

Abstract
Parkinson's disease (PD) is an age-related progressive neurodegenerative disease associated with selective loss of dopaminergic neurons. The characteristic hallmark of the disease is intracytoplasmic proteinacious inclusion bodies called Lewy bodies, primarily consisting of a presynaptic protein α-synuclein. Oxidative stress-mediated damage to macromolecules have been shown to occur frequently in PD. Oxidative damage to DNA in the form of oxidized guanine (8-oxodG) accumulates in both the mitochondrial and nuclear DNA of dopaminergic neurons of the substantia nigra in PD. 8-oxodG-mediated transcriptional mutagenesis has been shown to have the potential to alter phenotype of cells through production of mutant pool of proteins. This review comprehensively summarizes the role of oxidative stress-mediated damage incurred during neurodegeneration, and highlights the scope of transcriptional mutagenesis event in leading to α-synuclein aggregation as seen in PD.
Introduction
Parkinson's disease (PD) is an age-related progressive neurodegenerative disorder which is associated with selective loss of dopaminergic neurons from the substantia nigra pars compacta region of the midbrain.1 PD is broadly classified into a familial form (resulting from genetic alterations like mutations or multiplication in the SNCA gene encoding alpha-synuclein (α-SYN), early-onset form) and the idiopathic form with unknown etiology (late-onset form).2 The majority of idiopathic PD cases represent a late-onset sporadic form with cytoplasmic α-SYN aggregates which are the major component of Lewy bodies and Lewy neurites, the characteristic proteinacious cytoplasmic deposits that are pathological hallmark of the disease.3 Increasing evidence suggest that oxidative stress is a key contributor to the pathogenesis of PD, which causes damage to nucleic acids (both DNA and RNA), proteins, lipids and other cellular macromolecules whose functions are indispensable for cell survival. The metabolism of dopamine (DA) itself contributes to oxidative stress that renders the nigral neurons particularly vulnerable in PD.4, 5 The most frequent DNA lesion generated by oxidative stress is 8-oxo-7,8-dihydroguanine (8-oxodG), the oxidized form of guanine, often associated with neurodegenerative diseases including PD and Alzheimer's disease (AD).1, 5 8-oxodG, being a nonbulky DNA lesion, has high mutagenic potential by misincorporation of an adenine instead of cytosine causing G:C→T:A transversion mutation.6 8-oxodG has also been implicated in an event called transcriptional mutagenesis (TM), whereby a misincorporated adenine in the transcribing mRNA leads to the generation of mutated species of protein7, 8 (Figure 1). It is well documented that oxidative DNA damage accumulates in ageing brains and this accumulation is significantly increased in brains of patients with PD and AD compared to their age-matched controls.5, 9 These increased levels of DNA damage are also corroborated by decrease in the DNA repair capacity of specific enzymes such as 8-oxodG DNA glycosylase1 (OGG1).10 In addition to its involvement in tumor development, TM may have a very important role in the neurodegenerative disorders, in which a nucleation-dependent protein aggregation process has a pivotal role in neuronal degeneration as seen in PD and AD.11 As shown in α-SYN A53T mutant species that was reported in the familial form of PD,12 the pathologically misfolded proteins drive the template-directed misfolding of the native monomeric proteins, which contributes to the nucleation-dependent fibrillation process.13, 14 Moreover, compared with replicating cells, neurons that are post-mitotic cells might be even more vulnerable to 8-oxodG-mediated TM as pathogenic effects caused by mutant species generated through TM events could be accumulated over a lifetime.8 In the following review, we discuss the importance of oxidative damage in PD and its scope in the pathogenesis of the disease through 8-oxodG-mediated TM events.
Figure 1.
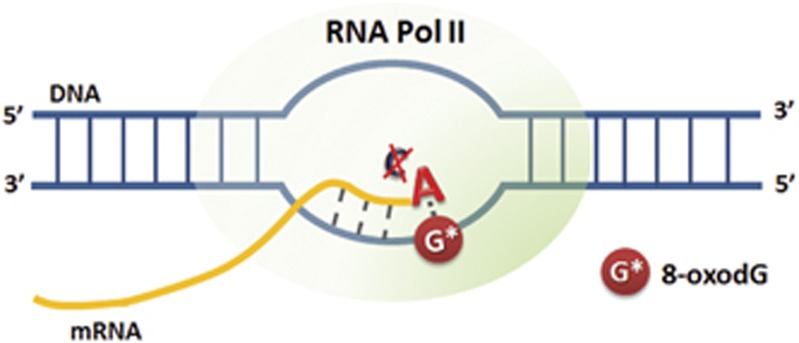
8-OxodG-mediated transcriptional mutagenesis event. TM event occurs when 8-oxodG present on the transcribing strand (3′ → 5′) of a gene can misinsert an adenine instead of a cytosine in the growing mRNA chain, thus introducing a mutation on the nascent mRNA strand.
