

Abstract
Background
Alcoholic liver disease (ALD) is commonly associated with intestinal barrier dysfunction. Alcohol-induced dysregulation of intestinal tight junction (TJ) proteins, such as Zonula Occludens-1 (ZO-1), plays an important role in alcohol-induced gut leakiness. However, the mechanism of alcohol-induced disruption of TJ proteins is not well established. The goal of this study was to elucidate this mechanism by studying the role of MicroRNA 212 (miR-212) and inducible nitric oxide synthase (iNOS) in alcohol-induced gut leakiness.
Methods
The permeability of the Caco-2 monolayer was assessed by transepithelial electrical resistance (TER) and flux of fluorescein sulfonic acid (FSA). miR-212 was measured by real time PCR. The wild type, iNOS knockout, and miR-212 knockdown mice were fed with alcohol diet (29% of total calories, 4.5% v/v) for 8 weeks. The LNA-anti-miR-212 was used to inhibit miR-212 expression in mice. The alcohol-induced intestinal permeability, miR-212 expression and liver injuries in mice were measured.
Results
Our in vitro monolayer and in vivo mice studies showed that: (1) alcohol-induced over-expression of the intestinal miR-212 and intestinal hyperpermeability is prevented by using miR-212 knock-down techniques; and (2). iNOS is upregulated in the intestine by alcohol and that iNOS signaling is required for alcohol-induced miR-212 over-expression, ZO-1 disruption, gut leakiness and steatohepatis.
Conclusions
These studies thus support a novel miR-212 mechanism for alcohol-induced gut leakiness and a potential target that could be exploited for therapeutic intervention to prevent leaky gut and liver injury in alcoholics.
Keywords: microRNA 212 (miR-212), inducible nitric oxide synthase (iNOS), intestinal permeability, Zonula Occludens-1 (ZO-1), alcoholic liver disease
Introduction
Alcoholic Liver Disease (ALD) occurs only in a subset (~30%) of alcoholics(Bode and Bode, 2005), indicating that excessive ethanol (EtOH) consumption is necessary but not sufficient to induce liver injury. Therefore, additional factors are required. We and others showed that gut-derived endotoxin appears to be this required co-factor. Thus gut leakiness appears to be the key cause of the endotoxemia in alcohol fed rodents and alcoholics(Keshavarzian et al., 2009; Keshavarzian et al., 1999). Therefore, a comprehensive understanding of the mechanism of alcohol-induced gut leakiness is essential for development of novel therapeutic interventions for prevention and/or treatment of alcohol-induced pathologies like ALD.
The integrity of the intestinal barrier depends on both healthy epithelial cells and on an intact paracellular pathway, which appears to be the main route for permeation of macromolecules such as endotoxin(Hollander, 1992). This pathway is a complex array of structures that includes tight junctions between gut epithelial cells. This dynamic tight junction is highly regulated and is able to change its size under various physiological and pathological conditions(Madara, 1990). Tight junctions function as gates that regulate intestinal permeability(Clayburgh et al., 2004). Cytoplasmic plaque proteins such as Zonula occludens-1 (ZO-1) constitute a major component of tight junctions(Sawada et al., 2003). ZO-1 may work as a scaffold in tight junctions because it interacts with several integral membrane proteins.
One potential mechanism for alcohol to induce gut leakiness is by disrupting tight junction protein homeostasis. Oxidative stress is one well established mechanism of disruption of tight junction proteins (Sawada et al., 2003; Unno et al., 1997). A significant potential source of oxidative stress is intestinal nitric oxide [NO]. Indeed, alcohol-induced overproduction of NO by inducible nitric oxide synthase [iNOS] disrupts barrier function, and prevention of alcohol-induced NO overproduction in rats and in Caco-2 monolayers restores normal barrier integrity(Unno et al., 1997). Moreover, we showed that alcohol-induced gut leakiness is caused by oxidative epithelial tissue damage due to upregulation of iNOS(Tang et al., 2009b). However, the mechanism of alcohol-induced, iNOS-mediated, gut leakiness is not fully understood.
Our recent findings reveal a new mechanism and suggest that microRNA (miRNA) may be an early and key factor in this signaling cascade. The miRNAs are small non-coding RNAs that are 18–25 nucleotides in length and can control gene expression. They target mRNAs, triggering either translational repression or RNA degradation(Carthew, 2006). miRNA regulation has been implicated in many cellular processes including cell proliferation, differentiation, apoptosis, and metabolism(Czech, 2006). Aberrant miRNA expression may be involved in many human diseases(Ambros, 2004). Recently we found that(Tang et al., 2008): (a) miR-212 is a miRNA expressed in intestinal epithelial cells, (b) ZO-1, one of the key tight junctional proteins involved in regulation of intestinal barrier(Sawada et al., 2003), is a target gene of miR-212, (c) miR-212 levels are higher in colon biopsy samples from ALD patients than in healthy controls. (d) Alcohol-induced miR-212 over-expression in Caco-2 intestinal epithelial cells is accompanied by reductions in ZO-1 protein expression, disruption of tight junction protein (ZO-1), and increased permeability of Caco-2 cell monolayers. This finding led to a new hypothesis that iNOS induced disruption of intestinal barrier might be mediated through miR-212 overexpression that in turn leads to down regulating its target gene, ZO-1. The aim of this study was to test this hypothesis using the in vitro Caco-2 cell model and in vivo animal model of alcohol-induced intestinal hyperpermeability.
Methods
1. Inhibition of miR-212 in Caco-2 cells
Caco-2 cells were obtained from American Type Culture Collection (Manassas, VA) at passage 15. We transfected Caco-2 cells with 30 nmol/L anti-miR-212 to inhibit miR-212 expression. Anti-miR-212 Inhibitor (Ambion, Cat #4385914) was designed to bind to, and inhibit the activity of endogenous miR-212 when introduced into cells. This RNA-based inhibitor is sequence-specific and chemically modified both to increase its stability and to improve its activity. Anti-miR-212 was delivered into cells by chemical transfection following the manufacture’s protocol.
2. iNOS knockdown with SiRNA
Caco-2 cells were treated with gene-specific siRNA directed at iNOS or control nontargeting siRNA to control for “off target” (non-gene-specific) effects of siRNA using a modification of our previously published methods(Forsyth et al., 2011).
