosolic Ca2+ levels. This coincides with decreased fat storage and decreased lipogenesis (Bi et al., 2014). In the same direction, it has been shown that genetic suppression of IP3R and RyR in Drosophila leads to increased fat accumulation, demonstrating that proper Ca2+ regulation is essential for fat tissue storage in the fly (Bi et al., 2014). Although the implications for higher organisms remain to be explored, the conservation of many critical molecules and pathways suggests that future work in this or other model organisms may be invaluable in the unraveling the role of Ca2+ homeostasis in metabolism and pave the way for exploring the functional consequences and mechanisms in other systems.
Inflammatory cells
As described in the introduction, the chronic metabolic diseases under consideration here are linked to metaflammation, and as such, immune cell function and phenotype make critical contributions to disease pathogenesis through integrated networks with metabolic and stromal cellular components. While many stromal and immune effector cells interact to generate and sustain metaflammation, macrophages are the most intensely studied and their infiltration of metabolic tissues such as adipose tissue and pancreatic islets is a hallmark of obesity (Ehses et al., 2007; Stefanovic-Racic et al., 2012; Weisberg et al., 2003; Xu et al., 2003). Metabolic signals and stimuli resulting from excess nutrient intake and metabolic stress drive adipose tissue macrophages toward a pro-inflammatory state (Lumeng et al., 2007). In parallel with the changes described above in liver and adipose tissue, organelle stress and alterations in Ca2+ homeostasis also play an important role in immune cell dysfunction and inflammatory activity in metabolic disease.
For example, macrophages isolated from genetically obese (ob/ob) mice display increased IP3R1 activity, which leads to higher Ca2+ release from ER to cytosol. This process seems to involve the regulation of IP3R1 by the oxi-reductase protein ERO1α, as silencing ERO1α or IP3R1 in macrophages restores ER homeostasis and reduces ER stress-induced apoptosis (Li et al., 2009). Macrophages from ob/ob mice and insulin- resistant IRS1 deficient mice also display reduced levels of SERCA2b mRNA and protein expression, and accordingly, a depleted ER Ca2+ pool. Interestingly, this effect is linked to reduced MEK signaling, as expression of constitutively active MEK in insulin-resistant macrophages induces SERCA mRNA expression and subsequently restores ER Ca2+ stores (Liang et al., 2012). These results suggest a direct role for defective SERCA activity, ER stress, and cytoplasmic Ca2+ signaling in the apoptosis and inflammatory phenotype of macrophages. Macrophage ER stress is also an important underlying factor in the pathogenesis of atherosclerosis (Hotamisligil, 2010a; Tabas, 2010). In vivo monitoring has documented activation of the unfolded protein response in macrophages during the progression of atherosclerosis (Thorp et al., 2011), and macrophage apoptosis and plaque instability are linked to ER stress induced by increased levels of circulating lipids (Devries-Seimon et al., 2005; Seimon et al., 2010). Furthermore, administration of chemical chaperones or suppression of a lipid binding protein to alleviate ER stress decreases lipid-induced macrophage apoptosis in vitro and the formation of atherosclerotic plaques in vivo (Erbay et al., 2009).
Alterations in Ca2+ signaling have also been tied to macrophage dysfunction in metabolic disease. For example, rising cytosolic Ca2+ (either coming from outside the cell or from the ER through IP3R-mediated Ca2+ release) has been shown to engage the NLRP3 inflammasome pathway, and the administration of IP3R and/or RyR inhibitors reverts this effect (Horng, 2014; Murakami et al., 2012; Rossol et al., 2012). However, how Ca2+ directly modulates NLRP3 is not well understood. It has been suggested that Ca2+ promotes the assembly of NLRP3 components (Lee et al., 2012), while another possible mechanism involves mitochondrial Ca2+ overload followed by increased ROS production and PTP formation as an intermediate step (Murakami et al., 2012). In addition, SOCE plays an important role in inflammatory function, as it is linked with to the production of reactive oxygen species and is necessary for phagocytosis by macrophages and neutrophils (Kang et al., 2012; Zhang et al., 2014a). Furthermore, the alterations in macrophage activity in obesity have been shown to involve CaMKK signaling. This protein is activated in macrophages derived from obese mice, and its deletion leads to impaired cytokine secretion and phagocytosis and induces morphological changes. Interestingly, obese animals lacking CaMKK are protected from high fat diet-induced glucose intolerance indicating the relevance in an immunometabolic context (Racioppi et al., 2012).