Oxidative stress-mediated damage in neurodegeneration
Oxygen is an essential component to the survival of all living beings. But the greatest paradox remains in the fact that production of reactive oxygen species (ROS) as a by-product of oxygen metabolism is highly toxic to cells. ROS are molecules that can react with cellular macromolecules and impair their functions. It can include both free radicals like superoxide, hydroxyl radical and nitric oxide (containing highly reactive unpaired electrons) and molecules like hydrogen peroxide and peroxynitrite. Post-mortem brain tissues from patients of PD, AD and amyotrophic lateral sclerosis (ALS) have clearly demonstrated higher amount of ROS in the selective areas that undergoes neurodegeneration.
Oxidative stress originates when the rate of ROS production is significantly higher compared with its elimination from the system. Several markers of the oxidized cellular macromolecules have been identified under conditions of neurodegeneration. For example, elevated levels of malondialdehyde and 4-hydroxynonenal, which are markers of oxidized lipids, have been observed in the cortex and hippocampus of patients with AD, in the substantia nigra of patients with PD and spinal fluid of patients with ALS.13, 14, 15, 16 Oxidative modification of unsaturated fatty acids can result in the generation of lipid peroxides which can further cause oxidation of the unsaturated fatty acids in a chain-like event, finally leading to the disruption of plasma membranes and membranes of other cellular organelles like mitochondria.17 The levels of protein carbonyls, a marker of protein oxidation, have been also reported to be consistently elevated in the hippocampus and neocortex of individuals with AD, in Lewy bodies in case of PD and motor neurons of ALS patients.18, 19, 20, 21 Oxidation of proteins can disrupt the active site of enzymes, lead to conformational change, disrupt protein–protein interactions, and alter their binding capacity to DNA, eventually threatening cell survival. In addition, increased levels of oxidative damage to DNA and RNA bases have been consistent with the neurodegenerative conditions like PD, AD and ALS. Although all DNA bases could be potentially oxidized, guanine is the most susceptible base to oxidative damage. It gets readily oxidized to form 8-oxodG and serves as a marker for oxidative damage.22 In AD, the level of nuclear DNA damage in the brain regions including frontal, parietal and temporal lobes is significantly higher compared with age-matched controls.23 Overall, it can be concluded that oxidative stress is commonly associated with neurodegenerative conditions and has a critical role in mediating the disease processes.
The origin of oxidative stress and subsequent accumulation of damage can not only be attributed to the overproduction of ROS but also to the inefficient cellular defense and repair machinery against oxidative stress.17, 24 The defense machinery refers to the antioxidant enzymes like superoxide dismutase, glutathione peroxidase, glutathione reductase and catalase among many others whose primary function is to scavenge ROS generated in the cells. For example, superoxide dismutase converts superoxide to hydrogen peroxide, which is subsequently converted to water by either catalase or glutathione peroxidase. Glutathione peroxidase detoxifies hydrogen peroxide using reduced glutathione. During this process, glutathione is oxidized and it can be subsequently reduced by glutathione reductase.17 A number of reports have shown reduced activity of the antioxidant machinery in AD.25, 26 In familial ALS, mutations in copper- and zinc-containing superoxide dismutase lead to a toxic gain of function, rendering superoxide dismutase itself to a pro-oxidant protein involved in ROS generation.27, 28 PD is also characterized by significant loss of the reducing agent glutathione in the substantia nigra, which is one of the earliest known indicators of nigral neuronal degeneration.29 Together, these evidences comprehensively indicate that reduced antioxidant potential might be a critical factor toward increased oxidative stress that is associated with neurodegenerative disorders.
As discussed previously, accumulation of ROS-induced DNA damage, like oxidation of bases and single strand breaks have been implicated in the etiology of AD, PD, ALS and other neurological disorders. This accumulation of the DNA damage may imply the defect in the DNA repair machinery of the cells. It has been shown that ROS-induced DNA damages are primarily repaired via highly conserved base excision repair pathway.30, 31 Neuronal dysfunctions have been linked to mutations or differential expression of base excision repair enzymes like OGG1, XRCC1,32, 33, 34 single-strand break repair enzymes like TDP1, aprataxin35, 36 and double-strand break repair enzymes like ATM and NBS1.37 Furthermore, it has been shown that Ogg1 knockout mice exhibit age-associated loss of nigrostriatal pathways and increased sensitivity to the dopaminergic neuronal toxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, than their wild-type littermates.38 Together, these evidences strongly suggest that oxidative stress-mediated damage to cellular macromolecules including DNA coupled with inefficient repair leads to progressive neurodegeneration as seen in AD, PD, ALS and others.