3. Real-time Quantification of miRNAs by Stem-loop RT-PCR
Total RNA from Caco-2 cells or colon tissue samples were isolated using mirVana RNA isolation kits (Ambion, Austin, TX) according to the manufacturer’s protocol. TaqMan microRNA assay kits with stem-loop RT primer (Applied Biosystems) were used for detection of miR-212. RNU6B was used as an internal control. Experiments and data analysis were done according to the manufacturer’s protocol. Relative quantities of miRNA expression were calculated as in our previous studies(Tang et al., 2008).
4. TER measurement for intestinal permeability in vitro
Intestinal permeability was measured as transepithelial electrical resistance (TER) using Caco-2 cells as previously described(Forsyth et al., 2011). TER was determined using a dual electrode system designed for cell culture insert analysis (EVOM; World Precision Instruments, Sarasota, FL) in which naked culture inserts were used to blank for baseline values, which are subtracted from all values using inserts with living cells.
5. FSA measurement for intestinal permeability in vitro
The permeability of the monolayer barrier was also assessed by a widely used and validated technique that measures the apical to basolateral paracellular flux of fluorescent markers such as fluorescein sulfonic acid (FSA, 200 μg/ml; 0.478 kDa) as we described (Banan et al., 2000b).
6. Western blots for ZO-1 Protein
The ZO-1 protein levels in Caco-2 cells were measured by using Western blotting (Bio-Rad mini-Protein system) as previously described(Forsyth et al., 2007). After blocking, membranes were blotted using antibody to ZO-1 (Zymed). Blots were washed, incubated with HRP-conjugated 2° Ab for 1 h at 4°C, washed, developed with enhanced chemiluminescence (ECL) solution (Amersham), exposed to film (Fuji) and finally scanned for analysis with Image J software(Forsyth et al., 2007).
7. Immunofluorescent Staining of ZO-1 and tight junction morphology
Cells from monolayers were fixed in cytoskeletal stabilization buffer and then postfixed in 95% ethanol at −20°C. Cells were subsequently processed for incubation with a primary antibody (monoclonal mouse anti-ZO-1(Zymed), 1:200 dilutions) for 1 h at 37°C and then incubated with a secondary antibody (fluorescein isothiocyanate-conjugated goat anti-mouse; Sigma-Aldrich, 1:50 dilution) for 1 h at room temperature. Slides were washed 3× in D-phosphate-buffered saline and mounted in Aquamount. Following staining, cells were observed with an Axiovert 100 microscope and Axiovision software (Carl Zeiss Inc., Thornwood, NY). The three-dimensional reconstruction of deconvoluted z-stacks was performed using Zeiss Axiovision software. Immunofluorescent microscopy analysis of Caco-2 monolayers has been previously described by us in detail(Forsyth et al., 2007).
8. Experimental diet and animals
Mice and Housing
Young adult (6–8 week old) wild-type C57BL/6J mice (WT) and C57BL/6 iNOS knockout (KO) mice were purchased from The Jackson Laboratory, Bar Harbor, ME. The animal facilities were accredited through the Association for Assessment and Accreditation of Laboratory Animal Care. All experiments were carried out in accordance with the conditions set forth by the National Institutes of Health and with the approval of the Institutional Animal Care and Use Committee (IACUC) at Rush University Medical Center.
Chronic Alcohol Consumption Protocol
Both WT and iNOS KO mice were fed with the Nanji diet alcohol protocol(Forsyth et al., 2011; Summa et al., 2013), which consisted of a two week introduction and gradual increase in alcohol dose, followed by eight weeks on the full alcohol concentration (29% of total calories, 4.5% v/v). Control mice were fed an isocaloric liquid diet in which the calories from alcohol were replaced with dextrose. The components of the liquid Nanji diet was described in previous study(Summa et al., 2013).
In Vivo Intestinal Permeability Testing
In vivo assessment of intestinal permeability was conducted as described previously(Forsyth et al., 2011; Summa et al., 2013) using a well-validated model to determine permeability across the epithelial barrier in the small intestine and colon(Forsyth et al., 2011; Summa et al., 2013). Briefly, mice were fasted for eight hours prior to the test, which was performed at the same time for all mice. A 200 μL solution containing lactulose (3.2 mg), sucrose (0.45 mg), sucralose (0.45 mg) and mannitol (0.9 mg) was administered via oral gavage, after which 2 mL 0.9% saline was administered subcutaneously to promote urine production. Urine produced over five hours was collected and the total volume was recorded. Intestinal permeability was determined by measuring urinary sugar concentration using gas chromatography, enabling calculation of the amount of orally administered sugar excreted in the urine over five hours. Intestinal permeability was measured at 8 weeks after initiating the full (i.e., 29%) alcohol concentration in the diet. Measurement of urinary sugars is used to calculate intestinal permeability and is expressed as percent oral dose excreted in the urine as we have described (Forsyth et al., 2011; Summa et al., 2013). The widely used 5 hour urinary L/M ratio represents small bowel permeability (L = lactulose, M=mannitol), whereas sucralose reflects small and large intestinal permeability. Sucrose is rapidly degraded after leaving the stomach, so increased sucrose excretion reflects gastric permeability (Forsyth et al., 2011; Summa et al., 2013).
Inhibition of miR-212 in vivo
The LNA-anti-miR-212 (Exiqon, Woburn, MA) was used to inhibit miR-212 expression in mice as previously described (Elmen et al., 2008) and modified in our current study. The mice were given two weekly intraperitoneal doses of 25 mg g−1 LNA-antimiR-212 or LNA mismatch control before starting the Chronic Alcohol Consumption Protocol as described in above. We selected the dose of 25 mg kg−1 based on the publications (Elmen et al., 2008; Krutzfeldt et al., 2007) and our preliminary data. Elment et al reported that intraperitoneal injections of LNA-antimiR at doses ranging from 1 to 200 mg kg−1 resulted in dose-dependent and sustained decrease in total plasma cholesterol with a median effective dose (ED50) of 10 mg g−1 (Elmen et al., 2008). Krutzfeldt, J. et al reported that they used much higher doses (40 mg kg−1) of antagomir-122 to silence efficiently miR-122 in mice (Krutzfeldt et al., 2007). Our preliminary data also indicated that LNA-anti-miR-212 at dose of 25 mg kg−1 significantly silenced miR-212 and did not show obvious side effects. Thus, we select the dose of 25 mg kg−1in our experiments. The LNA-antimiR-212 treatment was then continued weekly for 8 weeks. The inhibition of miR-212 on EtOH-induced miR-212 expression in colon mucosa and gut leakiness was studied.
