

Abstract
The outer kinetochore protein scaffold KNL1 is essential for error-free chromosome segregation during mitosis and meiosis. A critical feature of KNL1 is an array of repeats containing MELT-like motifs. When phosphorylated, these motifs form docking sites for the BUB1–BUB3 dimer that regulates chromosome biorientation and the spindle assembly checkpoint. KNL1 homologs are strikingly different in both the amount and sequence of repeats they harbor. We used sensitive repeat discovery and evolutionary reconstruction to show that the KNL1 repeat arrays have undergone extensive, often species-specific array reorganization through iterative cycles of higher order multiplication in conjunction with rapid sequence diversification. The number of repeats per array ranges from none in flowering plants up to approximately 35–40 in drosophilids. Remarkably, closely related drosophilid species have independently expanded specific repeats, indicating near complete array replacement after only approximately 25–40 Myr of evolution. We further show that repeat sequences were altered by the parallel emergence/loss of various short linear motifs, including phosphosites, which supplement the MELT-like motif, signifying modular repeat evolution. These observations point to widespread recurrent episodes of concerted KNL1 repeat evolution in all eukaryotic supergroups. We discuss our findings in the light of the conserved function of KNL1 repeats in localizing the BUB1–BUB3 dimer and its role in chromosome segregation.
Keywords: KNL1, kinetochore, repeat evolution, BUB1, mitosis
Introduction
Mitotic chromosome segregation in eukaryotes involves the capture and stable attachment of the plus ends of spindle microtubules by all chromosomes in a manner that connects sister chromatids to opposing spindle poles. Large multiprotein assemblies on centromeric DNA, known as kinetochores, facilitate such chromosome–spindle interactions (Santaguida and Musacchio 2009). In addition to providing a link between DNA and the spindle, kinetochores are the signaling hubs for the spindle assembly checkpoint (SAC) and the target of attachment-error correction mechanisms (Santaguida and Musacchio 2009; London and Biggins 2014; Sacristan and Kops 2015). The interplay between microtubule attachment, error-correction, and SAC signaling is centered on the KMN network (KNL1-C, MIS12-C, and NDC80-C), an outer-kinetochore multiprotein complex that forms the microtubule-binding interface of kinetochores (Foley and Kapoor 2012; Sacristan and Kops 2015). The focal point of this interplay is KNL1/CASC5/AF15q14/Blinkin (hereafter referred to as KNL1), a largely disordered protein that recruits various mitotic regulators to the kinetochore and is able to directly interact with microtubules (Welburn et al. 2011; Caldas and DeLuca 2014) (fig. 1).
Fig. 1.—
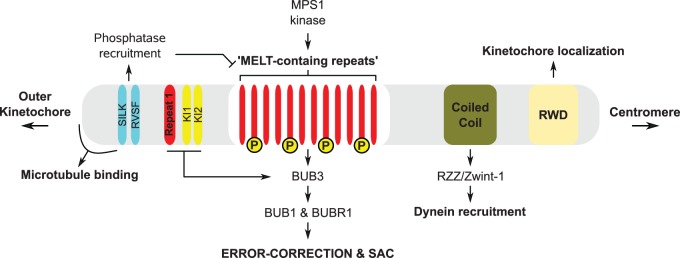
KNL1 is a hub for signaling at the kinetochore–microtubule interface. Schematic representation of the domain/motif architecture of human KNL1. Phospho motifs (MELTs) in the disordered middle region of KNL1 function as binding sites for various factors involved in SAC activation and error-correction (BUB3–BUB1/BUBR1). KI1 and KI2 increase the affinity of the BUB proteins for repeat 1. In addition this region harbors a basic patch involved in microtubule binding, as well as SILK/RVSF motifs for recruitment of PP1 phosphatase. PP1 can dephosphorylate the phospho-MELT motifs. The C-terminal region contains a tandem RWD (RING-WD40-DEAD) domain that localizes KNL1 to kinetochores and a coiled-coil that interacts with Zwint-1, a factor involved in recruiting the dynein adaptor RZZ–Spindly complex to kinetochores.
