

Abstract
Background
This study aimed to better understand the mechanisms underlying methotrexate (MTX)—resistance in osteosarcoma.
Methods
The raw transcription microarray data GSE16089 collected from three MTX-sensitive osteosarcoma (Saos-2) cell samples and three MTX-resistant osteosarcoma (Saos-2) cell samples were downloaded from Gene Expression Omnibus. After data processing, the differentially expressed genes (DEGs) were identified. Next, DEGs were submitted to DAVID for functional annotation based on the GO (Gene Ontology) database, as well as pathway enrichment analysis based on the KEGG (Kyoto Encyclopedia of Genes and Genomes) database. Transcription factors (TFs) and tumor-associated genes (TAGs) were identified with reference to TRANSFAC and TAG, and TSGene databases, respectively. The protein-protein interaction (PPI) network of the gene-encoded products was constructed, and the subnetwork with the highest score was also detected using Search Tool for the Retrieval of Interacting Genes and BioNet package.
Results
A total of 690 up-regulated genes and down-regulated 626 genes were identified. Up-regulated DEGs (including AARS and PARS2) were associated to transfer RNA (tRNA) aminoacylation while down-regulated DEGs (including AURKA, CCNB1, CCNE2, CDK1, and CENPA) were correlated with mitotic cell cycle. Totally, 13 TFs (including HMGB2), 13 oncogenes (including CCNA2 and AURKA), and 19 tumor suppressor genes (TSGs) (including CDKN2C) were identified from the down-regulated DEGs. Ten DEGs, including nine down-regulated genes (such as AURKA, CDK1, CCNE2, and CENPA) and one up-regulated gene (GADD45A), were involved in the highest score subnetwork.
Conclusion
AARS, AURKA, AURKB, CENPA, CCNB1, CCNE2, and CDK may contribute to MTX resistance via aminoacyl-tRNA biosynthesis pathway, cell cycle pathway, or p53 signaling pathway.
Keywords: Osteosarcoma, Methotrexate resistance, Microarray data
Background
Methotrexate (MTX) was first introduced to replace aminopterin to treat acute lymphocytic leukemia, which works via inhibiting dihydrofolate reductase (DHFR), a key enzyme required in intracellular folate metabolism, leading to decreased tretrahydrofolate coenzyme level, accordingly achieving the inhibition of thymidylate and the biosynthesis of DNA and purine. So far, different mechanisms have been presented to address the intrinsic and acquired MTX resistance: (1) decreased MTX transport, (2) impaired MTX polyglutamylation, (3) increased DHFR enzyme activity, (4) altered affinity of MTX for DHFR, and (5) increased MTX efflux due to elevated levels of the multidrug resistance protein (MRP) [1].
Osteosarcoma is a primary malignant, highly vascularized bone tumor, mainly occurring in adolescents and children [2, 3]. The unclear understanding of the underlying molecular mechanism greatly hinders the therapy of osteosarcoma [4]. Currently, multiagent chemotherapy, usually using doxorubicin, cisplatin, and high-dose MTX, has improved the survival of osteosarcoma patients from 11 to 70 % [5]. However, MTX resistance has become an issue of growing interest, as little information is available in this disease up to now [6]. TP53, a tumor suppressor gene (TSG), encodes a transcriptional regulator that responds to DNA damage or cellular stress and controls the progression and apoptosis of cell cycle. As previously reported, the accumulation of p53 protein is probably a predictor of response to methotrexate (MTX) [7]. p53 alterations increase the risk for the development of drug resistance by altering MTX transport [8].
Using the transcription profiles of three MTX-sensitive osteosarcoma (Saos-2) cell lines and three MTX-resistant Saos-2 cell lines and analyzing the network of the differentially expressed genes (DEGs), Selga et al. have found the alteration in the expression of a number of genes, such as eukaryotic translation elongation factor 1 alpha 1 (EEF1A1) in the MTX-resistant osteosarcomas (Saos-2) cell lines, pancreatic cancer, and erythroblastic leukemia cell lines [9]. However, further systematic analyses, including GO (Gene Ontology) functional and REACTOME pathway enrichment analysis, were not performed for DEGs concerning osteosarcoma cells in their study.
REACTOME is a knowledgebase of human reactions and pathways, which provides an integrated view of the molecular details of human biological processes ranging from metabolism to DNA replication and repair to signaling cascades [10], and has been used in various studies [11]. The transcription microarray data GSE16089 deposited in Gene Expression Omnibus (GEO), which includes three chips from MTX-sensitive and three from MTX-resistant osteosarcoma (Saos-2) cell lines [9], were downloaded and analyzed in this study so as to better understand the genetic etiology of osteosarcoma. The DEGs were identified, and the functional and pathway enrichment analysis was performed for them. The protein-protein interaction (PPI) network of the gene productions and its subnetwork were analyzed. These findings in this study will encourage us to investigate the anti-cancer effects of the DEGs or the pathways as well as the MTX resistance in osteosarcoma.
Material and methods
As the study did not involve any human or animal study, the ethical approval was not required.