Tissue Collection
At the conclusion of the experiment, mice were euthanized by conscious decapitation. Proximal colon and liver tissues were harvested for analyses. Samples were either snap-frozen in liquid nitrogen or placed into RNALater (Qiagen, Valencia, CA) and frozen at −80°C.
9. Liver Pathology
Formalin-fixed liver was stained with hematoxylin & eosin (H&E). Blinded assessment of samples was conducted by a gastrointestinal pathologist (SS). Histological analyses, including steatosis, inflammation, ballooning degeneration and the presence of acidophil bodies, were scored according to the following criteria by a GI pathologist [SS] who was blinded to the assignment of the experimental groups: Steatosis: severity was scored as percent hepatocyte involvement (0 = <5%, 1 = 5–33%, 2 = 34–66%, 3 = >67%), corresponding to the fraction of lipid-containing hepatocytes. Inflammation: severity was scored based on the number of inflammatory foci per 200× field (0 = no foci, 1 = 1 focus, 2 = 2–4 foci, 3 = >4 foci). Ballooning degeneration: scored based on the presence and frequency of ballooned cells (0 = none, 1 = few, 2 = prominent/many), as an indication of hepatocyte injury. Acidophil body presence: estimated by the presence and frequency of acidophil bodies (0 = absent, 1 = focal apoptosis (few acidophil bodies), 2 = many acidophil bodies, 3 = confluent necrosis), corresponding to injured hepatocytes demonstrating a feature of programmed cell death. These markers (steatosis, inflammation, ballooning degeneration and acidophil bodies) were selected because they are all well-established markers of steatohepatitis(Brunt and Tiniakos, 2010; Kleiner et al., 2005; Summa et al., 2013).
10. Statistical Analysis
The data are presented as means ± SE. The group means were compared by analysis of variance (ANOVA) and post-hoc tests since the data were normally distributed. P-values < 0.05 were considered statistically significant. All analyses were done using SPSS (SPSS Inc., Chicago, IL).
Results
1. EtOH-induced intestinal miR-212 over-expression and intestinal hyperpermeability
Our previous study showed that EtOH induced miR-212 overexpression and miR-212 over-expression was associated with EtOH-induced hyperpermeability of the CaCo2 cell monolayers (Tang et al., 2008), Thus, we hypothesized that miR-212 plays an important role in EtOH-induced intestinal hyperpermeability. To test this hypothesis, we transfected Caco-2 cells with 30 nM anti-miR-212 or controls. Cells were then treated with 0.2% EtOH for 24 h. Fluorescein sulfonic acid (FSA) clearance was measured as an indication of permeability of the monolayers. The results showed that anti-miR212 significantly inhibited [by 50%] EtOH-induced hyperpermeability of the intestinal epithelial cell monolayers (Fig 1). This suggests that EtOH-induced miR-212 overexpression promotes intestinal hyperpermeability.
Fig. 1. Anti-miR-212 inhibits EtOH-induced hyperpermeability in Caco-2 cells.

Caco-2 cells were transfected with 30nmol/L anti-miR-212 and relevant control. Then, the cells were treated with 0.2%EtOH for 24 h. Fluorescein sulfonic acid (FSA) clearance was measured as an indication of permeability of the monolayers. The results show that anti-miR212 significantly inhibited EtOH-induced hyperpermeability. Data are from triplicate cells from three independent experiments. Mean+SEM, *: p<0.05.
To translate our in vitro observation to an in vivo model to establish a key role of miRNA-212 in EtOH-induced gut leakiness, we turned to an animal model of alcohol-induced gut leakiness. This model has been established and validated in our previous studies(Keshavarzian et al., 2001). In this model, we inhibited miR-212 expression in vivo using LNA™ microRNA technology. Our results showed that intestinal miRNA-212 expression was significantly increased in alcohol fed mice and this over-expression of miRNA-212 was prevented in alcohol fed mice in which miR-212 was inhibited (Fig. 2a). Furthermore, EtOH significantly induced gut leakiness in alcohol fed control mice (mismatched LNA-anti-miR) and this EtOH-induced intestinal hyperpermeability was significantly inhibited in alcohol fed mice in which miR-212 was inhibited using LNA-anti-miR-212 (Fig. 2b).
Fig. 2. Silencing of miR-212 significantly inhibited EtOH-induced miR-212 expression in colon tissue (A) and prevented EtOH-induced intestinal hyperpermeability in mice (B).


(A). The mice were fed with a level of 29% calories from EtOH for 8 weeks. The EtOH significantly increased miR-212 expression levels in control (mismatched anti-miRNAs) mice (*: p<0.05 vs control). The EtOH-induced miR-212 expression was significantly inhibited in miR-212 silenced mice using LNA™ microRNA technology (#: p<0.05 vs Control+EtOH). The miR-212 expression levels were assayed by TaqMan real time PCR. (B). Control (mismatched) and miR-212 silenced mice were fed a liquid diet for 8 weeks as described in Methods and then intestinal permeability was determined by gavage of a sugar solution described in Methods followed by analysis of excreted urinesugar levels by GC using 5h urine samples. Intestinal permeability is presented as percent change from initial respective control baseline (set as 100%) of the L/M ratio (lactulose/mannitol ratio). EtOH significantly induced increase of intestinal permeability in control mice. This EtOH-induced hyperpermeability was significantly inhabited in miR-212 silenced mice using LNA microRNA. Data are expressed as mean±SEM. N=6, *: p<0.05 compared with Control group, #: p<0.05 compared with EtOH treated group.