Critical for KNL1’s role in ensuring high fidelity chromosome segregation is the recruitment of the paralogs BUBR1 and BUB1 (BUBs) to the outer kinetochore. Both BUBR1 and BUB1 are bifunctional proteins, being involved in the SAC as well as in regulating stability of kinetochore–microtubule interactions (Bolanos-Garcia and Blundell 2011). Their roles in these processes are, however, distinct. BUBR1 is a pseudokinase (Suijkerbuijk et al. 2012) that is a component of a diffusible anaphase inhibitor (Tang et al. 2001; Sudakin et al. 2001; Chao et al. 2012; Han et al. 2013) and regulates stability of kinetochore–microtubule attachments by localizing the phosphatase PP2A-B56 to kinetochores (Suijkerbuijk et al. 2012; Kruse et al. 2013; Xu et al. 2013). BUB1 regulates error-correction by localizing Aurora B kinase to the inner centromere through the phosphorylation of T120 on the Histone 2A tail (Kawashima et al. 2010; Yamagishi et al. 2010) and likely by localizing BUBR1/PP2A to kinetochores (Johnson et al. 2004; Klebig et al. 2009; Overlack et al. 2015), yet its role in the SAC is less well identified (Bolanos-Garcia and Blundell 2011). These two BUBs directly interact through their respective TPR (tetratricopeptide repeat) domains with two different KI motifs in the N-terminus of KNL1 (Bolanos-Garcia et al. 2011; Kiyomitsu et al. 2011; Krenn et al. 2012). These motifs are, however, not conserved beyond vertebrates and are not essential for BUB kinetochore binding in human cells (Vleugel et al. 2013; Krenn et al. 2014). Rather, the main BUB-recruitment site on KNL1 is an array of multiple so-called MELT repeats (Met-Glu-Leu-Thr). When phosphorylated by the mitotic kinase MPS1, they form phospho-docking sites for BUB3/BUB1 dimers, hence directly ensuring localization of BUB1 and indirectly of BUBR1 to kinetochores (London et al. 2012; Shepperd et al. 2012; Primorac et al. 2013).
We and others recently reported that the MELT repeats of human KNL1 are part of larger repeated units that contain (besides a central MELT-like motif) at least two other motifs required for function: A TΩ motif (TxxΩ; where Ω denotes aromatic residues), and a second phospho motif (SHT) C-terminal to the MELT motif (Vleugel et al. 2013, 2015; Krenn et al. 2014). Human KNL1 has approximately 20 of these larger repeats. We showed that only six repeats are capable of recruiting detectable BUB proteins to the kinetochore, which raises the question of the significance of the other 14 repeats. In addition, although pivotal for proper error-correction and SAC function, preliminary analyses hinted at a high degree of variation in KNL1 repeat evolution (Vleugel et al. 2012, 2013). Although MELT-like motifs were at the core of repeat units of most KNL1 orthologs we analyzed, the remainder of the repeat sequences diverged greatly, and instances were observed where even a MELT-like motif was indiscernible.
We performed phylogenetic analyses to reconstruct KNL1 repeat evolution with the aim to understand its highly divergent patterns and the possible implications for BUB kinetochore recruitment and chromosome segregation in eukaryotes.
Materials and Methods
Sequences
Classical homology searches using BLAST (Basic Local Alignment Search Tool) failed to detect sufficient homology for KNL1 genes. We therefore performed iterated sensitive homology searches with HMMer (Eddy 2011; Finn et al. 2011), using a permissive E value and bitscore cut-off to include diverged homologs. Given that we detected a single homolog per genome we considered them orthologs. We included orthologs based on the presence of a N-terminal PP1-recruitment motifs (SILK/RVSF), MELT-like repeats, conserved regions in the C-terminus including a recently discovered RWD domain (Petrovic et al. 2014), and a C-terminal coiled-coil region. Incompletely predicted genes were searched against whole-genome shotgun contigs (wgs; http://www.ncbi.nlm.nih.gov/genbank/wgs) using tBLASTn. Significant hits were manually predicted using AUGUST (Stanke et al. 2006) and GENESCAN (Burge and Karlin 1997). For the sequences that we used in this study, see supplementary sequence file, Supplementary Material online.