Oxidative DNA damage in the form of 8-oxodG in Parkinson's disease
Oxidative stress has been classically linked to the etiology of PD. The high metabolic activity of neurons along with their long life span makes them highly susceptible to oxidative damage.39, 40 Moreover, dopaminergic neurons in the substantia nigra, the most affected brain region by PD, are particularly vulnerable to oxidative stress. Although the exact causes for this selective vulnerability is yet to be elucidated, DA metabolism itself has been considered as a major culprit for selective degeneration. DA has the capacity to auto-oxidize at normal pH into toxic quinone species, producing superoxide and hydrogen peroxide.41 Monoamine oxidase can also enzymatically deaminate DA into nontoxic metabolite 3,4-dihydroxyphenylacetic acid and hydrogen peroxide.42 Hydrogen peroxide can in turn be broken down into hydroxyl radical in a reaction catalyzed by iron. The level of iron is reportedly higher in the nigral dopaminergic neurons as compared with the other regions of the brain, owning to its binding affinity to neuromelanin.14, 43, 44 Therefore, when it is synthesized or transported into cells from extracellular space, DA is rapidly stored into synaptic vesicles which provides a stable environment for DA without monoamine oxidase and low pH. Under conditions of PD, nigral neurons appear to be in an exaggerated oxidative stress, causing severe damage to cellular macromolecules.
Damage to nucleic acids is particularly very hazardous amongst all the cellular macromolecules, because it can change genetic information present in both nuclear and mitochondrial genome.45 DNA damage by oxidative stress can result in the production of either nonbulky DNA lesions like 8-oxodG that can be repaired by the base excision repair pathway or bulky DNA lesions which are generally repaired by the nucleotide excision repair pathway.46 8-oxodG, the most frequent DNA lesion caused by oxidative stress is often associated with neurodegenerative diseases including PD.5 Immunocytochemical analysis of 8-oxodG revealed a significant increase of this DNA oxidation marker in the substantia nigra of patients with PD although the extent of nuclear 8-oxodG accumulation is not as high as mitochondrial 8-oxodG.4, 5, 47 Despite the presence of 8-oxodG-specific DNA repair enzyme, OGG1, a significant percentage of this DNA lesion remains unrepaired and accumulated under disease conditions. Moreover, it is reported that the overall activity of OGG1 in brain decreases over ageing in a mouse model.48 The 8-oxodG generated by direct oxidation of DNA, can be base paired with adenine as well as cytosine during replication, and consequently lead to spontaneous G:C to T:A transversion mutation.49 Thus, 8-oxodG remains the extensively studied route for mutagenesis in proliferating cells. However, majority of the cells in our body, including neurons, exist in nonproliferating quiescent state. Neurons being post-mitotic cells, face a major challenge of DNA repair during transcription.50 Failure to maintain both transcriptional and translational fidelity is expected to result in functional impairment of the cells. Studies have shown that many of these nonbulky lesions present on the sense strand of DNA could be bypassed by RNA polymerase during transcription, leading to misinsertion of ribonucleotides to the growing mRNA strands, producing mutant transcripts.51, 52, 53 This phenomenon is referred as TM event in the cells. Since a significant increase of 8-oxodG was observed in the substantia nigra of patients with PD,4, 5, 47 it is highly possible that TM event might significantly contribute to the pathogenesis of PD. In the next few sections of this review, the perspective of TM in PD will be closely explored with the emphasis on α-SYN that is reckoned a major pathogenic molecule.