Finally, the production of cytokines and immunomodulators by immune cells can also affect Ca2+ signaling in other metabolic cells. This is evident in pancreas, for example, in the context of both type 1 and type 2 diabetes. In type 1 diabetes, cytokines such as IL-1 and IFN produced by immune cells (T cells and macrophages) act via NF-κB activation and nitric oxide production in beta cells to suppress SERCA2b expression and activity (Cardozo et al., 2005; Ramadan et al., 2011). A direct consequence is a decrease in ER luminal Ca2+ and ER stress, ultimately leading to defective adaptive responses and disease (Cardozo et al., 2005; Engin et al., 2013). In addition, chronic treatment of beta cells with a combination of cytokines up-regulates expression of voltage L-type Ca2+ channels resulting in increased intercellular Ca2+ (Kato et al., 1996). Taken together, these data support a model in which Ca2+ signaling plays an important role in mediating organelle dysfunction, and metaflammation, both by modulating immune or metabolic cell function and by altering the responses emanating from these cells on each other (Figure 4C).
Concluding Remarks and Perspectives
Multiple lines of evidence presented here demonstrate that obesity leads to dysregulation of cytosolic and organelle Ca2+ fluxes and disrupted equilibrium in metabolic tissues and immune cells, which in turn alters organelle homeostasis, signaling pathways, and autophagy. These changes at the cellular and organelle level translate directly into effects on hepatic glucose production, lipogenesis, inflammation and other processes that impact systemic metabolism and overall metabolic health. Therefore, Ca2+ handling could be considered a critical problem in metabolic disease.
As this field advances, we see some major open questions that will guide future research. Overall, these questions relate to gaps in the understanding of mechanisms that are dysfunctional during metabolic disease, for example the role of SOCE in organelle homeostasis and metabolic regulation, and how the balance between different components of Ca2+ homeostasis are impacted at different sites and processes of metabolic homeostasis. In this respect, an interesting question remains in exploring the role of Ca2+ signaling and control of organelle function in white and brown adipose tissue and possibly browning of white adipose tissue (Bartelt and Heeren, 2014). A second area for exploration is the immunometabolic abnormalities as a function of Ca2+ homeostasis in obesity, especially in adipose tissue but also in other organs, and how these alterations are related to specific metabolic pathologies such as insulin resistance, glucose production, lipogenesis, defective hormone production and secretion, and hyperglycemia. Third is the compelling possibility supported by recent studies that dysfunction of ER and mitochondria, crucial aspects of metabolism, may be integrated through Ca2+ homeostasis and fluxes regulating systemic metabolism. Details of the molecular components that support such integration as well as other components of Ca2+ handling by both ER and mitochondria will be important areas of future research. Finally, and perhaps most critically, studies are needed to determine how we can target Ca2+ signaling and homeostasis for therapeutic applications best. Since Ca2+ signaling is a crucial component of the normal physiological functions of every cell type, careful considerations are necessary to design the potential therapeutic strategies, regimens, and targets as well as designing the best pharmacological or other interventional approaches to engage or attenuate these pathways in obesity, diabetes and possibly many other chronic, immunometabolic diseases.
Acknowledgments
We regret the omission of many valuable contributions of our colleagues due to space limitations. We are very grateful to Dr. Kathryn Claiborn, Dr. Alexander Bartelt and Dr. Benedicte M. Pers for critical reading and editing of this manuscript. We thank Bruce Worden for assistance with the illustrations. Research in the Hotamisligil lab is supported by the National Institutes of Health (DK052539, DK064360, HL125753, AI116901), the Juvenile Diabetes Research Foundation, UCB and Servier Pharmaceuticals. A.P.A. is supported by PEW Charitable Trusts.
Footnotes
The authors have no conflicts of interest related to the contents of this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amaya MJ, Nathanson MH. Calcium signaling in the liver. Comprehensive Physiology. 2013;3:515–539. doi: 10.1002/cphy.c120013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KA, Lin F, Ribar TJ, Stevens RD, Muehlbauer MJ, Newgard CB, Means AR. Deletion of CaMKK2 from the liver lowers blood glucose and improves whole-body glucose tolerance in the mouse. Mol Endocrinol. 2012;26:281–291. doi: 10.1210/me.2011-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda AP, Pers BM, Parlakgul G, Guney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nature medicine. 2014;20:1427–1435. doi: 10.1038/nm.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nature reviews Endocrinology. 2014;10:24–36. doi: 10.1038/nrendo.2013.204. [DOI] [PubMed] [Google Scholar]