Repeat Discovery Pipeline
The MEME (Bailey et al. 2009) algorithm (option: anr) was used to search for gapless amino acid repeat sequences, which were aligned using MAFFT (Katoh and Standley 2013) (option: einsi). Sensitive profile HMM searches (permissive E value of 10) of the aligned repeats were iterated until convergence (Eddy 2011). Due to the sensitivity of the profile HMM searches, the results were manually scrutinized for obvious errors.
Sequence Logos and Similarity Matrices
The repeat consensus sequence was depicted as a sequence logo using Weblogo2 (MEME color scheme). To prevent over interpretation of gaps and infrequent amino acids, columns in the repeat alignment with less than 20% occupancy were removed. The deviation from the consensus of individual repeats was calculated by normalizing pairwise alignment scores (Smith–Waterman) for the highest average score of all repeats and corrected for their respective length. We visualized repeat evolution history by projection of the normalized and corrected Smith–Waterman scores onto a similarity matrix (as described by Björklund et al. [2006]). Subsequent clustering enabled the classification of repeats with shared ancestry. Due to incomplete and dispersed clustering, further manual assignment of clusters and thus repeat phylogeny was necessary. The short length and limited amount of conserved sites between repeat units did not allow us to fit the KNL1 repeat data to a model of sequence evolution (e.g., GTR [general time reversible]) to reconstruct its evolution due to lack of power and likely over –or under fitting of model parameters (at least need ∼50 amino acids per repeat unit for good results).
Results
KNL1 Orthologs are found in all eukaryotic supergroups
Despite extensive sequence variation we could define KNL1 orthologs (see Materials and Methods) in all eukaryotic supergroups. These include orthologs in the rhizarium (Bigelowiella natans), the excavate (Naegleria gruberi), archeaplastids (Galdiera sulphurea, Physcomitrella patens and other land plants) and the cryptophyte (Guillardia. theta), species in which no KNL1 orthologs were previously detected (Vleugel et al. 2012, 2013). A KNL1 ancestor was therefore likely part of the genome of the Last Eukaryotic Common Ancestor (LECA). In all, a total of 110 KNL1 orthologs, displaying a great variety of sequence properties, were used in this study (supplementary sequence file, Supplementary Material online).
Repeat arrays in KNL1 orthologs display rapid consensus sequence evolution and extensive number changes
To capture the evolutionary behavior of the repeated units in a systematic fashion, we built a framework for short sensitive repeat discovery (see Materials and Methods). The pipeline initiates with a probabilistic search for gapless repeats and in an iterative process refines a statistical sequence consensus profile (hidden Markov model) of the smallest possible single repeat unit. To facilitate the comparison between different taxa, we calculated both inter and intra species repeat unit variation in addition to the number of repeats per array. Our analyses of repeat units in the set of KNL1 orthologs revealed a number of striking observations, summarized in figure 2 and elaborated on thereafter. A brief summary: First, the number of MELT motif-containing repeats differs extensively between eukaryotic species, ranging from 0 in most land plants, up to approximately 35 in flies (fig. 2). Interestingly, we observed recurrent instances of repeat array expansion and/or regression between various taxa of the same clade throughout the eukaryotic tree of life. These include: vertebrates (clawed frog = 31 and zebra fish = 16), chordates (lancelet = 16 and the tunicates = 6-10), insects (silk worm = 8 and mosquito = 33) and fungi (Spizellomyces punctatis = 1 and Yarrowia lipolytica = 21) (fig. 2). Second, our classification method uncovered a high degree of variation in the repeat consensus sequence both within and between species. For example, expansion of a single repeat is apparent in the ascomycete fungus Blumeria graminis, while in zebra fish repeats have decayed and only the MELT motif has been conserved (fig. 2 and supplementary fig. S1d, Supplementary Material online). Similarly, repeats are highly divergent between KNL1 orthologs, displaying alterations to the canonical MELT motif as well as the presence of additionally conserved motifs, for example, TΩ, SHT and other potential phosphosites ([EDN]x[ST] or Rx[ST]) (e.g., insects in fig. 2). In addition, we observed that motifs that are part of one repeat evolve separately in other species (e.g., MELT and TΩ), which suggests different functions for these motifs and hinting at the modular nature of KNL1 repeat evolution (see “2nd” in fig. 2).