Microarray data
Gene expression microarray dataset deposited in the National Center of Biotechnology Information (NCBI) GEO (http://www.ncbi.nlm.nih.gov/geo/) with the accession number of GSE16089 was downloaded [9]. The annotation platform was GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array. According to contributors, three samples of either MTX-sensitive cells or MTX-resistant cells of the Saos-2 osteosarcoma cell line were used for gene expression analyses [9]. Saos-2 cell line was sensitive to MTX, and its MTX-resistant cells were obtained in the laboratory via incubation with stepwise concentrations of MTX (Lederle) as described previously [12].
Data processing
The raw probe profile data was downloaded from GEO. The processing of the raw microarray data was performed by robust multiarray average (RMA) using R/Bioconductor package Affy [13]. The preprocessing consisted of background correction, quantile normalization, and probe summarization of expression value. The gene expression matrixes were obtained for further analysis.
Identification of DEGs
Transcriptional sets were mapped to NCBI entrez genes using Gene ID converter [14]. The averaged value was calculated for further analysis if there were multiple probe sets corresponding to the same gene. Probes were filtered if they corresponded to multiple genes. The classical t test was performed among the samples to identify the genes specifically differentially expressed between MTX-sensitive and MTX-resistant Saos-2 cell lines. The cut-off criteria for the DEGs were set at p value <0.05 and |log2FC (fold change)| > 1.
Functional and pathway enrichment analysis
GO and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis provides prediction of gene function and informs people of how molecules or genes work [15, 16]. The DEGs were submitted to DAVID (Database for Annotation, Visualization, and Integrated Discovery) (http://david.abcc.ncifcrf.gov/) to find the significantly enriched biological process (BP) terms, molecular function (MF) terms, and cellular component (CC) terms based on the GO (Gene Ontology) database, as well as pathways based on the KEGG (Kyoto Encyclopedia of Genes and Genomes) database. For the identification of the significantly enriched biological processes in detail, the significantly altered DEGs were subjected to the REACTOME knowledgebase (http://www.reactome.org). The thresholds for the significant associated GO functional category and pathways were set at p value < 0.01.
Identification of transcription factors and tumor-associated genes
TRANSFAC (http://www.gene-regulation.com/index2) is a database on transcription factors, their genomic binding sites, and DNA-binding profiles [17]. To identify the DEGs that also act as transcription factors, transcription factor (TF) prediction was performed using the TRANSFAC database. TAG (tumor-associated gene) database (http://www.binfo.ncku.edu.tw/TAG/) is a semi-automatic information retrieving engine which collects specific information about genes from various resources. TSGene database (http://bioinfo.mc.vanderbilt.edu/TSGene/) is a resource of tumor suppressor genes (TSGs) that provides a comprehensive TSG catalog for advanced systems biology-based analysis for the cancer research community [18]. TAGs including oncogenes and TSGs were also identified from the DEGs using TAG and TSGene databases, respectively.
Construction of PPI network and the subnetwork analysis
STRING (Search Tool for the Retrieval of Interacting Genes) is a web server to retrieve and display the repeatedly occurring neighborhood of a gene which generalizes access to protein interaction data, by integrating known and predicted interactions from a variety of sources [19]. To describe the interactive network of DEGs, the STRING database was used to build the PPI network of encoding products of all of the DEGs. A STRING score of 0.4 was set as the reliability threshold. Cytoscape, a standard tool for integrated analysis and visualization of biological networks, was used to visualize the PPI network [20]. The connectivity degree analysis was performed, and hub nodes were obtained using the scale-free properties of PPI networks. The BioNet package is an R-Package for the functional analysis of biological networks and is used for the mining of the sub-networks in the PPI network [21]. The highest scoring subnetwork was obtained. The threshold of the given false discovery rate (FDR) value was 0.0001.
Results
Identification of DEGs
After data processing, a total of 4461 transcripts that were differentially expressed between MTX-sensitive and MTX-resistant Saos-2 cell lines were identified, 2300 up-regulated and 2161 down-regulated transcripts. Finally, 1316 DEGs were obtained, including 690 up-regulated DEGs (e.g., AARS, TARS, YARS, CCND1, PARS2, and GADD45A) and 626 down-regulated DEGs (e.g., AURKA, AURKB, CCNB1, CDK1, CDKN2C, CENPA, and HMGB2).
Functional and pathway enrichment analysis
According to the GO annotation, the up-regulated DEGs were functionally involved in BP terms such as response to endoplasmic reticulum stress (including AARS and CCND1) and transfer RNA (tRNA) aminoacylation for protein translation (including AARS, TARS, YARS, and PARS2), and MF terms such as aminoacyl-tRNA ligase activity (including AARS and PARS2), as well as CC terms such as intracellular membrane-bounded organelle (including CCND1 and GADD45A) (Table 1). Moreover, the up-regulated DEGs were mainly associated with the KEGG pathways such as aminoacyl-tRNA biosynthesis (including AARS and PARS2). The mainly related REACTOME pathways were cytosolic tRNA aminoacylation (including AARS) and tRNA aminoacylation (including AARS and PARS2) (Table 2).
Table 1.