8-oxodG-mediated transcriptional mutagenesis
DNA damage-mediated mutagenesis in a replication-based model has provided a wide range of information for better understanding of the origin of mutation and subsequently its contribution to the pathogenesis of human diseases such as cancers. However, as discussed briefly in the last section, majority of cells under normal physiological conditions are not frequently engaged in division and do not undergo continuous cycles of replication.54 Most of the multicellular organs of eukaryotes including brain or heart are mainly comprised of nondividing or terminally differentiated cells. Maintenance of complex physiological functions of these organs primarily depends on securing the high fidelity of both transcription and translation.7 Accumulation of TM-derived mutant transcripts and subsequently generated erroneous proteins has the potential to produce functional impairment of nonproliferating cells and organs.11 As the aging process may be accompanied by progressive deterioration of normal cellular functions such as DNA repair machinery and antioxidation processes, TM-mediated adverse effects on cells could be exacerbated over aging. In fact, under in vitro conditions allowing TM event, it has been shown that each round of transcription keeps producing a mutant transcript as long as 8-oxodG lesion is not repaired. This event is expected to generate a fairly large population of mutant transcripts which will be translated multiple times, leading to a relatively large amount of the mutant protein.7
A number of studies have shown that a plethora of DNA damage can lead to a TM event.11 The structural analysis of yeast RNA polymerase II at an 8-oxodG lesion revealed the possible mechanism of TM. In this study, it has been shown that 8-oxodG can mispair with adenine instead of cytosine through a Hoogsteen base-paring with the 8-oxodG lesion at the polymerase active center, thereby escaping the proofreading of the polymerase and maintained in the nascent RNA stand.55
The potential of 8-oxodG to cause TM event had been demonstrated in a bacterial system using Escherichia coli and its role in developing antibiotic resistance.7 Later, using a luciferase-based reporter system, TM-derived mutation event has been also proved in mammalian cells, demonstrating that TM event is strongly affected by factors, such as promoter strength of the gene, flanking sequence around the 8-oxodG lesion and position of the lesion with reference to the promoter.8 In the same study, OGG1 knockout cells showed more frequent TM event compared with the cells lacking a transcription-coupled repair machinery, suggesting that a 8-oxodG lesion is not efficiently recognized by transcriptional machinery for initiating repair and rather OGG1 has a critical role in preventing TM.8 Interestingly, TM event has been also linked to activation of oncogenic pathway.56 Replacement of a guanine with 8-oxodG at codon 61 of HRAS, a proto-oncogene, led to generation of a constitutively active mutant form of HRAS (Q61K) through TM event. Moreover, under condition of Ogg1 null background in mouse embryonic fibroblast, sufficient amount of the mutant protein was generated to activate the downstream MAPK pathway, leading to ERK phosphorylation.56
Together it suggests that 8-oxodG lesion is often bypassed by RNA polymerase II without efficient detection by the transcription-coupled repair or the base excision repair machinery, generating transcription mutant species and contributing to the pathogenesis of various diseases including cancer, neurodegeneration or cardiovascular disease.
Potential of TM event to cause α-SYN aggregation in Parkinson's disease
α-SYN is a 140 amino acid protein and natively unfolded. It can exist as random-coil state as well as β-sheet conformation upon aggregation or a α-helical conformation upon binding to membranes. Sequence of α-SYN can be divided into three regions with distinct characteristics (Figure 2): (1) the amphipathic lysine-rich N-terminus (residues 1–60), which is mainly involved in membrane interactions; (2) the middle hydrophobic region (non-Aβ component of amyloid plaques, residues 61–95), which is prone to β-sheet formation and fibrillization; and (3) the C-terminus (residues 96–140), which is a highly acidic and proline-rich region and primarily controls the nuclear localization and interaction with other proteins.57
Figure 2.
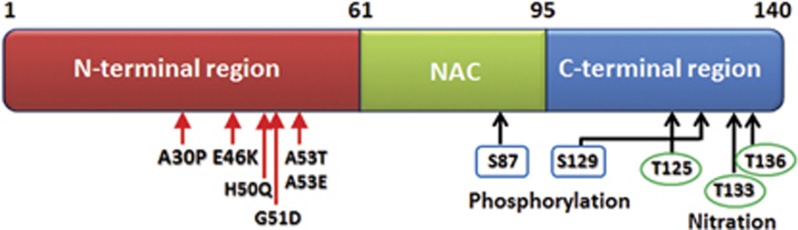
Structure of full length α-SYN protein and its functional components. N-terminal (1–60), non-Aβ component of amyloid plaques domain (61–95) and C-terminal (96–140); point mutations reported in familial PD (autosomal dominant form) are present in the N-terminal (red arrows). N-terminal contributes to the α-helical structure of α-SYN upon binding to lipid membranes. Non-Aβ component of amyloid plaques domain contains most hydrophobic residues and one phosphorylation site (blue rectangle, serine 87). Non-Aβ component of amyloid plaques domain promotes aggregation of the molecule by its propensity to form β-sheet structure. C-terminal has three nitration sites (green circle, tyrosine 125, 133, 136) and one phosphorylation site (blue rectangle, serine S129) and is mainly responsible for maintaining the protein solubility.