Fig. 2.—

Repeat analyses of KNL1 reveal recurrent patterns during 2 Gyr of eukaryotic evolution. Cartoon of the eukaryotic tree of life with selected species from all eukaryotic supergroups containing KNL1 orthologs. The proteins and repeats are represented on scale in the middle. The color of the repeats indicates the degree of similarity to the repeat consensus (see legend). The repeat sequence consensus is depicted as a sequence logo on the right (colors reflect distinct amino acid properties and height of the letters indicates conservation of amino acids). The number of repeats per species is indicated in the light red (MELT-containing repeats) and blue (second repeats). The location of the MELT motif within the repeat is underlined for each species.
Recurrent Episodes of Extensive Repeat Array Reorganization and Repeat Diversification in Vertebrates and Drosophilids
The widespread diversity in repeat arrays did not permit the reconstruction of a bona fide LECA MELT-repeat array, but instead hinted at lineage-specific drivers and/or functions to explain this pattern of evolution. To determine the evolutionary relationship between the repeats, we resorted to a pairwise similarity matrix approach (Björklund et al. 2006), as the short and divergent nature of the repeats did not allow for the use of common model-guided phylogenetic methods (e.g., GTR using RaxML; see Materials and Methods). Subsequent clustering of the similarity matrices allowed for the visualization and (partial) reconstruction of evolutionary events that gave rise to arrays of both individual and closely related species. We focused on vertebrates and drosophilids because of the optimal sampling of closely related species and well-annotated genomes within these taxa, which allowed for tracing diverse patterns of repeat array reorganization up to single repeat resolution. We observe the following:
(1) Short multiplex (2–6) block duplications. Block duplication is the main mechanism through which arrays are reorganized. For human KNL1, we found a triplet block duplication of the repeats 12–14 and 16–18 (fig. 3A and supplementary fig. S1a, Supplementary Material online) (Vleugel et al. 2013). With the exception of the Chinese tree shrew (which had an additional duplication, supplementary fig. S2, Supplementary Material online), all placental mammals share the human array topology (see supplementary alignment S4, Supplementary Material online), which was therefore likely part of their common ancestor (∼65 Ma) (O’Leary et al. 2013). Comparison with orthologs of the nonplacental mammals opossum, Tasmanian devil (marsupials) and platypus (monotreme), revealed multiple block duplications of different size (2–6) in approximately the same region as the placental mammal duplication (fig. 3 and supplementary fig. S1b, see dynamic region in supplementary fig. S2, and alignment S1, Supplementary Material online).
(2) Homogenization. We observed additional instances of very recent single-copy repeat expansion that resulted in an almost complete overwriting of the array (hereafter referred to as homogenization). Most notably in lamprey (Perkinsus marinus; supplementary fig. S1c, Supplementary Material online) and the ascomycete B. graminis (fig. 2), the repeat arrays are highly similar within one species. The low number of substitutions in the DNA hints to a recent and rapid repeat regeneration event (supplementary fig. S3, Supplementary Material online).
(3) Array size maintenance and repeat loss. We noticed incomplete repeat units and discontinuous patterns of overlapping block duplication indicating that the repeats in the dynamic region of mammalian KNL1 were partially overwritten (see “+” signs in supplementary fig. S1a–c and the gaps in supplementary fig. S4, Supplementary Material online). In addition, we observed that repeats in the middle of the dynamic region in platypus were more similar to each other compared with repeats at the outside of the array, indicating unequal crossover as a potential mechanism for array maintenance (fig. 3B and supplementary fig. S1b, Supplementary Material online). Some of the repeat units in mammals exhibit divergence from the repeat consensus (“* signs” in supplementary fig. S1a–c, Supplementary Material online), acquiring multiple mutations in important residues, leading to decay and ultimately loss of these repeats. Strikingly, similarity between repeat 1,7 and 11 and those within the duplicated triplet block in human KNL1 correlates with their capacity to recruit BUB proteins, suggesting that diverged repeats loose their function (Vleugel et al. 2013, 2015). In zebra fish, no order in which duplications were generated could be inferred and decay has occurred at multiple repeats, as both the TΩ and the SHT motif are lost (fig. 1 and supplementary fig. S1d, Supplementary Material online).