2. EtOH-induced iNOS upregulation mediated miR-212 expression, ZO-1 protein down regulation and barrier disruption in intestinal epithelial cell monolayers
Our previous studies showed that iNOS hyperactivity & NO overproduction mediate EtOH-induced intestinal oxidative stress and disruption of intestinal epithelial cell monolayer barrier integrity. We also showed that EtOH increased miR-212 expression, decreased ZO-1 protein levels, disrupted tight junctions, and increased the permeability of monolayers of Caco-2 cells and that miR-212 overexpression correlated with hyperpermeability of the monolayer barrier(Tang et al., 2008). miR-212 levels were higher in colon biopsy samples in ALD patients than in healthy controls and ZO-1 protein levels were lower(Tang et al., 2008). Here we attempted to determine whether iNOS-induced disruption of intestinal barrier integrity is mediated through miR-212. To this end, we knocked down iNOS in Caco-2 cells using specific SiRNA technology and then exposed the monolayer to 0.2% alcohol for 2 hours and then measured miR-212. We found that EtOH significantly increased miR-212 expression (p<0.05, Fig. 3). Knocking down of iNOS significantly prevented EtOH-induced miR-212 overexpression in Caco-2 cells (p<0.05, Fig. 3). These data support our hypothesis that iNOS mediates the EtOH-induced miR-212 overexpression.
Fig. 3. Knocking down iNOS significantly inhibited EtOH-induced miR-212 expression in Caco-2 cells.

miR-212 levels in Caco-2 cells were measured by Real time PCR. The miR-212 levels in Caco-2 cells treated with 0.2% EtOH for 24 h were increased compared with control. Knocking down of iNOS using siRNA technology significantly inhibits 0.2%EtOH-induced miR-212 increase. Data are expressed as mean±SEM. N=6, *: p<0.05 compared with Control group, #: p<0.05 compared with 0.2 % EtOH treated group.
To determine whether iNOS is also mediating miRNA induced downregulation of ZO-1 in alcohol exposed monolayers, Caco-2 cells were treated with 0.2% EtOH for 24 hours and then harvested. Western blots showed that EtOH significantly decreased ZO-1 protein levels compared to controls (Fig. 4). ZO-1 bands of Western blots were scanned and assessed using Image J densitometry software. ZO-1 levels were normalized by beta-actin levels. The data show that 0.2% EtOH decreased ZO-1 protein levels by 71% (p<0.05, Fig. 4). Knocking down of iNOS significantly inhibited EtOH-induced ZO-1 protein down regulation in Caco-2 cells (p<0.05, Fig. 4). We also assessed ZO-1 morphology using Immunofluorescent microscopy after 24 hour exposure of Caco-2 monolayers to 0.2% EtOH.Immunofluorescent staining of ZO-1 showed that EtOH caused disruption of the tight junctions of intestinal epithelial cells, including extensive disorganization, kinking, condensation, and beading of its normal ring structure (Fig. 5). Knocking down iNOS significantly inhibited EtOH-induced disruption of ZO-1 tight junction morphology in Caco-2 cells (Fig. 5). These data suggest that iNOS mediates EtOH-induced ZO-1 protein down regulation.
Fig. 4. Knocking down iNOS significantly inhibited EtOH-induced ZO-1 protein down regulation in Caco-2 cells.

Effect of EtOH on ZO-1 expression. (A): Role of of iNOS in EtOH-induced ZO-1 protein down regulation. ZO-1 protein levels in Caco-2 cells treated with 0.2% EtOH for 24 h were decreased compared with control. Knocking down of iNOS using siRNA technology significantly inhibits 0.2 %EtOH-induced ZO-1 decrease. Data are expressed as mean±SEM. N=6, *: p<0.05 compared with Control group, #: p<0.05 compared with 0.2 % EtOH treated group.
Fig. 5. Effect of siRNA for iNOS on EtOH-induced ZO-1 morphology changes in Caco-2 cells.

Immunoflurescent staining for ZO-1 shows that EtOH (0.2% treated for 24 hrs) disrupts intercellular ZO-1 tight junctions. The EtOH-induced displacement of ZO-1 tight junction protein including extensive disorganization, kinking, condensation, and beading of its normal ring structure. Knocking down of iNOS using siRNA significantly inhibited EtOH-induced disruption of ZO-1 tight junction. Representative images from three independent experiments were shown. The magnification for results presented in the figure is 100 folds
To determine whether iNOS is also mediating miR-212-induced intestinal hyperpermeability in alcohol exposed monolayers, we then used two methods (TER and FSA) to measure the intestinal epithelial cell monolayer permeability. We incubated Caco-2 monolayers with 0.2% EtOH for 30 minutes to 2 hours and found that EtOH significantly decreased TER (increased barrier permeability) in a time-dependent manner (p<0.05, Fig. 6A). Knocking down iNOS significantly inhibited EtOH-induced hyperpermeability in monolayers of Caco-2 cells (p<0.05, Fig. 6A). FSA measurement further confirmed that EtOH significantly increased FSA (increased barrier permeability) in Caco-2 monlayers (p<0.05, Fig. 6B) which was prevented by knocking down iNOS using SiRNA (p<0.05, Fig6B). The results suggest that iNOS is involved in the EtOH-induced and miR-212 associated disruption of intestinal barrier integrity.
Fig. 6. Knocking down iNOS significantly inhibited EtOH-induced hyperpermeability in monolayer of Caco-2 cells.


(A). Effect of iNOS on the EtOH-induced TER changes in Caco-2 cells. Decrease of TER means increase of permeability. (B). Effect of iNOS on the EtOH-induced FSA changes in Caco-2 cells. Increase of FSA means increase of permeability. Data are expressed as mean±SEM. N=6, *: p<0.05 compared with Control group, #: p<0.05 compared with 0.2 % EtOH treated group.