The top five terms of Gene Ontology category analysis for the up-regulated differentially expressed genes
| Category | Term | Name | Counts | Gene symbol | p value |
|---|---|---|---|---|---|
| Biological process | GO:0034976 | Response to endoplasmic reticulum stress | 21 | AARS, CCND1, FAM129A, GSK3B, … | 3.17E−10 |
| GO:0006418 | tRNA aminoacylation for protein translation | 12 | AARS, PARS2, TARS, YARS, … | 3.41E−08 | |
| GO:0043038 | Amino acid activation | 12 | AARS, PARS2, TARS, YARS, … | 7.23E−08 | |
| GO:0043039 | tRNA aminoacylation | 12 | AARS, PARS2, TARS, YARS, … | 7.23E−08 | |
| GO:0034620 | Cellular response to unfolded protein | 14 | AARS, CCND1, SERP1, STC2, … | 1.35E−06 | |
| Molecular function | GO:0004812 | Aminoacyl-tRNA ligase activity | 12 | AARS, PARS2, TARS, YARS, … | 6.72E−09 |
| GO:0016875 | Ligase activity, forming carbon-oxygen bonds | 12 | AARS, PARS2, TARS, YARS, … | 6.72E−09 | |
| GO:0016876 | Ligase activity, forming aminoacyl-tRNA and related compounds | 12 | AARS, PARS2, TARS, YARS, … | 6.72E−09 | |
| GO:0015175 | Neutral amino acid transmembrane transporter activity | 7 | SLC1A4, SLC1A5, SLC3A2, SLC7A5, … | 2.70E−06 | |
| GO:0015171 | Amino acid transmembrane transporter activity | 9 | SLC1A4, SLC1A5, SLC3A2, SLC7A5, … | 1.74E−4 | |
| Cellular components | GO:0043231 | Intracellular membrane-bounded organelle | 370 | CCND1, COL1A2, EEF1A2, GADD45A, … | 5.07E−06 |
| GO:0043227 | Membrane-bounded organelle | 371 | CCND1, COL1A2, EEF1A2, GADD45A, … | 5.19E−06 | |
| GO:0005622 | Intracellular | 448 | AARS, CCND1, EEF1A2, GADD45A, … | 3.09E−05 | |
| GO:0044424 | Intracellular part | 443 | AARS, CCND1, EEF1A2, GADD45A, … | 4.49E−05 | |
| GO:0005737 | Cytoplasm | 346 | AARS, CD9, CCND1, EEF1A2, … | 8.23E−05 |
Table 2.
Pathway enrichment of the up-regulated differentially expressed genes
| Pathway | Term | Name | Count | Gene symbol | p value |
|---|---|---|---|---|---|
| KEGG pathway | 970 | Aminoacyl-tRNA biosynthesis | 12 | AARS, PARS2, TARS, YARS, … | 6.95E−07 |
| 260 | Glycine, serine, and threonine metabolism | 5 | CBS, CTH, PHGDH, PSAT1, SHMT2 | 3.5E−03 | |
| 532 | Glycosaminoglycan biosynthesis-chondroitin sulfate | 4 | CHPF, CHST15, UST, XYLT1 | 5.2E−03 | |
| 565 | Ether lipid metabolism | 5 | ENPP2, LPCAT1, PLA2G12A, PLA2G4A, PPAP2B | 6.0E−03 | |
| REACTOME pathway (top ten terms) | 379716 | Cytosolic tRNA aminoacylation | 10 | AARS, PARS2, TARS, YARS, … | 1.50E−09 |
| 379724 | tRNA aminoacylation | 12 | AARS, PARS2, TARS, YARS, … | 5.17E−09 | |
| 352230 | Amino acid transport across the plasma membrane | 8 | SLC1A4, SLC1A5, SLC3A2, SLC7A5, … | 4.75E−06 | |
| 425374 | Amino acid and oligopeptide SLC transporters | 9 | SLC1A4, SLC1A5, SLC3A2, SLC7A5, … | 2.39E−05 | |
| 380994 | Activation of genes by ATF4 | 4 | ASNS, ATF3, DDIT3, HERPUD1 | 3.61E−05 | |
| 70614 | Amino acid synthesis and interconversion (transamination) | 5 | ASNS, GLS, GPT2, PHGDH, PSAT1 | 0.000160459 | |
| 381042 | PERK regulated gene expression | 4 | ASNS, ATF3, DDIT3, HERPUD1 | 4.48E−04 | |
| 1614603 | Cysteine formation from homocysteine | 2 | CBS, CTH | 1.1E−03 | |
| 73943 | Reversal of alkylation damage by DNA dioxygenases | 2 | ALKBH2, ALKBH3 | 1.1E−03 | |
| 73942 | DNA damage reversal | 2 | ALKBH2, ALKBH3 | 3.1E−03 |
KEGG Kyoto Encyclopedia of Genes and Genomes
The down-regulated DEGs were significantly enriched in BP terms such as microtubule cytoskeleton organization and mitotic cell cycle (including AURKA, AURKB, CCNB1, CDK1, and CENPA) and in MF terms such as protein binding and nucleotide binding (including AURKA, AURKB, CCNB1, CDK1, and CENPA), as well as in CC terms related to chromosome, centromeric region (including AURKB, CCNB1, CENPA, and CENPH), and kinetochore (including CCNB1, CENPA, and CENPH) (Table 3). Also, the down-regulated DEGs were significantly involved in KEGG pathways of cell cycle, oocyte meiosis, and p53 signaling pathway (including CCNB1, CCNE2, and CDK1). The relevant REACTOME pathways were resolution of sister chromatid cohesion and mitotic M-M/G1 phases (including AURKB, CCNB1, CDK1, CENPA, and CENPH) (Table 4).