Fig. 3.—

Patterns of repeat array reorganization in mammals and drosophilids. Individual repeats are scored based on similarity to the repeat consensus (similar to fig. 2). The example matrix at the top depicts the duplication of a twin repeat block (1,2–4,5). Similarity matrices (clustered [bottom-left] and unclustered [upper-right]) show patterns of repeat duplication; above the matrices scaled linear representations of the repeat array. Repeat numbers are colored according to their shared ancestry. (A) A single block duplication of repeat triplet 12–13–14 or 16–17–18 shaped human KNL1. (B) Overlapping twin block multiplications point to a complex history of platypus KNL1 evolution. (C ) Pseudohomogenization and near full array replacement in four Drosophila species. Colors below the matrix indicate which repeat in the matrix belongs to which species. Colored numbers correspond to position in amplicon of the respective species. Alignment of sequence logos indicates species-specific changes in consensus sequence. Anopheles quadriannulatus is a species of mosquito and is used to show Drosophila-specific increase in duplication rate.
All types of repeat evolution described also occurred within the drosophilid genus (25–40 Ma). (supplementary fig. S5 and alignment S3, Supplementary Material online). Four species (Drosophila pseudoobscura, Drosophila virilis, Drosophila kikkawai, and Drosophila willistoni) diverged their arrays to such extent, that we could only infer 2 one-to-one orthologous repeats (D. pseudoobscure 2–3 and D. kikkawai 11–12). Strikingly, each of these four species independently expanded specific repeats through subsequent rounds of extensive multiplication resulting in (partial) homogenization. This significantly altered the length of the array as well as the species-specific consensus sequence (see fig. 3C and supplementary alignment S2, Supplementary Material online).
Modular Evolution of Short Conserved Motifs in the Repeats
Recurrent episodes of array reorganization (expansion and contraction) may well be rooted in the selection for changes of the repeat consensus. To understand how the contents of the repeats such as those of the drosophilids have diverged, we tracked the behavior of the repeat consensus over approximately 550 Ma of arthropod evolution (Misof et al. 2014). To that end, the repeat consensus sequence logos of 50 arthropods were manually aligned and centered at the MELT-like motif and other recognizable motifs such as the N-terminal TΩ (fig. 4A). We found that the MELT-like motif is altered at position 0, −1, −2 (relative to the Thr), intermediately changing from ME[LF]T in most species to DMSLT in moths, butterflies and the beetle Dendroctonus ponderosae, MEET in mosquitos (Anophelini), and finally EP[MI]EEE in drosophilids. The phosphoconsensus of TΩ switched between predominantly basic residues [KR] and acidic residue [DE] (see Hymenoptera) at position −2 relative to Thr. This creates a potential phosphorylation site for Aurora B-like basophilic or PLK1/MPS1-like acidophilic kinases, respectively. KNL1 is a known substrate for such kinases in opisthokont model organisms (Vleugel et al. 2012). We also noticed a conserved proline at +4 (relative to Thr), which was also present in the repeats of the fungus Y. lipolytica and red algae G. sulphurea (fig. 2), indicating parallel gain and a potential shared functionality. The differential loss and emergence of conserved short motifs, (for example TΩ and other phosphosites) signifies the modular character of the KNL1 repeat evolution. To reconstruct the repeat consensus evolution of all eukaryotes, we abstracted the repeats into a presence/absence pattern of frequently conserved short motifs, divided over four regions within repeats (supplementary fig. S6, Supplementary Material online). We traced the origin of the TΩ motif to the base of the opisthokonts, with parallel loss in most fungi and early branching animals (Trichoplax, sea anemone, and sponges). Furthermore, we observe additional parallel events similar to those in arthropods (fig. 4B), such as TΩ phosphorylation consensus switching, MELT to MSLT/MEET and frequent changes of downstream conserved sites (glycines, proline, cysteine, and hydrophobic stretches) (see * signs for parallel events in supplementary fig. S6, Supplementary Material online).