- Baumbach J, Hummel P, Bickmeyer I, Kowalczyk KM, Frank M, Knorr K, Hildebrandt A, Riedel D, Jackle H, Kuhnlein RP. A Drosophila in vivo screen identifies store-operated calcium entry as a key regulator of adiposity. Cell metabolism. 2014;19:331–343. doi: 10.1016/j.cmet.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Calcium signalling remodelling and disease. Biochem Soc Trans. 2012;40:297–309. doi: 10.1042/BST20110766. [DOI] [PubMed] [Google Scholar]
- Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12526–12534. doi: 10.1073/pnas.1302455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi J, Wang W, Liu Z, Huang X, Jiang Q, Liu G, Wang Y, Huang X. Seipin promotes adipose tissue fat storage through the ER Ca(2)(+)-ATPase SERCA. Cell metabolism. 2014;19:861–871. doi: 10.1016/j.cmet.2014.03.028. [DOI] [PubMed] [Google Scholar]
- Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, Cheung P, Merali S. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes. 2008;57:2438–2444. doi: 10.2337/db08-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollen M, Kee SM, Graves DJ, Soderling TR. Substrate specificity of phosphorylase kinase: effects of heparin and calcium. Archives of biochemistry and biophysics. 1987;254:437–447. doi: 10.1016/0003-9861(87)90122-6. [DOI] [PubMed] [Google Scholar]
- Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira J, et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. Journal of cell science. 2011;124:2143–2152. doi: 10.1242/jcs.080762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugrim AE. Regulation of Ca2+ release by cAMP-dependent protein kinase. A mechanism for agonist-specific calcium signaling? Cell calcium. 1999;25:219–226. doi: 10.1054/ceca.1999.0027. [DOI] [PubMed] [Google Scholar]
- Byyny RL, LoVerde M, Lloyd S, Mitchell W, Draznin B. Cytosolic calcium and insulin resistance in elderly patients with essential hypertension. American journal of hypertension. 1992;5:459–464. doi: 10.1093/ajh/5.7.459. [DOI] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Choi S, Maleth JJ, Park S, Ahuja M, Muallem S. The ER/PM microdomain, PI(4,5)P and the regulation of STIM1-Orai1 channel function. Cell calcium. 2015 doi: 10.1016/j.ceca.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramelo JJ, Parodi AJ. Getting in and out from calnexin/calreticulin cycles. The Journal of biological chemistry. 2008;283:10221–10225. doi: 10.1074/jbc.R700048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, Tonnesen M, Van Eylen F, Mandrup-Poulsen T, Herchuelz A, Eizirik DL. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes. 2005;54:452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- Carrasco S, Meyer T. STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annual review of biochemistry. 2011;80:973–1000. doi: 10.1146/annurev-biochem-061609-165311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal K, Balderas-Villalobos J, Bello-Sanchez MD, Phillips-Farfan B, Molina-Munoz T, Aldana-Quintero H, Gomez-Viquez NL. Ca mishandling and cardiac dysfunction in obesity and insulin resistance: Role of oxidative stress. Cell calcium. 2014;56:408–415. doi: 10.1016/j.ceca.2014.08.003. [DOI] [PubMed] [Google Scholar]
- Chaudhuri D, Sancak Y, Mootha VK, Clapham DE. MCU encodes the pore conducting mitochondrial calcium currents. eLife. 2013;2:e00704. doi: 10.7554/eLife.00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe CU, Ehrlich BE. The inositol 1, 4,5-trisphosphate receptor (IP3R) and its regulators: sometimes good and sometimes bad teamwork. Science’s STKE: signal transduction knowledge environment. 2006:re15. doi: 10.1126/stke.3632006re15. [DOI] [PubMed] [Google Scholar]
- Choi MS, Brines RD, Holman MJ, Klaus GG. Induction of NF-AT in normal B lymphocytes by anti-immunoglobulin or CD40 ligand in conjunction with IL-4. Immunity. 1994;1:179–187. doi: 10.1016/1074-7613(94)90096-5. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Colombini M. VDAC structure, selectivity, and dynamics. Biochimica et biophysica acta. 2012;1818:1457–1465. doi: 10.1016/j.bbamem.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland DE, Dalton AJ. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Biophys Biochem Cytol. 1959;5:393–396. doi: 10.1083/jcb.5.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornea RL, Gruber SJ, Lockamy EL, Muretta JM, Jin D, Chen J, Dahl R, Bartfai T, Zsebo KM, Gillispie GD, et al. High-throughput FRET assay yields allosteric SERCA activators. Journal of biomolecular screening. 2013;18:97–107. doi: 10.1177/1087057112456878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Thomas AP, Hajnoczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. The EMBO journal. 1999;18:96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Vance JE, Chen MH, Voelker DR, Vance DE. Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. A specific biochemical and cytological marker for a unique membrane fraction in rat liver. The Journal of biological chemistry. 1993;268:16655–16663. [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- de Meis L, Vianna AL. Energy interconversion by the Ca2+-dependent ATPase of the sarcoplasmic reticulum. Annual review of biochemistry. 1979;48:275–292. doi: 10.1146/annurev.bi.48.070179.001423. [DOI] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Rizzuto R. Molecular control of mitochondrial calcium uptake. Biochemical and biophysical research communications. 2014;449:373–376. doi: 10.1016/j.bbrc.2014.04.142. [DOI] [PubMed] [Google Scholar]
- Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 1961;47:1744–1750. doi: 10.1073/pnas.47.11.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, Davis RJ, Flavell R, Tabas I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. The Journal of cell biology. 2005;171:61–73. doi: 10.1083/jcb.200502078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- Egger G. In search of a germ theory equivalent for chronic disease. Preventing chronic disease. 2012;9:E95. doi: 10.5888/pcd9.110301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egnatchik RA, Leamy AK, Jacobson DA, Shiota M, Young JD. ER calcium release promotes mitochondrial dysfunction and hepatic cell lipotoxicity in response to palmitate overload. Molecular metabolism. 2014;3:544–553. doi: 10.1016/j.molmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, Gueripel X, Ellingsgaard H, Schneider MK, Biollaz G, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–2370. doi: 10.2337/db06-1650. [DOI] [PubMed] [Google Scholar]
- Eissing L, Scherer T, Todter K, Knippschild U, Greve JW, Buurman WA, Pinnschmidt HO, Rensen SS, Wolf AM, Bartelt A, et al. De novo lipogenesis in human fat and liver is linked to ChREBP-beta and metabolic health. Nature communications. 2013;4:1528. doi: 10.1038/ncomms2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine reviews. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- Engin F, Nguyen T, Yermalovich A, Hotamisligil GS. Aberrant islet unfolded protein response in type 2 diabetes. Scientific reports. 2014;4:4054. doi: 10.1038/srep04054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL, Mathis D, Hotamisligil GS. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Science translational medicine. 2013;5:211ra156. doi: 10.1126/scitranslmed.3006534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, Fazio S, Wiest MM, Watkins SM, Linton MF, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nature medicine. 2009;15:1383–1391. doi: 10.1038/nm.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JR. Mechanisms of CaMKII Activation in the Heart. Frontiers in pharmacology. 2014;5:59. doi: 10.3389/fphar.2014.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertunc ME, Sikkeland J, Fenaroli F, Griffiths G, Daniels MP, Cao H, Saatcioglu F, Hotamisligil GS. Secretion of fatty acid binding protein aP2 from adipocytes through a nonclassical pathway in response to adipocyte lipase activity. Journal of lipid research. 2015;56:423–434. doi: 10.1194/jlr.M055798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshima H, Poole DC, Kano Y. In vivo calcium regulation in diabetic skeletal muscle. Cell calcium. 2014;56:381–389. doi: 10.1016/j.ceca.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Exton JH. Mechanisms of hormonal regulation of hepatic glucose metabolism. Diabetes/metabolism reviews. 1987;3:163–183. doi: 10.1002/dmr.5610030108. [DOI] [PubMed] [Google Scholar]
- Fernandez-Velasco M, Ruiz-Hurtado G, Gomez AM, Rueda A. Ca handling alterations and vascular dysfunction in diabetes. Cell calcium. 2014;56:397–407. doi: 10.1016/j.ceca.2014.08.007. [DOI] [PubMed] [Google Scholar]
- Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proceedings of the National Academy of Sciences of the United States of America; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiological reviews. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- Foskett JK, Philipson B. The mitochondrial Ca(2+) uniporter complex. Journal of molecular and cellular cardiology. 2015;78:3–8. doi: 10.1016/j.yjmcc.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiological reviews. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Fan J, Blanco J, Gimenez-Cassina A, Danial NN, Watkins SM, Hotamisligil GS. Polysome profiling in liver identifies dynamic regulation of endoplasmic reticulum translatome by obesity and fasting. PLoS genetics. 2012a;8:e1002902. doi: 10.1371/journal.pgen.1002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell metabolism. 2012b;15:623–634. doi: 10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Fu S, Yalcin A, Lee GY, Li P, Fan J, Arruda AP, Pers BM, Yilmaz M, Eguchi K, Hotamisligil GS. Phenotypic assays identify a small molecule modulator of the unfolded protein response with anti-diabetic activity. Science translational medicine. 2015 doi: 10.1126/scitranslmed.aaa9134. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473:528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilon P, Chae HY, Rutter GA, Ravier MA. Calcium signaling in pancreatic beta-cells in health and in Type 2 diabetes. Cell calcium. 2014;56:340–361. doi: 10.1016/j.ceca.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Giordano A, Murano I, Mondini E, Perugini J, Smorlesi A, Severi I, Barazzoni R, Scherer PE, Cinti S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. Journal of lipid research. 2013;54:2423–2436. doi: 10.1194/jlr.M038638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C, De Stefani D, Bononi A, Rizzuto R, Pinton P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. The international journal of biochemistry & cell biology. 2009;41:1817–1827. doi: 10.1016/j.biocel.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell metabolism. 2012;15:635–645. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxidants & redox signaling. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Thematic review series: Adipocyte Biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. Journal of lipid research. 