Table 3.
The top five terms of Gene Ontology category analysis for the down-regulated differentially expressed genes
| Category | Term | Name | Counts | Gene symbol | p value |
|---|---|---|---|---|---|
| Biological process | GO:0000226 | Microtubule cytoskeleton organization | 46 | AURKA, AURKB, CCNB1 CDK1, CENPA, FBXO5, … | 0 |
| GO:0000278 | Mitotic cell cycle | 119 | AURKA, AURKB, CCNE2, CDK1, CENPA, CENPH, FBXO5, RRM2, … | 0 | |
| GO:0000280 | Nuclear division | 68 | AURKA, AURKB, CCNB1, CCNB2, CENPE, … | 0 | |
| GO:0006996 | Organelle organization | 164 | AURKA, AURKB, CCNB1, CCNB2, CDK1, CENPA, CENPE, CENPH, … | 0 | |
| GO:0007017 | Microtubule-based process | 59 | AURKA, AURKB, CCNB1, CDK1, CENPA, FBXO5, … | 0 | |
| Molecular function | GO:0005515 | Protein binding | 334 | AURKA, AURKB, CCNB1, CCNB2, CCNE2, CDK1, HMGB2, … | 8.15E−13 |
| GO:0000166 | Nucleotide binding | 122 | AURKA, AURKB, CDK1, CENPE, EEF1A1, … | 1.32E−07 | |
| GO:1901265 | Nucleoside phosphate binding | 122 | AURKA, AURKB, CDK1, CENPE, EEF1A1, … | 1.35E−07 | |
| GO:0036094 | Small molecule binding | 127 | AURKA, AURKB, CDK1, CENPE, EEF1A1, … | 2.24E−07 | |
| GO:0008017 | Microtubule binding | 19 | CENPE, KIF14, KIF15, PLK1, … | 8.25E−07 | |
| Cellular components | GO:0000775 | Chromosome, centromeric region | 37 | AURKB, BUB1, CCNB1, CENPA, CENPH, … | 0 |
| GO:0000776 | Kinetochore | 29 | CCNB1, CENPA, CENPE CENPH, KIF22, … | 0 | |
| GO:0000777 | Condensed chromosome kinetochore | 27 | CCNB1, CENPA, CENPE CENPH, KIF2C, … | 0 | |
| GO:0000779 | Condensed chromosome, centromeric region | 28 | AURKB, CCNB1, CENPA, CENPE, CENPH, … | 0 | |
| GO:0000793 | Condensed chromosome | 42 | AURKB, CCNB1, CENPA, CENPH, KIF2C, … | 0 |
Table 4.
Pathway enrichment analysis of the down-regulated differentially expressed genes
| Pathway | Term | Name | Count | Gene symbol | p value |
|---|---|---|---|---|---|
| KEGG pathway | 4110 | Cell cycle | 21 | CCNA2, CCNB1, CCNB2, CCNE2, CDK1, CDKN2C, … | 2.35E−09 |
| 5322 | Systemic lupus erythematosus | 15 | H2AFX, H2AFY, HIST1H2AE, HIST1H2AM, … | 1.07E−04 | |
| 4114 | Oocyte meiosis | 13 | AURKA, CCNB1, CCNB2, CCNE2, CDK1, FBXO5, … | 1.86E−04 | |
| 4115 | p53 signaling pathway | 9 | CCNB1, CCNB2, CCNE2, CDK1, GADD45G, … | 7.05E−04 | |
| 30 | Pentose phosphate pathway | 5 | ALDOC, DERA, PFKP, PGM1, PRPS1 | 2.5E−03 | |
| 4914 | Progesterone-mediated oocyte maturation | 9 | CCNA2, CCNB1, CCNB2, CDK1, PLK1, … | 3.7E−03 | |