Fig. 4.—

Repeat sequence consensus evolution of arthropods. (A) Alignment of repeat consensus sequences (weblogo) of arthropods based on the TΩ and MELT motif (red shaded). (B) Abstraction of conserved features indicates that repeats in arthropods consist of blocks that can be lost and gained. The repeat is subdivided into four “slots” (N-term, middle, MELT, and C-term) that contain all the observed motifs in arthropod evolution. Letters in blocks indicate the conservation of that amino acid or motif (P, proline; C, cysteine; GG, (double) glycine; “–,” aspartate or glutamate; Φ, bulky hydrophobic residues; Ω, aromatic residues; phenylalanine or tyrosine).
No Clear Indication for Positive Selection on Primate KNL1 Repeat Sequences
As the evolutionary reconstruction reveals episodes of repeat array rearrangement and diversification, we wondered whether repeats in closely related species would be under positive selection (higher nonsynonymous vs. synonymous substitution rate). We therefore fitted a concatenated alignment of the KNL1 repeats of 13 primates to various models of sequence evolution to estimate the dN/dS ratio using PAML (Ziheng Yang 2007) (see Materials and Methods and Results, supplementary fig. S7, Supplementary Material online). Although there seem to be different selective pressures impinging on the KNL1 repeat arrays in different species (supplementary fig. S7a and b, Supplementary Material online), we could not detect significant positive selection on different sites (supplementary fig. S7b, Supplementary Material online). Considering all sites, primate KNL1 repeats appear to be under weak purifying selection (dN/dS = 0.55).
Discussion
Our analyses and reconstructions reveal great diversity in the evolution of KNL1 repeat sequences. This diversity is the result of a myriad of mutations (repeat point mutation, loss, and duplication) further acted upon by selective forces. Together the interplay of these processes has driven a multitude of compound outcomes such as repeat homogenization and changes in repeat array length and consensus between closely related species (fig. 5). Similar patterns of rapid repeat evolution have been observed for proteins involved in adaptive evolution, for example in VERL, a protein involved in egg-sperm interaction in abalones (Panhuis et al. 2006), in PRDM9, a protein involved in homologous recombination during meiosis (Oliver et al. 2009), and in the arms race between zinc-finger proteins and retrotransposons (Jacobs et al. 2014). Repeats in some core cellular proteins such as structural BRC repeats in the DNA-damage-related protein BRCA2 (Bennett and Noor 2009; Lou et al. 2014) and a phosphomotif in the C-terminal domain of RNA polymerase (Chunlin Yang and Stiller 2014) have likewise undergone striking repeat evolution in specific clades. To our knowledge however, our study is the first to trace such extensive dynamic repeat evolution for a disordered signaling protein across all eukaryotic supergroups.
Fig. 5.—

Model of repeat evolution in KNL1. KNL1 repeat units (black bars) are depicted as having four “motif slots.” The color white indicates the ancestral state of the repeat; black the loss of the respective slot; and further coloring signifies subsequent mutations. Arrays are subjected to continuous repeat turnover (gain/loss) through iterative cycles of unequal crossover (II) in combination with repeat point mutation (I) leading to repeat diversification, potential decay (loss), and de novo motif emergence. Repeat arrays are stabilized by purifying selection to maintain a sufficient number of functional repeats (dark red). Intermittent episodes of extensive single copy expansion allow for rapid evolution of the consensus and/or array length, which is reminiscent of adaptive evolution (dark blue). Species names indicate which type of behavior is seen for that species.
Patterns and Mechanisms of Extensive Array Reorganization
Single-repeat or block-repeat multiplication is the result of duplications iterating in relatively quick succession. We find duplications undergoing no further dynamics, for example, approximately 65 Ma of evolution (placental mammals). In contrast, we also find cases where a block or single repeat underwent very recent iterating duplications (lamprey and drosophilids), indicating the episodic nature of KNL1 repeat evolution. Scars of overlapping block multiplications and a higher similarity of repeats in the middle of arrays (fig. 3B and supplementary figs. S1b and S4, Supplementary Material online) point to unequal crossover to maintain stable repeat arrays (fig. 5), similar to what was described for centromeric DNA repeat evolution (Melters et al. 2013). Interestingly, high numbers of repeated units increase local sequence homology and thereby the chance of replication slippage and unequal crossover (Ellegren 2004). It is however unclear why the arrays never appear to be longer than approximately 35 units. This may have to do with the potential negative impact on chromosome segregation by a large number of BUB1–BUB3 recruitment modules, or of problematic protein folding/aggregation in case of extended unstructured regions. In any case, the array size limitation is indicative of purifying selection against excessive multiplications.