3. EtOH-induced intestinal iNOS upregulation resulted in EtOH-induced miR-212 overexpression and intestinal hyper-permeability in alcohol fed mice
The above in vitro data established an important role for iNOS upregulation in EtOH-induced barrier disruption and suggested that iNOS mediated EtOH-induced hyperpermeability is through changes in miR-212 expression and its target gene, ZO-1. To further investigate this hypothesis, we used iNOS knock out (KO) mice in our animal model of alcohol-induced hyperpermeability. The wild type (WT) mice and iNOS knockout (KO) mice were fed with a diet containing alcohol [29% of calories from EtOH] for 8 weeks. The miR-212 expression levels in colon tissues were assayed by TaqMan real time PCR. The miR-212 expression levels in intestinal mucosa were increased in EtOH fed wild type mice (p<0.05, compared with WT Control group, Fig. 7), but not in iNOS KO alcohol fed mice (p<0.05 compared with WT with EtOH treated group, Fig. 7). We also measured intestinal permeability at 8 weeks after feeding with the alcohol containing diet. Intestinal permeability was assessed by measuring urinary sucralose in 5 hour urine following gavage of sugar cocktail containing poorly absorbed sugars-including sucralose as we previously reported (Forsyth et al., 2011). We found that urinary sucralose was significantly increased in alcohol fed wild type mice (p<0.05, compared with WT Control group, Fig. 8); but not in alcohol fed iNOS KO mice (Fig. 8).
Fig. 7. Role of iNOS in EtOH-induced miR-212 expression levels in vivo.
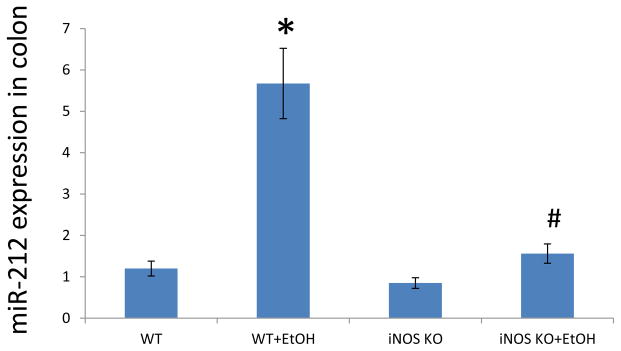
The wild type (WT) mice and iNOS knockout (KO) mice were fed with a level of 29% calories from EtOH for 8 weeks. The miR-212 expression levels were assayed by TaqMan real time PCR. The miR-212 expression levels in intestinal mucosa were increased in EtOH treated mice, but not in iNOS KO mice. N=10, *: p<0.05 compared with WT Control group, #: p<0.05 compared with WT with EtOH treated group.
Fig. 8. EtOH-induced intestinal hyperpermeability in wild type (WT) mice but not in iNOS KO mice.

The wild type (WT) mice and iNOS knockout (KO) mice were fed with a level of 29% calories from EtOH for 8 weeks. Intestinal permeability was determined by sugar gavage bolus as our previous report. N=10, *: p<0.05 compared with WT Control group, #: p<0.05 compared with WT with EtOH treated group.
We also assessed liver injury using histology. As expected, and has been shown by several other groups(Mathews et al., 2014), alcohol fed mice had histological evidence of steatohepatitis (Fig. 9). Alcohol-induced steatosis was significantly blunted and liver cell injury and death were prevented in iNOS KO mice (figure 9) that also had normal intestinal permeability.
Fig. 9. Knockout of iNOS significantly improves alcohol-induced liver pathology.

(A) Histological assessment of liver steatosis; (B) lobular inflammation; (C) ballooning degeneration; (D) acidophil bodies; all revealed significant effects of EtOH in WT mice. The EtOH-induced liver pathology in iNOS KO mice were less serious comparing with WT mice treated with EtOH group. N=10, *: p<0.05 compared with WT Control group, #: p<0.05 compared with WT with EtOH treated group. Histological assessment of liver samples was performed by a blinded gastrointestinal pathologist. Steatosis score was based on % hepatocyte involvement: 0=<5%, 1=5–33%, 2=34–66%, 3=>67%. Lobular inflammation score was based on the number of foci/200× field: 0=none, 1=1, 2=2–4, 3=>4. Ballooning degeneration score was based on the presence and frequency of ballooned cells: 0=none, 1=few, 2=prominent/many. Acidophil body score was based on the presence of acidophil bodies: 0=absent, 1=focal apoptosis (few), 2=many, 3=confluent necrosis.
Discussion
Alcoholic liver disease (ALD) is one of the most common and serious complications of heavy drinking. It is a major health problem in the US, consuming 15% of total health care dollars(Burbige et al., 1984; Galambos, 1973; Grant et al., 1988; Maher, 2002a; O’Connor and Schottenfeld, 1998), and associated with 20% mortality(Maher, 2002b). It is now well established that gut derived endotoxins are the required co-factor for ALD and gut leakiness is one of the primary causes of endotoxemia in alcoholics with liver disease (Keshavarzian et al., 2009; Keshavarzian et al., 1999). However, the mechanisms through which alcohol consumption causes gut leakiness are not completely understood. Our in vitro, in vivo animal, and ex-vivo human studies reported here and previously provide compelling evidence for the central involvement of iNOS activation in EtOH-induced gut leakiness. Indeed, several of our studies have shown that iNOS activation is required for EtOH-induced gut leakiness. We reported that:1) EtOH increases iNOS activity and NO levels in intestinal monolayers and increases monolayer permeability (Keshavarzian and Fields, 2003; Keshavarzian et al., 1999; Tang et al., 2008). A specific iNOS inhibitor (L-NIL) prevented EtOH-induced monolayer leakiness(Banan et al., 2000a); 2) EtOH no longer can cause leakiness in monolayers incapable of upregulating iNOS (i.e., transfected with dominant negative iNOS antisense)(Banan et al., 2000a); 3) iNOS is increased in intestinal mucosa of alcoholics with ALD and in alcohol-treated rats with gut leakiness and endotoxemia(Keshavarzian et al., 2009) 4) Daily gavage of the specific iNOS inhibitor L-NIL prevented iNOS upregulation and oxidative stress in the intestinal mucosa of alcohol-fed rats and also prevented alcohol-induced gut leakiness(Tang et al., 2009b); 5) Daily gavage of Lactobacillus GG(Forsyth et al., 2009) or supplementation of the diet with oats(Keshavarzian et al., 2001; Tang et al., 2009a) prevented nitration of intestinal mucosal proteins, oxidative stress, and gut leakiness in alcohol-fed rats.