2007;48:1905–1914. doi: 10.1194/jlr.R700007-JLR200. [DOI] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual review of immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- Gregor MF, Misch ES, Yang L, Hummasti S, Inouye KE, Lee AH, Bierie B, Hotamisligil GS. The role of adipocyte XBP1 in metabolic regulation during lactation. Cell reports. 2013;3:1430–1439. doi: 10.1016/j.celrep.2013.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, Klein S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Hernandez A, Verkhratsky A. Calcium signalling in diabetes. Cell calcium. 2014;56:297–301. doi: 10.1016/j.ceca.2014.08.009. [DOI] [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nature reviews Molecular cell biology. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, Gelinas C, Neems DS, Sasaki Y, Feske S, et al. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Molecular and cellular biology. 2008;28:5209–5222. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Sigma-1 receptors (sigma(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. The Journal of pharmacology and experimental therapeutics. 2003;306:718–725. doi: 10.1124/jpet.103.051284. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Herman MA, Peroni OD, Villoria J, Schon MR, Abumrad NA, Bluher M, Klein S, Kahn BB. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484:333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Ho IC, Kim JH, Rooney JW, Spiegelman BM, Glimcher LH. A potential role for the nuclear factor of activated T cells family of transcriptional regulatory proteins in adipogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15537–15541. doi: 10.1073/pnas.95.26.15537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annual review of immunology. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends in immunology. 2014;35:253–261. doi: 10.1016/j.it.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nature medicine. 2010a;16:396–399. doi: 10.1038/nm0410-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010b;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Ishiguro K, Green T, Rapley J, Wachtel H, Giallourakis C, Landry A, Cao Z, Lu N, Takafumi A, Goto H, et al. Ca2+/calmodulin-dependent protein kinase II is a modulator of CARMA1-mediated NF-kappaB activation. Molecular and cellular biology. 2006;26:5497–5508. doi: 10.1128/MCB.02469-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BH, Kim JH, Zemel MB, Woychik RP, Michaud EJ, Wilkison WO, Moustaid N. Upregulation of adipocyte metabolism by agouti protein: possible paracrine actions in yellow mouse obesity. The American journal of physiology. 1996;270:E192–196. doi: 10.1152/ajpendo.1996.270.1.E192. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Kang J, Park KH, Kim JJ, Jo EK, Han MK, Kim UH. The role of CD38 in Fcgamma receptor (FcgammaR)-mediated phagocytosis in murine macrophages. The Journal of biological chemistry. 2012;287:14502–14514. doi: 10.1074/jbc.M111.329003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Ishida H, Tsuura Y, Tsuji K, Nishimura M, Horie M, Taminato T, Ikehara S, Odaka H, Ikeda I, et al. Alterations in basal and glucose-stimulated voltage-dependent Ca2+ channel activities in pancreatic beta cells of non-insulin-dependent diabetes mellitus GK rats. The Journal of clinical investigation. 1996;97:2417–2425. doi: 10.1172/JCI118688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. The Journal of clinical endocrinology and metabolism. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- Khan H, Metra M, Blair JE, Vogel M, Harinstein ME, Filippatos GS, Sabbah HN, Porchet H, Valentini G, Gheorghiade M. Istaroxime, a first in class new chemical entity exhibiting SERCA-2 activation and Na-K-ATPase inhibition: a new promising treatment for acute heart failure syndromes? Heart failure reviews. 2009;14:277–287. doi: 10.1007/s10741-009-9136-z. [DOI] [PubMed] [Google Scholar]
- Khan MT, Wagner L, 2nd, Yule DI, Bhanumathy C, Joseph SK. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. The Journal of biological chemistry. 2006;281:3731–3737. doi: 10.1074/jbc.M509262200. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free radical biology & medicine. 2009;47:333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Leaver EV, Pappone PA. Beta-adrenergic potentiation of endoplasmic reticulum Ca(2+) release in brown fat cells. American journal of physiology Cell physiology. 2002;282:C1016–1024. doi: 10.1152/ajpcell.00204.2001. [DOI] [PubMed] [Google Scholar]
- Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–127. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR, Tabas I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. The Journal of cell biology. 2009;186:783–792. doi: 10.1083/jcb.200904060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CP, Han S, Li G, Tabas I, Tall AR. Impaired MEK signaling and SERCA expression promote ER stress and apoptosis in insulin-resistant macrophages and are reversed by exenatide treatment. Diabetes. 2012;61:2609–2620. doi: 10.2337/db11-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of clinical investigation. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. The Journal of clinical investigation. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Anderson ME. Mechanisms of altered Ca(2)(+) handling in heart failure. Circulation research. 2013;113:690–708. doi: 10.1161/CIRCRESAHA.113.301651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nature reviews. Drug discovery. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free radical biology & medicine. 2008;44:1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Wiedmann M. Calcineurin enhances MEF2 DNA binding activity in calcium-dependent survival of cerebellar granule neurons. The Journal of biological chemistry. 1999;274:31102–31107. doi: 10.1074/jbc.274.43.31102. [DOI] [PubMed] [Google Scholar]
- Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, Pinton P. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell death & disease. 2012;3:e304. doi: 10.1038/cddis.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiological reviews. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annual review of physiology. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina A, Reina S, Guarino F, De Pinto V. VDAC isoforms in mammals. Biochimica et biophysica acta. 2012;1818:1466–1476. doi: 10.1016/j.bbamem.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. The Biochemical journal. 2009;417:651–666. doi: 10.1042/BJ20081847. [DOI] [PubMed] [Google Scholar]
- Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:11282–11287. doi: 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K, Cooper TJ, Thomas G, Simmen T. The subcellular distribution of calnexin is mediated by PACS-2. Molecular biology of the cell. 2008;19:2777–2788. doi: 10.1091/mbc.E07-10-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson MH, Gautam A, Bruck R, Isales CM, Boyer JL. Effects of Ca2+ agonists on cytosolic Ca2+ in isolated hepatocytes and on bile secretion in the isolated perfused rat liver. Hepatology. 1992;15:107–116. doi: 10.1002/hep.1840150119. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Cristina de Souza J, Harari AA, Backs J, Olson EN, Tabas I. Activation of calcium/calmodulin-dependent protein kinase II in obesity mediates suppression of hepatic insulin signaling. Cell metabolism. 2013;18:803–815. doi: 10.1016/j.cmet.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan L, Wong CC, Li G, Xu T, Pajvani U, Park SK, Wronska A, Chen BX, Marks AR, Fukamizu A, et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell metabolism. 2012;15:739–751. doi: 10.1016/j.cmet.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Palty R, Hershfinkel M, Sekler I. Molecular identity and functional properties of the mitochondrial Na+/Ca2+ exchanger. The Journal of biological chemistry. 2012;287:31650–31657. doi: 10.1074/jbc.R112.355867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Lee JH. Calcium channel blockers as potential therapeutics for obesity-associated autophagy defects and fatty liver pathologies. Autophagy. 2014;0 doi: 10.4161/15548627.2014.984268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HW, Park H, Semple IA, Jang I, Ro SH, Kim M, Cazares VA, Stuenkel EL, Kim JJ, Kim JS, et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nature communications. 2014;5:4834. doi: 10.1038/ncomms5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, Zhou Y, Lee J, Lee J, Ozcan U. Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19320–19325. doi: 10.1073/pnas.1012044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Annals of the New York Academy of Sciences. 2010;1201:183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- Pereira L, Ruiz-Hurtado G, Rueda A, Mercadier JJ, Benitah JP, Gomez AM. Calcium signaling in diabetic cardiomyocytes. Cell calcium. 2014;56:372–380. doi: 10.1016/j.ceca.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Petersen OH, Tepikin A, Park MK. The endoplasmic reticulum: one continuous or several separate Ca(2+) stores? Trends Neurosci. 2001;24:271–276. doi: 10.1016/s0166-2236(00)01787-2. [DOI] [PubMed] [Google Scholar]
- Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiological reviews. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- Prasad V, Okunade GW, Miller ML, Shull GE. Phenotypes of SERCA and PMCA knockout mice. Biochemical and biophysical research communications. 2004;322:1192–1203. doi: 10.1016/j.bbrc.2004.07.156. [DOI] [PubMed] [Google Scholar]
- Prins D, Michalak M. Organellar calcium buffers. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a004069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes & development. 2007;21:1443–1455. doi: 10.1101/gad.1550907. [DOI] [PubMed] [Google Scholar]
- Racioppi L, Noeldner PK, Lin F, Arvai S, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2 regulates macrophage-mediated inflammatory responses. The Journal of biological chemistry. 2012;287:11579–11591. doi: 10.1074/jbc.M111.336032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala MW, Scherer PE. Minireview: The adipocyte--at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–3773. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- Ramadan JW, Steiner SR, O’Neill CM, Nunemaker CS. The central role of calcium in the effects of cytokines on beta-cell function: implications for type 1 and type 2 diabetes. Cell calcium. 2011;50:481–490. doi: 10.1016/j.ceca.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimessi A, Bonora M, Marchi S, Patergnani S, Marobbio CM, Lasorsa FM, Pinton P. Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy. 2013;9:1677–1686. doi: 10.4161/auto.24795. [DOI] [PubMed] [Google Scholar]