Patterns of Repeat Unit Consensus Evolution
The KNL1 repeat consensus sequence evolved in a modular fashion. It consists of several short conserved motifs, which are recurrently gained (indicative of convergent motif evolution) and lost at both up- and downstream positions relative to the MELT motif. The KNL1 repeat thus serves as a unit that contains multiple motif slots. This unit is dynamic in the motif content of its slots as well as dynamic in duplication and losses. Although the motifs slots seem to evolve dynamically on large time scales, on shorter time scales species-specific alignments of repeats units reveal conservation of each motif consensus by purifying selection—allowing us in fact to detect them as such (see sequence logos). Simultaneously, episodes of extensive array reorganization could lead to the expansion of specific repeat isoforms (signified by homogenization events), indicating how species have rapidly evolved their repeat consensus sequence.
Drivers of Repeat Evolution: A Role for BUB1–BUB3?
The wide array of evolutionary processes impinging on the KNL1 repeat array raises the question what function of the repeats is driving these processes? We envision two distinct but nonmutually exclusive possibilities: 1) The altering number of repeats signifies different requirements for the number of BUB1–BUB3 molecules needed on a kinetochore or the length of the protein. As the number of functional repeats in human KNL1 dictates the efficiency of attachment error-correction (Vleugel et al. 2013), selective pressures may have called for rapid adaptability of the number of BUB molecules that can bind kinetochores. In such a scenario, the appearance of additional motifs could reflect differences in the BUB3 structure and/or regulatory pathways that impinge on BUB3 kinetochore recruitment. Recent work from our lab on human KNL1 showed that a vertebrate-specific SHT motif, C-terminal to the MELT motif, is an additional phosphomotif that interacts with a basic patch on the surface of BUB3 (Vleugel et al. 2015). This patch is present in numerous Bub3 homologs of nonvertebrates, indicating co-option of pre-existing BUB3 features for interaction with the SHT motif in the ancestor of vertebrates. It is therefore possible that the various motifs in diverse eukaryotes bind to various conserved core features of the Bub3 structure. Of interest is also the loop region within BUB1 that stabilizes the interaction, which diversifies rapidly throughout eukaryotic evolution. Finally, some of the motifs may have evolved to accommodate different cell division kinases/phosphatases, possibly explaining changes in phospho-motif sequences. Further detailed molecular and functional analyses of the repeat motifs and their mode of interaction with the BUB3–BUB1 dimer, kinases, and –or phosphatases will be required to understand the repeat evolution. 2) A minimal requirement for BUB3 binding is maintained through purifying selection on the core MELT-like motif and the changes in number and sequence of additionally conserved motifs (e.g., the additional phosphosites) signify other, yet unknown functions of KNL1 repeat divergence. The observed repeat (pseudo) homogenization events in B. graminis, lamprey, and several drosophilids are reminiscent of genetic conflicts, such as the compensatory evolution of centromere sequences and centromere-binding proteins to prevent genetic conflict during asymmetric meiosis, known as centromere drive (Henikoff et al. 2001). The centromere-drive hypothesis describes an arms race between centromere sequence variants with higher probabilities of being retained in the oocyte (rather than the evolutionary invisible polar bodies) and centromere-binding proteins that negate this bias (Malik and Henikoff 2009; Chmátal et al. 2014). Interestingly, in nematodes KNL1 is involved in biorientation of acentrosomal meiosis (Dumont and Desai 2012) and KNL1 protein expression is highest at the sperm acrosome in humans (Sasao et al. 2004). Nevertheless, there is currently no evidence that KNL1 binds centromere sequences directly, and rapid evolution of its repeats occurs also in species with symmetric meiosis. Other forms of genetic conflict that may explain KNL1 repeat evolution include defense against supernumerary/selfish (B-) chromosomes that utilize kinetochore proteins and the mitotic spindle to segregate (Werren 2011), or in the evasion of hijacking of the mitotic machinery by intracellular pathogens.