Our current study shows that an alcohol-containing (Nanji) diet for 8 weeks caused gut leakiness in wild type mice but not in iNOS knockout mice. Our study also indicates that iNOS is required for EtOH-induced miR-212 overexpression. We first showed that miRNA 212 expression is increased in alcohol treated monolayers and in the intestine of alcohol fed mice who had gut leakiness; inhibition of miRNA 212 over-expression in alcohol treated Caco-2 cell monolayers and in alcohol fed mice by microRNA knockdown technology prevented alcohol-induced intestinal hyperpermeability. Furthermore, we found that alcohol-induced miR-212 over-expression and disruption of ZO-1 morphology in Caco-2 cells were significantly inhibited when iNOS was knocked down. Our in vivo data confirmed our in vitro findings and also show that chronic daily alcohol feeding caused intestinal miR-212 overexpression, gut leakiness and steatohepatitis in wild type alcohol fed mice but not in iNOS knockout alcohol fed mice. These results suggest that iNOS plays an important role in alcohol-induced miR-212 over-expression which disrupts intestinal barrier integrity by inhibiting ZO-1 translation. Our studies indicate a novel mechanism for alcohol-induced gut leakiness. This mechanism provides a potential therapeutic target for preventing the leaky gut in patients with ALD and other alcohol-induced disorders associated with intestinal hyperpermeability and endotoxin-mediated inflammation and tissue injury.
Our results in showed that LNA-anti-miR-212 significantly decreased the miR-212 expression level that was detected by specific real time PCR. This is consistent with previous reports that LNA-anti-miRNAs efficiently silenced miRNAs in mice (Elmen et al., 2008; Krutzfeldt et al., 2007). However, the real time PCR may detects the artifact, Northern blot and measurement of target genes of miRNAs may be useful to further confirm the in vivo effect of LNA-anti-miRNAs on the miRNAs expression levels assayed by real time PCR (Torres et al., 2011).
One unanswered question is how does iNOS mediate an EtOH-induced increase of miR-212 expression in intestinal epithelial cells? Recent studies have shown that cAMP response element-binding protein (CREB) activation leads to the transcription of the miR-212 locus through an ERK1-dependent pathway in BDNF-stimulated neurons(Hollander et al., 2010). Other studies also showed that that alcohol induced cAMP and CREB activation(Soares-Simi et al., 2013). Thus, we hypothesize that iNOS mediates EtOH-induced upregulation of miR-212 expression and down-regulation of ZO-1 via a CREB-regulated pathway. This hypothesis will be tested in our future studies.
Although our study indicates that iNOS is required for EtOH-induced miR-212 overexpression, the EtOH-induced miR-212 overexpression is not 100% dependent on iNOS signaling. Other signaling pathways may also mediate EtOH-induced miR-212 expression directly or indirectly. Indeed, our recent research shows that alcohol-induced activation of Snail, through PAK1 and Epidermal Growth Factor Receptor (EGRF) signaling, plays an important role in the regulation of tight junction proteins and intestinal permeability(Forsyth et al., 2007; Forsyth et al., 2011). NF-kB is another possible mediating factor for iNOS-induced miR-212 expression because NF-kB has been shown to be involved in regulation of expression of several miRNAs and we and others have shown the important role of NF-kB in alcohol-induced iNOS upregulation and gut leakiness (Banan et al., 2007; Taganov et al., 2006). For example, it was reported that NF-kB-dependent induction of miR-146 regulated Toll-like receptor and cytokine signaling through a negative feedback regulation loop(Taganov et al., 2006). Thus, further studies are needed to investigate the interaction between NF-kB activation, iNOS upregulation and miR-212 expression involved in the mechanism by which EtOH induces intestinal dysfunction.
Our current study indicates that iNOS mediates EtOH-induced intestinal hyperpermeability through miR-212/ZO-1 pathway. However, according to the miRNA databases(Kiriakidou et al., 2004; Krek et al., 2005; Lewis et al., 2003; Rajewsky, 2006) and other publications, there are hundreds of predicted target genes for miR-212, such as: CYP2E1, which associates with oxidative stress (Shukla et al., 2013); p300, PTEN and FOXO3a, this group of target genes of miR-212 are associated with inflammation, intestinal epithelial cell proliferation, differentiation, and apoptosis(Wong et al., 2013). Thus, iNOS or miR-212 may also affect these target genes to fine tune the intestinal permeability during the process of EtOH induced tissue damage.
This current study focused on miR-212 and demonstrates that iNOS is required for EtOH-induced miR-212 overexpression. However, EtOH also induces other miRNAs up or down regulation in brain, liver and other tissues(Lippai et al., 2013; Miranda et al., 2010; Prins et al., 2014; Shukla and Lim, 2013). Whether iNOS mediates these miRNA changes due to alcohol needs further study.
One of the limitations of this study is that we used global iNOS KO mice in the experiments. Although our study showed that the alcohol-induced intestinal hyperpermeability and liver injury are significantly less than that in the wild type mice, we could not rule out the possibility that lack of liver injury in iNOS KO mice could be due to the iNOS KO in the liver cells. Indeed, some investigators have shown that iNOS KO protect liver against alcohol-induced injury(Deng and Deitrich, 2007; Jaeschke et al., 2002). Here, our study show that the protection of alcohol-induced liver injury in iNOS KO mice is associated with lack of gut leakiness and the alcohol-induced gut leakiness is associated with the regulation of miR-212 and disruption of intestinal barrier through targeting ZO-1 tight junction protein. To overcome this limitation, intestinal tissue specific iNOS KO mice may be useful in future studies (Beck et al., 2007; Krieglstein et al., 2007).
As the discussion about iNOS, the alcohol-induced liver injury was decreased in miR-212 silencing mice may be due to local (gut leakiness) or system (liver) or both miR-212 silencing. Intestinal tissue specific inhibition or knockout of miR-212 mice are needed to investigate the mechanisms in the future study.
In summary, we found that iNOS plays an important role in EtOH-induced intestinal miR-212 over-expression and gut leakiness. Knockout of iNOS inhibits EtOH-induced miR-212 overexpression which prevents disruption of intestinal paracellular tight junctions by down-regulation of the translation of ZO-1, one of its target genes. Investigating the role of iNOS and miR-212 in EtOH-induced gut leakiness could lead to: a) the development of novel therapeutic strategies to prevent and/or treat alcohol-induced gut leakiness, endotoxemia and its consequences like ALD; b) novel approaches to prevent the potential deleterious effects of alcohol on gene regulation and function (epigenomics) with a potential positive impact on the course of cancers and even for prevention of cancers that are associated with alcoholism (e.g. esophageal, colon, pancreatic, breast); c) new therapy for many other human diseases associated with disturbances of intestinal barrier function and intestinal derived inflammation.