| 3030 | DNA replication | 5 | FEN1, LIG1, POLA2, POLE, POLE2 | 9.0E−03 | |
| REACTOME pathway (top ten terms) | 1640170 | Cell cycle | 90 | AURKA, AURKB, CCNB1, CCNE2, CDK1, CENPA, CENPH, … | 0 |
| 2500257 | Resolution of sister chromatid cohesion | 34 | AURKB, CCNB1, CCNB2, CDK1, CENPA, CENPH, … | 0 | |
| 453277 | Mitotic M-M/G1 phases | 57 | AURKB, CCNB1, CCNB2, CDK1, CENPA, CENPH, … | 0 | |
| 68877 | Mitotic prometaphase | 39 | AURKB, CCNB1, CCNB2, CDK1, CENPA, CENPH, … | 0 | |
| 68886 | M phase | 48 | AURKB, CCNB1, CCNB2, CDK1, CENPA, CENPH, … | 0 | |
| 69278 | Cell cycle, mitotic | 79 | AURKA, AURKB, CCNB1, CCNE2, CDK1, CENPA, CENPH, … | 0 | |
| 2555396 | Mitotic metaphase and anaphase | 38 | AURKB, CENPA, CENPH, FBXO5, PLK1, PSMD1, … | 1.11E−16 | |
| 2467813 | Separation of sister chromatids | 36 | AURKB, CENPA, CENPH, PLK1, PSMD1, … | 4.44E−16 | |
| 68882 | Mitotic anaphase | 37 | AURKB, CENPA, CENPH, KIF2C, PSMD1, … | 4.44E−16 | |
| 69275 | G2/M transition | 23 | AURKA, CCNA2, CCNB1, CCNB2, CDK1, PLK1, PLK4, … | 6.87E−10 |
KEGG Kyoto Encyclopedia of Genes and Genomes
Identification of TFs, oncogenes, and TSGs
After the annotation, 13 TFs (TAF9, RUNX3, RUNX2, PBX1, MYBL1, MSX2, MEIS2, HMGB2, GTF2H2, FOXM1, FOXD1, EZH2, and BRIP1), 13 oncogenes (WHSC1, SERTAD1, RUNX2, RBM3, PTTG1, PBX1, MYBL1, MLLT11, KIT, HMMR, CEP55, CCNA2, and AURKA), and 19 TSGs (TGFBI, S100A2, RUNX3, PPP1R1B, PLK2, MSH2, ING3, IGFBP7, IGFBP3, HTRA1, GADD45G, FANCG, EGLN3, EEF1A1, CLU, CDKN2C, CDH13, CCDC136, BUB1B) were identified. However, no TFs, oncogenes, or TSGs were identified from the up-regulated DEGs.
PPI network and the subnetwork analysis
After construction and visualization for the PPI network of the DEG products, a total of 375 nodes were included, such as CDK1 (degree = 82), CCNA2 (degree = 64), CCNB1 (degree = 62), and CENPE (degree = 56) (Fig. 1). The subnetwork with the highest score contained ten gene-encoding proteins, namely CDK1, RRM2, CENPA, AURKA, FBXO5, CCNE2, CENPH, ASF1B, PSMD1, and GADD45A (Fig. 2).
Fig. 1.
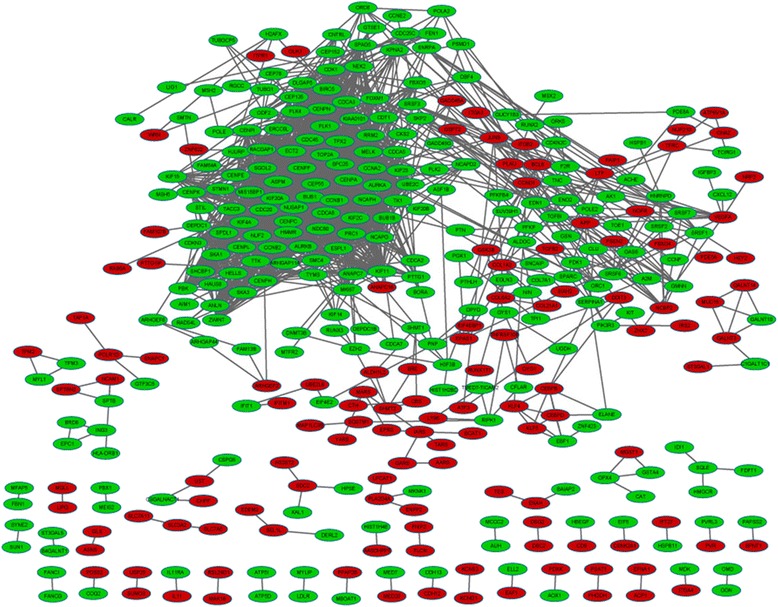
The protein-protein interaction network for the differentially expressed genes (DEGs). The gene productions are indicated by ellipse dots and the linkages among them are indicated by edges. Red stands for the productions of up-regulated DEGs and green for productions of down-regulated DEGs
Fig. 2.