- Riquelme CA, Magida JA, Harrison BC, Wall CE, Marr TG, Secor SM, Leinwand LA. Fatty acids identified in the Burmese python promote beneficial cardiac growth. Science. 2011;334:528–531. doi: 10.1126/science.1210558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Bernardi P, Favaron M, Azzone GF. Pathways for Ca2+ efflux in heart and liver mitochondria. The Biochemical journal. 1987;246:271–277. doi: 10.1042/bj2460271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nature reviews Molecular cell biology. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–327. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell metabolism. 2013;18:685–697. doi: 10.1016/j.cmet.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, Schubert K, Schoneberg T, Schaefer M, Krugel U, et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nature communications. 2012;3:1329. doi: 10.1038/ncomms2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nature reviews Molecular cell biology. 2012;13:607–625. doi: 10.1038/nrm3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlottmann I, Ehrhart-Bornstein M, Wabitsch M, Bornstein SR, Lamounier-Zepter V. Calcium-dependent release of adipocyte fatty acid binding protein from human adipocytes. International journal of obesity. 2014;38:1221–1227. doi: 10.1038/ijo.2013.241. [DOI] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR, 3rd, Takemori H, et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. 2004;119:61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Secor SM, Diamond J. A vertebrate model of extreme physiological regulation. Nature. 1998;395:659–662. doi: 10.1038/27131. [DOI] [PubMed] [Google Scholar]
- Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell metabolism. 2010;12:467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah RD, Gonzales F, Golez E, Augustin D, Caudillo S, Abbott A, Morello J, McDonough PM, Paolini PJ, Shubeita HE. The antidiabetic agent rosiglitazone upregulates SERCA2 and enhances TNF-alpha- and LPS-induced NF-kappaB-dependent transcription and TNF-alpha-induced IL-6 secretion in ventricular myocytes. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2005;15:41–50. doi: 10.1159/000083637. [DOI] [PubMed] [Google Scholar]
- Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- Shi H, Halvorsen YD, Ellis PN, Wilkison WO, Zemel MB. Role of intracellular calcium in human adipocyte differentiation. Physiological genomics. 2000;3:75–82. doi: 10.1152/physiolgenomics.2000.3.2.75. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Ben-Hail D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion. 2012;12:24–34. doi: 10.1016/j.mito.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Shulman GI. Cellular mechanisms of insulin resistance. The Journal of clinical investigation. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Magi R, Strawbridge RJ, Pers TH, Fischer K, Justice AE, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–196. doi: 10.1038/nature14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. The EMBO journal. 2005;24:717–729. doi: 10.1038/sj.emboj.7600559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nature reviews Molecular cell biology. 2012;13:549–565. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood A, Jeyaraju DV, Prudent J, Caron A, Lemieux P, McBride HM, Laplante M, Toth K, Pellegrini L. A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:16017–16022. doi: 10.1073/pnas.1408061111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic-Racic M, Yang X, Turner MS, Mantell BS, Stolz DB, Sumpter TL, Sipula IJ, Dedousis N, Scott DK, Morel PA, et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes. 2012;61:2330–2339. doi: 10.2337/db11-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone SJ, Levin MC, Zhou P, Han J, Walther TC, Farese RV., Jr The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. The Journal of biological chemistry. 2009;284:5352–5361. doi: 10.1074/jbc.M805768200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian M, Metya SK, Sadaf S, Kumar S, Schwudke D, Hasan G. Altered lipid homeostasis in Drosophila InsP3 receptor mutants leads to obesity and hyperphagia. Disease models & mechanisms. 2013;6:734–744. doi: 10.1242/dmm.010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. The Journal of clinical investigation. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. The Journal of cell biology. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai G, Csordas G, Hantash BM, Thomas AP, Hajnoczky G. Calcium signal transmission between ryanodine receptors and mitochondria. The Journal of biological chemistry. 2000;275:15305–15313. doi: 10.1074/jbc.275.20.15305. [DOI] [PubMed] [Google Scholar]
- Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circulation research. 2010;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorp E, Iwawaki T, Miura M, Tabas I. A reporter for tracking the UPR in vivo reveals patterns of temporal and cellular stress during atherosclerotic progression. Journal of lipid research. 2011;52:1033–1038. doi: 10.1194/jlr.D012492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. The Journal of clinical investigation. 2009;119:2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobar N, Oliveira AG, Guadagnini D, Bagarolli RA, Rocha GZ, Araujo TG, Santos-Silva JC, Zollner RL, Boechat LH, Carvalheira JB, et al. Diacerhein improves glucose tolerance and insulin sensitivity in mice on a high-fat diet. Endocrinology. 2011;152:4080–4093. doi: 10.1210/en.2011-0249. [DOI] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A. The importance of being subtle: small changes in calcium homeostasis control cognitive decline in normal aging. Aging cell. 2007;6:267–273. doi: 10.1111/j.1474-9726.2007.00296.x. [DOI] [PubMed] [Google Scholar]