Multiplication as well as various single-nucleotide polymorphisms of SNCA have been reported in dominantly inherited early-onset PD.12, 58, 59, 60, 61 α-SYN mutant species, A30P, A53T and E46K have been shown to alter the aggregation process and interfere with oligomerization, fibril formation and the distribution in cellular compartments.12, 62, 63, 64, 65, 66, 67, 68 More recently, three additional mutations of α-SYN, H50Q, G51D and A53E, have been identified in PD patients.63, 64, 65 Increasing body of evidence from various experimental models has shown that elevated levels of wild-type and various mutant species are prone to accelerate the aggregation process. In misfolded state, α-SYN is characterized by twisted, nonbranched filaments of β-sheets.69 The mutant α-SYN proteins are transformed into amyloid fibrillar species consisting of β-sheets, which have properties to serve as templates to drive soluble proteins to adopt similar structural changes, leading to the formation of highly ordered aggregated structure.69, 70, 71 Increased oxidative stress is a key contributor to the pathogenesis of PD and several in vitro and in vivo experiments have linked oxidative stress to α-SYN aggregation. Exposure of neuronal cells to various oxidative stressors including ferrous ions, hydrogen peroxide, MPP+ (1-methyl-4-phenylpyridinium), rotenone, nitric oxide and superoxide all promoted the aggregation process.72, 73, 74, 75 In vivo studies also corroborated the same idea that chronic and systemic exposure of rodents to rotenone, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine or paraquat leads to selective nigrostriatal dopaminergic lesions accompanying degeneration with α-SYN aggregation.76, 77, 78, 79
Thus, how the 8-oxodG-mediated TM event might have a critical role in the process of α-SYN fibrillogenesis depends on whether the mutant species generated by this process can acquire a misfolded state by themselves, which will eventually act as a seed in the nucleation-dependent aggregation process as seen for reported mutant α-SYN proteins.80 If the 8-oxodG-mediated TM mutant species are more stable in the β-sheet form, then a limited amount of TM species could be enough to promote prion-like nucleation of α-SYN.80, 81 Over the past few years, increasing number of studies have provided evidence that α-SYN aggregates can propagate from one brain region to another in a prion-like manner.82, 83 This self-perpetuating cycle of α-SYN fibrillation and propagation could be initiated by the addition of a minute amount of pathogenic proteins that can potentially serve as aggregate seeding. 8-oxodG-derived TM species may trigger this process, leading to the pathogenesis of PD.
In a paraquat-based animal model of PD, it has been shown that nuclear 8-oxodG accumulation in the substantia nigra is clearly correlated with increase in proteinase-K resistant species of α-SYN aggregates.78 A recent study has pointed that genomic distribution of 8-oxodG is not a random event, instead it is localized preferentially to specific areas of the chromosome and is negatively correlated with transcriptional activity of a gene.84 This fact implies that TM could affect various genes in the context of chromatin structure, and mutant species originating from TM event should be outnumbered by the normal physiological form. Therefore, for most proteins, generation of small portion of nonfunctional TM species would not be likely to cause entire functional impairment. However, α-SYN could become cytotoxic in the nucleation-dependent oligomerization process, in which small addition of mutant α-SYN species to wild-type population may initiate the seeding process and fibrillogenesis80, 85. This unique biochemical feature of α-SYN would strongly support the feasibility of the proposed model.
All possible mutant α-SYN proteins which could be generated through TM-mediated replacement of cytosine with adenine in the mRNA strand are listed in Figure 3. Among TM-generated α-SYN mutants, the following mutants, A30E, H50N and A53E, have mutations at the same amino acid position as the familial forms of mutants, A30P, H50Q and both A53E and A53T, respectively. It would strongly suggest that those TM mutants could similarly cause a nucleation-dependent α-SYN fibrillation. Apart from these mutant forms, there are several other residues that might disrupt the native structure of the protein when they get mutated and make them more prone to aggregation. To predict the aggregation propensity of expected TM species, we used TANGO, a statistical mechanics algorithm, which enables identifying β-aggregating regions within a protein based on the sequence information.86 TANGO algorithm indicated that a couple of expected TM proteins has significantly higher β-aggregation scores than wild-type α-SYN, which include L38I, S42Y, H50N and Q62K in the N-terminal or the non-Aβ component of amyloid plaques domain, predicting increases in aggregation property (Figure 4). Post-translational modifications such as phosphorylation on serine 87 and 129 may also affect the α-SYN aggregation process.87, 88 TM event can replace the serine at 129 position with a tyrosine. Together we propose a hypothetical model by which 8-oxodG lesions in the protein coding region of SNCA could yield a small amount of TM protein species with higher aggregation propensity that potentially serve as a seeding for accelerating the aggregation of wild-type α-SYN, leading to the self-propagation of aggregates and causing degeneration of the nigrostriatal pathway in PD (Figure 5).
Figure 3.