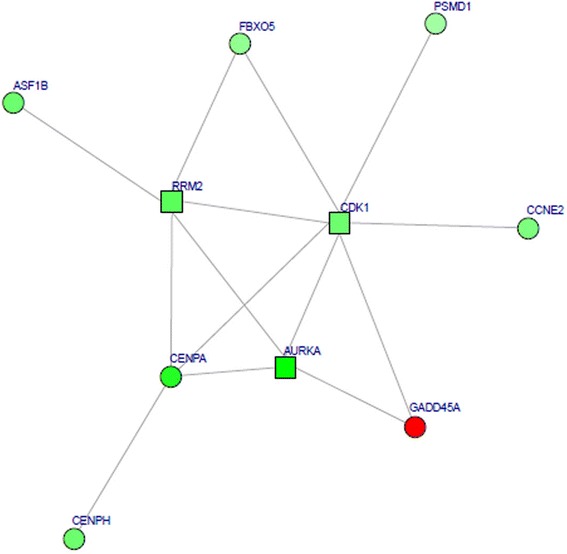
The highest score subnetwork for the differentially expressed genes (DEGs). Red stands for the productions of up-regulated DEGs and green for productions of down-regulated DEGs. Round dot indicates the highly associated DEGs with osteosarcoma and square indicates less relevant DEGs
Discussion
In this study, a total of 690 up-regulated DEGs and 626 down-regulated DEGs were identified in MTX-resistant osteosarcoma cells. According to the functional and pathway enrichment analysis, the up-regulated DEGs such as AARS, TARS, YARS, and PARS2 were mainly associated with tRNA aminoacylation, and the down-regulated DEGs such as AURKA, AURKB, CCNB1, CDK1, and CENPA were mainly correlated with mitotic cell cycle.
The up-regulated DEGs, including AARS, TARS, YARS, and PARS2, were biologically related to tRNA aminoacylation. AARS, TARS, and YARS encode aminoacyl-tRNA synthetases alanyl-tRNA synthetase, threonyl-tRNA synthetase, and tyrosyl-tRNA synthetase, respectively, which catalyze the aminoacylation of tRNA by their cognate amino acid, and thus are necessary for protein synthesis. Aminoacyl-tRNA synthetases usually take distinct roles in inflammation and transcriptional regulation [22]. The aberrant expression and cellular localization of aminoacyl-tRNA synthetases disturb normal cell regulatory networks and cause abnormalities through multiple routes [23]. For example, inhibition of osteosarcoma cell migration might be related to the extracellular functions of TARS [24]. MTX has been used to treat antisynthetase syndrome, a type of heterogeneous autoimmune disorder, in which autoantibodies target anti-aminoacyl-transfer RNA synthetase for specific amino acid. The up-regulation of aminoacyl-tRNA synthetases in MTX-resistant cells may indicate that this type of cells has developed an ability to improve the expression level of aminoacyl-tRNA synthetases in the presence of MTX that offset the effect of MTX, leading to MTX resistance.
The down-regulated genes were dominantly related to mitotic cell cycle or nuclear division, for example, AURKA, AURKB, CENPA, CDK1, CCNB1, and CCNB2. AURKA is known as an oncogene. AURKA is an important regulator to G2/M transition [25]. AURKA protein is mainly located at the microtubule organizing center at the metaphase I (M1) of oocytes [26]. Further investigations have shown that the localization of AURKA in the area of aligned chromosomes is consistent with the AURKA-dependent phosphorylation of kinetochore component centromere protein A (CENPA). CENPA phosphorylation requires the enrichment of AURKB to maintain the phosphorylation on Ser7 at inner centromeres and for kinetochore function [26–28]. Jiang et al. [29] showed that silencing of AURKA expression in osteosarcoma cells significantly decreased both colony formation ability in vitro and tumorigenesis ability in vivo as well as induced cell apoptosis and G2/M cell cycle arrest in osteosarcoma cells. In addition, the median survival time was significantly longer in patients with low-CENPA expression osteosarcomas than in those with high-CENPA expression osteosarcomas. AURKA and CENPA had, respectively, been identified as a susceptibility gene and an independent poor prognostic factor for osteosarcoma [25, 30]. The down-regulation of mitosis-related genes seems contrary to the commonly known fact that mitosis-related genes are usually up-regulated in tumors. However, up-regulation of mitosis-related genes is just recognized in tumors, not in drug-resistant tumor cells. Actually, we know little about the anti-drug mechanisms in tumors. It is possible that down-regulation of mitosis-related genes is a protection mechanism in MTX-resistant cells, or just at certain time point. Apparently, this finding has to be validated experimentally, since the sample number is so small.
Furthermore, CDK1, CCNB1, and CCNB2 which were enriched in mitotic cell cycle, oocyte meiosis, and p53 signaling pathways were inactivated by MTX treatment in osteosarcoma cell lines. As we know, deregulated cell proliferation and tumor-associated cell cycle always propel the complexity and idiopathy of cancer [31, 32]. Tumor-associated cell cycle is often mediated by the alterations of cyclin-dependent kinase (CDK) activities [33]. Cyclin B1 (CCNB1) to which p53 is directly bonded mediates G2/M progression and inhibits cell division. As reported, the inhibition of cyclin B and CDK1 led to the arrest of osteosarcoma cell division [34]. Inactivation of CDK1 and CDK2 triggered the apoptosis of osteosarcoma cells [35]. Moreover, MTX prevents tumor cells from proliferating by inhibiting dihydrofolate reductase (DHFR) [36]. The inhibition of CDK reduces the expression of both DHFR mRNA and protein thus enhancing sensitivity of human osteosarcoma cell lines to MTX [36, 37]. These studies concluded that the usage of combination of cyclin-CDK inhibitors and MTX which regulated mitotic cell cycle and p53 signaling pathway might overcome MTX resistance in osteosarcoma cells.