- Tong Q, Dalgin G, Xu H, Ting CN, Leiden JM, Hotamisligil GS. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science. 2000;290:134–138. doi: 10.1126/science.290.5489.134. [DOI] [PubMed] [Google Scholar]
- Toyoshima C, Inesi G. Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annual review of biochemistry. 2004;73:269–292. doi: 10.1146/annurev.biochem.73.011303.073700. [DOI] [PubMed] [Google Scholar]
- Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Araujo EP, Vassallo J, Curi R, Velloso LA, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- Tubbs E, Theurey P, Vial G, Bendridi N, Bravard A, Chauvin MA, Ji-Cao J, Zoulim F, Bartosch B, Ovize M, et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes. 2014;63:3279–3294. doi: 10.2337/db13-1751. [DOI] [PubMed] [Google Scholar]
- Vallerie SN, Hotamisligil GS. The role of JNK proteins in metabolism. Science translational medicine. 2010;2:60rv65. doi: 10.1126/scitranslmed.3001007. [DOI] [PubMed] [Google Scholar]
- Van Petegem F. Ryanodine receptors: structure and function. The Journal of biological chemistry. 2012;287:31624–31632. doi: 10.1074/jbc.R112.349068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. The Journal of biological chemistry. 1990;265:7248–7256. [PubMed] [Google Scholar]
- Vandecaetsbeek I, Vangheluwe P, Raeymaekers L, Wuytack F, Vanoevelen J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi A, Lebel L, Hashim Y, Mehta Z, Ashcroft SJ, Turner R. Sequence variants of the sarco(endo)plasmic reticulum Ca(2+)-transport ATPase 3 gene (SERCA3) in Caucasian type II diabetic patients (UK Prospective Diabetes Study 48) Diabetologia. 1999;42:1240–1243. doi: 10.1007/s001250051298. [DOI] [PubMed] [Google Scholar]
- Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell death and differentiation. 2012;19:1880–1891. doi: 10.1038/cdd.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li G, Goode J, Paz JC, Ouyang K, Screaton R, Fischer WH, Chen J, Tabas I, Montminy M. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature. 2012;485:128–132. doi: 10.1038/nature10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Vera L, Fischer WH, Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460:534–537. doi: 10.1038/nature08111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. The Journal of clinical investigation. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westrate LM, Lee JE, Prinz WA, Voeltz GK. Form Follows Function: The Importance of Endoplasmic Reticulum Shape. Annual review of biochemistry. 2015 doi: 10.1146/annurev-biochem-072711-163501. [DOI] [PubMed] [Google Scholar]
- Wu KD, Lee WS, Wey J, Bungard D, Lytton J. Localization and quantification of endoplasmic reticulum Ca(2+)-ATPase isoform transcripts. The American journal of physiology. 1995;269:C775–784. doi: 10.1152/ajpcell.1995.269.3.C775. [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of clinical investigation. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell metabolism. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang TT, Suk HY, Yang X, Olabisi O, Yu RY, Durand J, Jelicks LA, Kim JY, Scherer PE, Wang Y, et al. Role of transcription factor NFAT in glucose and insulin homeostasis. Molecular and cellular biology. 2006;26:7372–7387. doi: 10.1128/MCB.00580-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Kleiner S, Wu J, Sah R, Gupta RK, Banks AS, Cohen P, Khandekar MJ, Bostrom P, Mepani RJ, et al. TRPV4 is a regulator of adipose oxidative metabolism, inflammation, and energy homeostasis. Cell. 2012;151:96–110. doi: 10.1016/j.cell.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemel MB, Kim JH, Woychik RP, Michaud EJ, Kadwell SH, Patel IR, Wilkison WO. Agouti regulation of intracellular calcium: role in the insulin resistance of viable yellow mice. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:4733–4737. doi: 10.1073/pnas.92.11.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemel MB, Shi H, Greer B, Dirienzo D, Zemel PC. Regulation of adiposity by dietary calcium. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2000;14:1132–1138. [PubMed] [Google Scholar]
- Zhang H, Clemens RA, Liu F, Hu Y, Baba Y, Theodore P, Kurosaki T, Lowell CA. STIM1 calcium sensor is required for activation of the phagocyte oxidase during inflammation and host defense. Blood. 2014a;123:2238–2249. doi: 10.1182/blood-2012-08-450403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li Y, Jiang S, Yu H, An W. Enhanced endoplasmic reticulum SERCA activity by overexpression of hepatic stimulator substance gene prevents hepatic cells from ER stress-induced apoptosis. American journal of physiology Cell physiology. 2014b;306:C279–290. doi: 10.1152/ajpcell.00117.2013. [DOI] [PubMed] [Google Scholar]
- Zhang M, Tanaka T, Ikura M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nature structural biology. 1995;2:758–767. doi: 10.1038/nsb0995-758. [DOI] [PubMed] [Google Scholar]
- Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18:444–452. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]