All the possible mutant amino acid sequences of α-SYN generated by 8-oxodG-driven TM. Upper line (wild type) represents the reference amino acid sequence of the wild-type protein: lower line (TM) shows amino acid changes can be caused by 8-oxodG- mediated transcriptional mutagenesis arising from misincorporated adenine instead of cytosine in the mRNA sequence during transcription (red); green letters designate reported a-SYN mutants (A30P, E46K, H50Q, G51D, A53E & A53T) in familial PD; and serine 87 and129 (blue) is the site of phosphorylation as seen during aggregation of the molecule.
Figure 4.
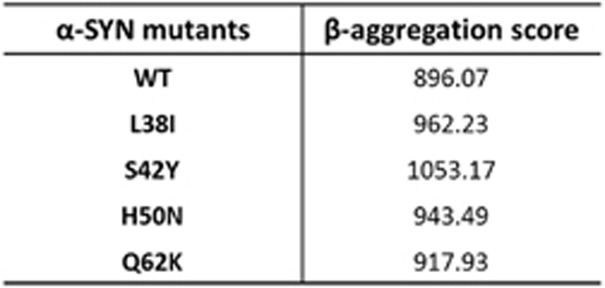
Predicted score for wild-type and some of the possible mutant α-SYN species generated by TM event to form β-aggregate by TANGO. β-aggregation score for wild-type and some of the possible mutant α-SYN species which can be generated through TM event as predicted by the structure analyzing algorithm TANGO (see text for reference). Abbreviations: L38I, leucine to isoleucine; S42Y, serine to tyrosine; H50N, histidine to asparagine; Q62K, glutamine to lysine; WT, wild type.
Figure 5.

Proposed model for aggregation of wild-type α-SYN protein by TM-generated mutant α-SYN species and propagation. Mutant α-SYN generated through TM event may trigger a nucleation-dependent aggregation of the predominantly higher amount of wild-type species and lead to propagation of α-SYN aggregation.
Conclusion
The majority of idiopathic PD cases are a late-onset sporadic form with cytoplasmic α-SYN aggregates, which indicates that increasing degree of aggregation does not depend only on genetic mutations in SNCA. However, till date, the approaches to understand the molecular mechanism of α-SYN aggregation have focused primarily on the biochemical properties of mutant protein species that were identified in rare familial form of PD and their behavior within the cells. The proposed model will give an insight into a novel mechanism called ‘transcriptional mutagenesis' caused by the accumulation of oxidatively damaged DNA lesions, 8-oxodG, in the SNCA gene. Comprehensive investigation on age-dependent changes in α-SYN mRNA profiles as well as identification of TM species supported by functional studies on mutant proteins will definitely add a new dimension to the understanding of α-SYN pathology in conjunction with oxidative stress.
Acknowledgments
We thank Dr Tong H Joh for his critical reading of the manuscript and suggestion. GJ acknowledges support from BK21 MD/PhD program of Kyunghee University College of Medicine. This work is supported by the University of Florida College of Medicine grant program and Michael J Fox Foundation to YSK.
The authors declare no conflict of interest.
References
- Lotharius J, Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- Kruger R, Eberhardt O, Riess O, Schulz JB. Parkinson's disease: one biochemical pathway to fit all genes. Trends Mol Med. 2002;8:236–240. doi: 10.1016/s1471-4914(02)02333-x. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Lee VM, Trojanowski JQ. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol. 2002;52:205–210. doi: 10.1002/ana.10279. [DOI] [PubMed] [Google Scholar]
- Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, et al. Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, et al. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, et al. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregeon D, Doddridge ZA, You HJ, Weiss B, Doetsch PW. Transcriptional mutagenesis induced by uracil and 8-oxoguanine in Escherichia coli. Mol Cell. 2003;12:959–970. doi: 10.1016/s1097-2765(03)00360-5. [DOI] [PubMed] [Google Scholar]
- Bregeon D, Peignon PA, Sarasin A. Transcriptional mutagenesis induced by 8-oxoguanine in mammalian cells. PLoS Genet. 2009;5:e1000577. doi: 10.1371/journal.pgen.1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell MA, Gabbita SP, Markesbery WR. Increased DNA oxidation and decreased levels of repair products in Alzheimer's disease ventricular CSF. J Neurochem. 1999;72:771–776. doi: 10.1046/j.1471-4159.1999.0720771.x. [DOI] [PubMed] [Google Scholar]