However, the DEGs identified in the present study were not the same with those identified to be associated with MTX resistance by Selga et al. using seven cell lines of different types of cancer [9]. This discrepancy may be attributed to the fact that genes identified by bioinformatics methods often vary with the criteria you adopt for analysis, and that Selga et al. focused on seeking genes commonly expressed in different MTX-resistant tumors, whereas we only paid attention to those specifically related to MTX resistance developed in osteosarcoma cell line. However, since there are only three samples for either MTX-sensitive or MTX-resistant cells, a very small sample, the universality and applicability of our findings is impaired, and further experimental proofs are needed to validate the findings.
Conclusions
In conclusion, this study identified several potential molecular targets that might contribute to the MTX resistance in osteosarcoma cells, such as GADD45A, AARS, AURKA, AURKB, CENPA, CCNB1, CCNE2, and CDK1, which may function via aminoacyl-tRNA biosynthesis pathway, cell cycle pathway, or p53 signaling pathway. However, the finding here should be taken prudently.
Acknowledgements
None.
Footnotes
Xiao-rong Yang and Yan Xiong are first authors.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XY and YX participated in the design of this study, and they both performed the statistical analysis. HD carried out the study and collected important background information. RG drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Xiao-rong Yang, Email: jafsdfdfs@163.com.
Yan Xiong, Email: gfsgrtsgtfdg@126.com.
Hong Duan, Phone: +86-28-8542-2570, Email: hongduanduan223@163.com.
Ren-rong Gong, Email: dfvcgrsgt@163.com.
References
- 1.Longo-Sorbello G. Current understanding of methotrexate pharmacology and efficacy in acute leukemias. Use of newer antifolates in clinical trials. Haematologica. 2001;86(2):121–7. [PubMed] [Google Scholar]
- 2.Kunz P, Fellenberg J, Moskovszky L, Sápi Z, Krenacs T, Machado I et al. Improved survival in osteosarcoma patients with atypical low vascularization. Annals Surg Oncol. 2015;22(2):489-96. [DOI] [PubMed]
- 3.Savage SA, Mirabello L, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, et al. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nat Genet. 2013;45(7):799–803. doi: 10.1038/ng.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rainusso N, Kurenbekova L, Donehower L, Rosen J, Yustein J. Abstract C80: characterization of metastatic cancer stem cells in osteosarcoma. Cancer Res. 2013;73(3 Supplement):C80–C. doi: 10.1158/1538-7445.TIM2013-C80. [DOI] [Google Scholar]
- 5.Zheng S-e, Xiong S, Lin F, G-l Q, Feng T, Shen Z, et al. Pirarubicin inhibits multidrug-resistant osteosarcoma cell proliferation through induction of G2/M phase cell cycle arrest. Acta Pharmacol Sin. 2012;33(6):832–8. doi: 10.1038/aps.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo W, Healey JH, Meyers PA, Ladanyi M, Huvos AG, Bertino JR, et al. Mechanisms of methotrexate resistance in osteosarcoma. Clin Cancer Res. 1999;5(3):621–7. [PubMed] [Google Scholar]
- 7.Elledge RM, Gray R, Mansour E, Yu Y, Clark GM, Ravdin P, et al. Accumulation of p53 protein as a possible predictor of response to adjuvant combination chemotherapy with cyclophosphamide, methotrexate, fluorouracil, and prednisone for breast cancer. J Natl Cancer Inst. 1995;87(16):1254–6. doi: 10.1093/jnci/87.16.1254. [DOI] [PubMed] [Google Scholar]
- 8.Yeager TR, Reznikoff CA. Methotrexate resistance in human uroepithelial cells with p53 alterations. J Urol. 1998;159(2):581–5. doi: 10.1016/S0022-5347(01)63988-0. [DOI] [PubMed] [Google Scholar]
- 9.Selga E, Oleaga C, Ramírez S, de Almagro MC, Noé V. Networking of differentially expressed genes in human cancer cells resistant to methotrexate. Genome Med. 2009;1(9):83. doi: 10.1186/gm83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Eustachio P. Reactome knowledgebase of human biological pathways and processes. Bioinformatics for Comparative Proteomics. Springer; 2011;694:49–61. [DOI] [PubMed]
- 11.Navab R, Strumpf D, Bandarchi B, Zhu C-Q, Pintilie M, Ramnarine VR, et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc Natl Acad Sci. 2011;108(17):7160–5. doi: 10.1073/pnas.1014506108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selga E, Noé V, Ciudad CJ. Transcriptional regulation of aldo-keto reductase 1C1 in HT29 human colon cancer cells resistant to methotrexate: role in the cell cycle and apoptosis. Biochem Pharmacol. 2008;75(2):414–26. doi: 10.1016/j.bcp.2007.08.034. [DOI] [PubMed] [Google Scholar]