Acknowledgments
The studies were supported by NIH grants: AA018729 (YT), AA019405 (AK), and AA020216 (AK and CBF), and AA023417 (AK).
References
- Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Banan A, Fields JZ, Decker H, Zhang Y, Keshavarzian A. Nitric oxide and its metabolites mediate ethanol-induced microtubule disruption and intestinal barrier dysfunction. J Pharmacol Exp Ther. 2000a;294(3):997–1008. [PubMed] [Google Scholar]
- Banan A, Keshavarzian A, Zhang L, Shaikh M, Forsyth CB, Tang Y, Fields JZ. NF-kappaB activation as a key mechanism in ethanol-induced disruption of the F-actin cytoskeleton and monolayer barrier integrity in intestinal epithelium. Alcohol. 2007;41(6):447–60. doi: 10.1016/j.alcohol.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Banan A, Zhang Y, Losurdo J, Keshavarzian A. Carbonylation and disassembly of the F-actin cytoskeleton in oxidant induced barrier dysfunction and its prevention by epidermal growth factor and transforming growth factor alpha in a human colonic cell line. Gut. 2000b;46(6):830–7. doi: 10.1136/gut.46.6.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck PL, Li Y, Wong J, Chen CW, Keenan CM, Sharkey KA, McCafferty DM. Inducible nitric oxide synthase from bone marrow-derived cells plays a critical role in regulating colonic inflammation. Gastroenterology. 2007;132(5):1778–90. doi: 10.1053/j.gastro.2007.01.032. [DOI] [PubMed] [Google Scholar]
- Bode C, Bode JC. Activation of the innate immune system and alcoholic liver disease: effects of ethanol per se or enhanced intestinal translocation of bacterial toxins induced by ethanol? Alcohol Clin Exp Res. 2005;29(11 Suppl):166S–71S. doi: 10.1097/01.alc.0000189280.19073.28. [DOI] [PubMed] [Google Scholar]
- Brunt EM, Tiniakos DG. Histopathology of nonalcoholic fatty liver disease. World J Gastroenterol. 2010;16(42):5286–96. doi: 10.3748/wjg.v16.i42.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbige EJ, Lewis DR, Jr, Halsted CH. Alcohol and the gastrointestinal tract. Med Clin North Am. 1984;68(1):77–89. doi: 10.1016/s0025-7125(16)31242-1. [DOI] [PubMed] [Google Scholar]
- Carthew RW. Gene regulation by microRNAs. Curr Opin Genet Dev. 2006;16(2):203–8. doi: 10.1016/j.gde.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84(3):282–91. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- Czech MP. MicroRNAs as therapeutic targets. N Engl J Med. 2006;354(11):1194–5. doi: 10.1056/NEJMcibr060065. [DOI] [PubMed] [Google Scholar]
- Deng XS, Deitrich RA. Ethanol metabolism and effects: nitric oxide and its interaction. Curr Clin Pharmacol. 2007;2(2):145–53. doi: 10.2174/157488407780598135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjarn M, Hansen HF, Berger U, Gullans S, Kearney P, Sarnow P, Straarup EM, Kauppinen S. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452(7189):896–9. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- Forsyth CB, Banan A, Farhadi A, Fields JZ, Tang Y, Shaikh M, Zhang LJ, Engen PA, Keshavarzian A. Regulation of oxidant-induced intestinal permeability by metalloprotease-dependent epidermal growth factor receptor signaling. J Pharmacol Exp Ther. 2007;321(1):84–97. doi: 10.1124/jpet.106.113019. [DOI] [PubMed] [Google Scholar]
- Forsyth CB, Farhadi A, Jakate SM, Tang Y, Shaikh M, Keshavarzian A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol. 2009;43(2):163–72. doi: 10.1016/j.alcohol.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth CB, Tang Y, Shaikh M, Zhang L, Keshavarzian A. Role of snail activation in alcohol-induced iNOS-mediated disruption of intestinal epithelial cell permeability. Alcohol Clin Exp Res. 2011;35(9):1635–43. doi: 10.1111/j.1530-0277.2011.01510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galambos J. Alcoholic hepatitis: its therapy and prognosis. Prog Liv Dis. 1973;4:567–588. [PubMed] [Google Scholar]
- Grant BF, Dufour MC, Harford TC. Epidemiology of alcoholic liver disease. Semin Liver Dis. 1988;8(1):12–25. doi: 10.1055/s-2008-1040525. [DOI] [PubMed] [Google Scholar]
- Hollander D. The intestinal permeability barrier. A hypothesis as to its regulation and involvement in Crohn’s disease. Scand J Gastroenterol. 1992;27(9):721–6. doi: 10.3109/00365529209011172. [DOI] [PubMed] [Google Scholar]
- Hollander JA, Im HI, Amelio AL, Kocerha J, Bali P, Lu Q, Willoughby D, Wahlestedt C, Conkright MD, Kenny PJ. Striatal microRNA controls cocaine intake through CREB signalling. Nature. 2010;466(7303):197–202. doi: 10.1038/nature09202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65(2):166–76. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- Keshavarzian A, Choudhary S, Holmes EW, Yong S, Banan A, Jakate S, Fields JZ. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J Pharmacol Exp Ther. 2001;299(2):442–8. [PubMed] [Google Scholar]
- Keshavarzian A, Farhadi A, Forsyth CB, Rangan J, Jakate S, Shaikh M, Banan A, Fields JZ. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol. 2009;50(3):538–47. doi: 10.1016/j.jhep.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshavarzian A, Fields J. Alcoholic liver disease: is it an “extraintestinal” complication of alcohol-induced intestinal injury? J Lab Clin Med. 2003;142(5):285–7. doi: 10.1016/S0022-2143(03)00140-9. [DOI] [PubMed] [Google Scholar]
- Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol. 1999;94(1):200–7. doi: 10.1111/j.1572-0241.1999.00797.x. [DOI] [PubMed] [Google Scholar]
- Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z, Hatzigeorgiou A. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18(10):1165–78. doi: 10.1101/gad.1184704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–21. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Krieglstein CF, Anthoni C, Cerwinka WH, Stokes KY, Russell J, Grisham MB, Granger DN. Role of blood- and tissue-associated inducible nitric-oxide synthase in colonic inflammation. Am J Pathol. 2007;170(2):490–6. doi: 10.2353/ajpath.2007.060594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, Manoharan M, Stoffel M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007;35(9):2885–92. doi: 10.1093/nar/gkm024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Lippai D, Bala S, Csak T, Kurt-Jones EA, Szabo G. Chronic alcohol-induced microRNA-155 contributes to neuroinflammation in a TLR4-dependent manner in mice. PLoS One. 2013;8(8):e70945. doi: 10.1371/journal.pone.0070945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madara JL. Warner-Lambert/Parke-Davis Award lecture. Pathobiology of the intestinal epithelial barrier. Am J Pathol. 1990;137(6):1273–81. [PMC free article] [PubMed] [Google Scholar]
- Maher JJ. Alcoholic steatosis and steatohepatitis. Semin Gastrointest Dis. 2002a;13(1):31–9. [PubMed] [Google Scholar]
- Maher JJ. Treatment of alcoholic hepatitis. J Gastroenterol Hepatol. 2002b;17(4):448–55. doi: 10.1046/j.1440-1746.2002.02722.x. [DOI] [PubMed] [Google Scholar]
- Mathews S, Xu M, Wang H, Bertola A, Gao B. Animal Models of Alcohol-induced liver disease: Pathophysiology, Translational Relevance and Challenges. Am J Physiol Gastrointest Liver Physiol. 2014 doi: 10.1152/ajpgi.00041.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda RC, Pietrzykowski AZ, Tang Y, Sathyan P, Mayfield D, Keshavarzian A, Sampson W, Hereld D. MicroRNAs: master regulators of ethanol abuse and toxicity? Alcohol Clin Exp Res. 2010;34(4):575–87. doi: 10.1111/j.1530-0277.2009.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor PG, Schottenfeld RS. Patients with alcohol problems. N Engl J Med. 1998;338(9):592–602. doi: 10.1056/NEJM199802263380907. [DOI] [PubMed] [Google Scholar]
- Prins SA, Przybycien-Szymanska MM, Rao YS, Pak TR. Long-Term Effects of Peripubertal Binge EtOH Exposure on Hippocampal microRNA Expression in the Rat. PLoS One. 2014;9(1):e83166. doi: 10.1371/journal.pone.0083166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38(Suppl):S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- Sawada N, Murata M, Kikuchi K, Osanai M, Tobioka H, Kojima T, Chiba H. Tight junctions and human diseases. Med Electron Microsc. 2003;36(3):147–56. doi: 10.1007/s00795-003-0219-y. [DOI] [PubMed] [Google Scholar]
- Shukla SD, Lim RW. Epigenetic effects of ethanol on the liver and gastrointestinal system. Alcohol Res. 2013;35(1):47–55. [PMC free article] [PubMed] [Google Scholar]
- Shukla U, Tumma N, Gratsch T, Dombkowski A, Novak RF. Insights into insulin-mediated regulation of CYP2E1: miR-132/-212 targeting of CYP2E1 and role of phosphatidylinositol 3-kinase, Akt (protein kinase B), mammalian target of rapamycin signaling in regulating miR-132/-212 and miR-122/-181a expression in primary cultured rat hepatocytes. Drug Metab Dispos. 2013;41(10):1769–77. doi: 10.1124/dmd.113.052860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares-Simi SL, Pastrello DM, Ferreira ZS, Yonamine M, Marcourakis T, Scavone C, Camarini R. Changes in CREB activation in the prefrontal cortex and hippocampus blunt ethanol-induced behavioral sensitization in adolescent mice. Front Integr Neurosci. 2013;7:94. doi: 10.3389/fnint.2013.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summa KC, Voigt RM, Forsyth CB, Shaikh M, Cavanaugh K, Tang Y, Vitaterna MH, Song S, Turek FW, Keshavarzian A. Disruption of the Circadian Clock in Mice Increases Intestinal Permeability and Promotes Alcohol-Induced Hepatic Pathology and Inflammation. PLoS One. 2013;8(6):e67102. doi: 10.1371/journal.pone.0067102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481–6. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Banan A, Forsyth CB, Fields JZ, Lau CK, Zhang LJ, Keshavarzian A. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol Clin Exp Res. 2008;32(2):355–64. doi: 10.1111/j.1530-0277.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- Tang Y, Forsyth CB, Banan A, Fields JZ, Keshavarzian A. Oats supplementation prevents alcohol-induced gut leakiness in rats by preventing alcohol-induced oxidative tissue damage. J Pharmacol Exp Ther. 2009a doi: 10.1124/jpet.108.148643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Forsyth CB, Farhadi A, Rangan J, Jakate S, Shaikh M, Banan A, Fields JZ, Keshavarzian A. Nitric oxide-mediated intestinal injury is required for alcohol-induced gut leakiness and liver damage. Alcohol Clin Exp Res. 2009b;33(7):1220–30. doi: 10.1111/j.1530-0277.2009.00946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AG, Fabani MM, Vigorito E, Gait MJ. MicroRNA fate upon targeting with anti-miRNA oligonucleotides as revealed by an improved Northern-blot-based method for miRNA detection. Rna. 2011;17(5):933–43. doi: 10.1261/rna.2533811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno N, Wang H, Menconi MJ, Tytgat SH, Larkin V, Smith M, Morin MJ, Chavez A, Hodin RA, Fink MP. Inhibition of inducible nitric oxide synthase ameliorates endotoxin-induced gut mucosal barrier dysfunction in rats. Gastroenterology. 1997;113(4):1246–57. doi: 10.1053/gast.1997.v113.pm9322519. [DOI] [PubMed] [Google Scholar]
- Wong HK, Veremeyko T, Patel N, Lemere CA, Walsh DM, Esau C, Vanderburg C, Krichevsky AM. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer’s disease. Hum Mol Genet. 2013;22(15):3077–92. doi: 10.1093/hmg/ddt164. [DOI] [PubMed] [Google Scholar]