- Xu G, Herzig M, Rotrekl V, Walter CA. Base excision repair, aging and health span. Mech Ageing Dev. 2008;129:366–382. doi: 10.1016/j.mad.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregeon D, Doetsch PW. Transcriptional mutagenesis: causes and involvement in tumour development. Nat Rev Cancer. 2011;11:218–227. doi: 10.1038/nrc3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, et al. Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. J Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, et al. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson's disease. J Neurochem. 1989;52:1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Fu W, Keller JN, Markesbery WR, Appel S, Smith RG, et al. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann Neurol. 1998;44:819–824. doi: 10.1002/ana.410440518. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiol Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence. Nat Med. 2004;10:S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good PF, Hsu A, Werner P, Perl DP, Olanow CW. Protein nitration in Parkinson's disease. J Neuropathol Exp Neurol. 1998;57:338–342. doi: 10.1097/00005072-199804000-00006. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Matsubara K, Fujikawa Y, Nagahiro Y, Shimizu K, Umegae N, et al. Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann Neurol. 2000;47:524–527. [PubMed] [Google Scholar]
- Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- Kasai H, Nishimura S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984;12:2137–2145. doi: 10.1093/nar/12.4.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40:405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- Zemlan FP, Thienhaus OJ, Bosmann HB. Superoxide dismutase activity in Alzheimer's disease: possible mechanism for paired helical filament formation. Brain Res. 1989;476:160–162. doi: 10.1016/0006-8993(89)91550-3. [DOI] [PubMed] [Google Scholar]
- Pappolla MA, Omar RA, Kim KS, Robakis NK. Immunohistochemical evidence of oxidative [corrected] stress in Alzheimer's disease. Am J Pathol. 1992;140:621–628. [PMC free article] [PubMed] [Google Scholar]
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, Hayden DL, et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol. 1997;41:210–221. doi: 10.1002/ana.410410212. [DOI] [PubMed] [Google Scholar]
- Perry TL, Godin DV, Hansen S. Parkinson's disease: a disorder due to nigral glutathione deficiency. Neurosci Lett. 1982;33:305–310. doi: 10.1016/0304-3940(82)90390-1. [DOI] [PubMed] [Google Scholar]
- Mitra S, Izumi T, Boldogh I, Bhakat KK, Hill JW, Hazra TK. Choreography of oxidative damage repair in mammalian genomes. Free Radic Biol Med. 2002;33:15–28. doi: 10.1016/s0891-5849(02)00819-5. [DOI] [PubMed] [Google Scholar]
- Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppede F, Ceravolo R, Migheli F, Fanucchi F, Frosini D, Siciliano G, et al. The hOGG1 Ser326Cys polymorphism is not associated with sporadic Parkinson's disease. Neurosci Lett. 2010;473:248–251. doi: 10.1016/j.neulet.2010.02.059. [DOI] [PubMed] [Google Scholar]
- Dogru-Abbasoglu S, Aykac-Toker G, Hanagasi HA, Gurvit H, Emre M, Uysal M. The Arg194Trp polymorphism in DNA repair gene XRCC1 and the risk for sporadic late-onset Alzheimer's disease. Neurol Sci. 2007;28:31–34. doi: 10.1007/s10072-007-0744-x. [DOI] [PubMed] [Google Scholar]
- Qian Y, Chen W, Wu J, Tao T, Bi L, Xu W, et al. Association of polymorphism of DNA repair gene XRCC1 with sporadic late-onset Alzheimer's disease and age of onset in elderly Han Chinese. J Neurol Sci. 2010;295:62–65. doi: 10.1016/j.jns.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, et al. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Hartsuiker E, Caldecott KW. TDP1 facilitates repair of ionizing radiation-induced DNA single-strand breaks. DNA Repair (Amst) 2007;6:1485–1495. doi: 10.1016/j.dnarep.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- Cardozo-Pelaez F, Sanchez-Contreras M, Nevin AB. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem Int. 2012;61:721–730. doi: 10.1016/j.neuint.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC. Emerging links between premature ageing and defective DNA repair. Mech Ageing Dev. 2008;129:503–505. doi: 10.1016/j.mad.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, Hagen TM, et al. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci USA. 2002;99:2356–2361. doi: 10.1073/pnas.261709299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- Maker HS, Weiss C, Silides DJ, Cohen G. Coupling of dopamine oxidation (monoamine oxidase activity) to glutathione oxidation via the generation of hydrogen peroxide in rat brain homogenates. J Neurochem. 1981;36:589–593. doi: 10.1111/j.1471-4159.1981.tb01631.x. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D, Riederer P, Youdim MB. Iron-melanin interaction and lipid peroxidation: implications for Parkinson's disease. J Neurochem. 1991;57:1609–1614. doi: 10.1111/j.1471-4159.1991.tb06358.x. [DOI] [PubMed] [Google Scholar]
- Riederer P, Sofic E, Rausch WD, Schmidt B, Reynolds GP, Jellinger K, et al. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don't divide what's to repair. Mutat Res. 2007;614:24–36. doi: 10.1016/j.mrfmmm.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Shimura-Miura H, Hattori N, Kang D, Miyako K, Nakabeppu Y, Mizuno Y. Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson's disease. Ann Neurol. 1999;46:920–924. [PubMed] [Google Scholar]
- Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27:1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- Nouspikel T, Hanawalt PC. DNA repair in terminally differentiated cells. DNA Repair (Amst) 2002;1:59–75. doi: 10.1016/s1568-7864(01)00005-2. [DOI] [PubMed] [Google Scholar]
- Morreall JF, Petrova L, Doetsch PW. Transcriptional mutagenesis and its potential roles in the etiology of cancer and bacterial antibiotic resistance. J Cell Physiol. 2013;228:2257–2261. doi: 10.1002/jcp.24400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan A, You HJ, Doetsch PW. Phenotypic change caused by transcriptional bypass of uracil in nondividing cells. Science. 1999;284:159–162. doi: 10.1126/science.284.5411.159. [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Endou M, Yamaguchi Y, Wada T, Handa H, Tanaka K. Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II. Implications for transcription-coupled DNA repair and transcriptional mutagenesis. J Biol Chem. 2003;278:7294–7299. doi: 10.1074/jbc.M208102200. [DOI] [PubMed] [Google Scholar]
- Holmquist GP. Cell-selfish modes of evolution and mutations directed after transcriptional bypass. Mutat Res. 2002;510:141–152. doi: 10.1016/s0027-5107(02)00259-2. [DOI] [PubMed] [Google Scholar]
- Damsma GE, Cramer P. Molecular basis of transcriptional mutagenesis at 8-oxoguanine. J Biol Chem. 2009;284:31658–31663. doi: 10.1074/jbc.M109.022764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxowsky TT, Meadows KL, Klungland A, Doetsch PW. 8-Oxoguanine-mediated transcriptional mutagenesis causes Ras activation in mammalian cells. Proc Natl Acad Sci USA. 2008;105:18877–18882. doi: 10.1073/pnas.0806464105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn TB, Kim SY, Kim JY, Park SS, Lee DS, Min HJ, et al. alpha-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology. 2008;70:43–49. doi: 10.1212/01.wnl.0000271080.53272.c7. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord. 2013;28:811–813. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. 2013;73:459–471. doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, et al. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease-type pathology. Neurobiol Aging. 2014;35:2180 e1–2180 e5. doi: 10.1016/j.neurobiolaging.2014.03.024. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci USA. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredenburg RA, Rospigliosi C, Meray RK, Kessler JC, Lashuel HA, Eliezer D, et al. The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry. 2007;46:7107–7118. doi: 10.1021/bi7000246. [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 2015;16:109–120. doi: 10.1038/nrn3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, et al. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci. 2001;21:8053–8061. doi: 10.1523/JNEUROSCI.21-20-08053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci. 2000;20:6048–6054. doi: 10.1523/JNEUROSCI.20-16-06048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakimura J, Kitamura Y, Takata K, Kohno Y, Nomura Y, Taniguchi T. Release and aggregation of cytochrome c and alpha-synuclein are inhibited by the antiparkinsonian drugs, talipexole and pramipexole. Eur J Pharmacol. 2001;417:59–67. doi: 10.1016/s0014-2999(01)00902-5. [DOI] [PubMed] [Google Scholar]
- Borland MK, Trimmer PA, Rubinstein JD, Keeney PM, Mohanakumar K, Liu L, et al. Chronic, low-dose rotenone reproduces Lewy neurites found in early stages of Parkinson's disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol Neurodegener. 2008;3:21. doi: 10.1186/1750-1326-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Cristovao AC, Guhathakurta S, Bok E, Je G, Yoo SD, Choi DH, et al. NADPH oxidase 1 mediates alpha-synucleinopathy in Parkinson's disease. J Neurosci. 2012;32:14465–14477. doi: 10.1523/JNEUROSCI.2246-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003;179:9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J Biol Chem. 1999;274:19509–19512. doi: 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci USA. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Jiang L, Akatsuka S, Suyama M, Toyokuni S. Genome-wide profiling of 8-oxoguanine reveals its association with spatial positioning in nucleus. DNA Res. 2014;21:603–612. doi: 10.1093/dnares/dsu023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem. 2010;285:34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T. Pathological biochemistry of alpha-synucleinopathy. Neuropathology. 2007;27:474–478. doi: 10.1111/j.1440-1789.2007.00785.x. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]