- 13.Irizarry RA, Hobbs B, Collin F, Beazer‐Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 14.Alibes A, Yankilevich P, Canada A, Diaz-Uriarte R. IDconverter and IDClight: conversion and annotation of gene and protein IDs. BMC Bioinformatics. 2007;8:9. doi: 10.1186/1471-2105-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Consortium GO. The gene ontology (GO) project in 2006. Nucleic Acids Res. 2006;34(suppl 1):D322–6. doi: 10.1093/nar/gkj021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heinemeyer T, Chen X, Karas H, Kel AE, Kel O, Liebich I, et al. Expanding the TRANSFAC database towards an expert system of regulatory molecular mechanisms. Nucleic Acids Res. 1999;27(1):318–22. doi: 10.1093/nar/27.1.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao M, Sun J, Zhao Z. TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res. 2013;41(D1):D970–6. doi: 10.1093/nar/gks937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Von Mering C, Jensen LJ, Kuhn M, Chaffron S, Doerks T, Krüger B, et al. STRING 7—recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007;35(suppl 1):D358–62. doi: 10.1093/nar/gkl825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beisser D, Klau GW, Dandekar T, Müller T, Dittrich MT. BioNet: an R-Package for the functional analysis of biological networks. Bioinformatics. 2010;26(8):1129–30. doi: 10.1093/bioinformatics/btq089. [DOI] [PubMed] [Google Scholar]
- 22.Park SG, Ewalt KL, Kim S. Functional expansion of aminoacyl-tRNA synthetases and their interacting factors: new perspectives on housekeepers. Trends Biochem Sci. 2005;30(10):569–74. doi: 10.1016/j.tibs.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Kim D, Kwon NH, Kim S. Association of aminoacyl-tRNA synthetases with cancer. 2013. [DOI] [PubMed] [Google Scholar]
- 24.Williams TF, Mirando AC, Wilkinson B, Francklyn CS, Lounsbury KM. Secreted threonyl-tRNA synthetase stimulates endothelial cell migration and angiogenesis. Scientific Reports. 2013;3. [DOI] [PMC free article] [PubMed]
- 25.Cox DG, Hankinson SE, Hunter DJ. Polymorphisms of the AURKA (STK15/Aurora kinase) gene and breast cancer risk (United States) Cancer Causes Control. 2006;17(1):81–3. doi: 10.1007/s10552-005-0429-9. [DOI] [PubMed] [Google Scholar]
- 26.Saskova A, Solc P, Baran V, Kubelka M, Schultz RM, Motlik J. Aurora kinase A controls meiosis I progression in mouse oocytes. Cell Cycle. 2008;7(15):2368–76. doi: 10.4161/cc.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kunitoku N, Sasayama T, Marumoto T, Zhang D, Honda S, Kobayashi O, et al. CENP-A phosphorylation by Aurora-A in prophase is required for enrichment of Aurora-B at inner centromeres and for kinetochore function. Dev Cell. 2003;5(6):853–64. doi: 10.1016/S1534-5807(03)00364-2. [DOI] [PubMed] [Google Scholar]
- 28.Marumoto T, Zhang D, Saya H. Aurora-A—a guardian of poles. Nat Rev Cancer. 2005;5(1):42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Z, Jiang J, Yang H, Ge Z, Wang Q, Zhang L, et al. Silencing of Aurora kinase A by RNA interference inhibits tumor growth in human osteosarcoma cells by inducing apoptosis and G2/M cell cycle arrest. Oncol Rep. 2014;31(3):1249–54. doi: 10.3892/or.2014.2986. [DOI] [PubMed] [Google Scholar]
- 30.Gu X-M, Fu J, Feng X-J, Huang X, Wang S-M, Chen X-F, et al. Expression and prognostic relevance of centromere protein A in primary osteosarcoma. Pathol Res Pract. 2014;210(4):228–33. doi: 10.1016/j.prp.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 31.Williams GH, Stoeber K. The cell cycle and cancer. J Pathol. 2012;226(2):352–64. doi: 10.1002/path.3022. [DOI] [PubMed] [Google Scholar]
- 32.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–8. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 33.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–66. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 34.Han X-R, Sun Y, Bai X-Z. The anti-tumor role and mechanism of integrated and truncated PDCD5 proteins in osteosarcoma cells. Cell Signal. 2012;24(8):1713–21. doi: 10.1016/j.cellsig.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 35.Fu W, Ma L, Chu B, Wang X, Bui MM, Gemmer J, et al. The cyclin-dependent kinase inhibitor SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma cells. Mol Cancer Ther. 2011;10(6):1018–27. doi: 10.1158/1535-7163.MCT-11-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uchiyama H, Sowa Y, Wakada M, Yogosawa M, Nakanishi R, Horinaka M, et al. Cyclin-dependent kinase inhibitor SU9516 enhances sensitivity to methotrexate in human T-cell leukemia Jurkat cells. Cancer Sci. 2010;101(3):728–34. doi: 10.1111/j.1349-7006.2009.01449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee DH, Thoennissen NH, Goff C, Iwanski GB, Forscher C, Doan NB, et al. Synergistic effect of low-dose cucurbitacin B and low-dose methotrexate for treatment of human osteosarcoma. Cancer Lett. 2011;306(2):161–70. doi: 10.1016/j.canlet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]