Supplementary Material
Supplementary sequence file, alignment PAML analysis, alignments S1–S4, references, and figures S1–S7 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank the members of the Kops and Snel lab for fruitful discussion and critical reading of the manuscript. The authors also thank members of the Malik, Henikoff and Biggins labs at FHCRC for discussions. This work is supported by a grant from the Netherlands Organization for Scientific Research (NWO-Vici 865.12.400).
Literature Cited
- Bailey TL, et al. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Research 37:202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett SM, Noor MAF. 2009. Molecular evolution of a Drosophila homolog of human BRCA2. Genetica 137:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björklund AK, Ekman D, Elofsson A. 2006. Expansion of protein domain repeats. PLoS Comput Biol. 2:e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolanos-Garcia VM, Blundell TL. 2011. BUB1 and BUBR1: multifaceted kinases of the cell cycle. Trends Biochem Sci. 36:141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolanos-Garcia VM, et al. 2011. Structure of a Blinkin-BUBR1 complex reveals an interaction crucial for kinetochore-mitotic checkpoint regulation via an unanticipated binding site. Structure 19:1691–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C, Karlin S. 1997. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 268:78–94. [DOI] [PubMed] [Google Scholar]
- Caldas GV, DeLuca JG. 2014. KNL1: bringing order to the kinetochore. Chromosoma 123:169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao WCH, Kulkarni K, Zhang Z, Kong EH, Barford D. 2012. Structure of the mitotic checkpoint complex. Nature 484:208–213. [DOI] [PubMed] [Google Scholar]
- Chmátal L, et al. 2014. Centromere strength provides the cell biological basis for meiotic drive and karyotype evolution in mice. Curr Biol. 24:2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont J, Desai A. 2012. Acentrosomal spindle assembly and chromosome segregation during oocyte meiosis. Trends Cell Biol. 22:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput Biol. 7 (10):e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H. 2004. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 5:435–445. [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39:W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley EA, Kapoor TM. 2012. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol. 14:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, et al. 2013. Catalytic assembly of the mitotic checkpoint inhibitor BubR1-Cdc20 by a Mad2-induced functional switch in Cdc20. Mol Cell. 51:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Malik HS. 2001. The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293:1098–1102. [DOI] [PubMed] [Google Scholar]
- Jacobs FMJ, et al. 2014. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 516:242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VL, Scott MIF, Holt SV, Hussein D, Taylor SS. 2004. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J Cell Sci. 117:1577–1589. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima SA, Yamagishi Y, Honda T, Ishiguro K-I, Watanabe Y. 2010. Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science 327:172–177. [DOI] [PubMed] [Google Scholar]
- Kiyomitsu T, Murakami H, Yanagida M. 2011. Protein interaction domain mapping of human kinetochore protein Blinkin reveals a consensus motif for binding of spindle assembly checkpoint proteins Bub1 and BubR1. Mol Cell Biol. 31:998–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebig C, Korinth D, Meraldi P. 2009. Bub1 regulates chromosome segregation in a kinetochore-independent manner. J Cell Biol. 185:841–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn V, Overlack K, Primorac I, van Gerwen S, Musacchio A. 2014. KI motifs of human Knl1 enhance assembly of comprehensive spindle checkpoint complexes around MELT repeats. Curr Biol. 24:29–39. [DOI] [PubMed] [Google Scholar]
- Krenn VV, Wehenkel AA, Li XX, Santaguida SS, Musacchio AA. 2012. Structural analysis reveals features of the spindle checkpoint kinase Bub1-kinetochore subunit Knl1 interaction. J Cell Biol. 196:451–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse T, et al. 2013. Direct binding between BubR1 and B56-PP2A phosphatase complexes regulate mitotic progression. J Cell Sci. 126:1086–1092. [DOI] [PubMed] [Google Scholar]
- London N, Biggins S. 2014. Signalling dynamics in the spindle checkpoint response. Nat Rev Mol Cell Biol. 15:736–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, Ceto S, Ranish JA, Biggins S. 2012. Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr Biol. 22:900–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou DI, et al. 2014. Rapid evolution of BRCA1 and BRCA2 in humans and other primates. BMC Evol Biol. 14:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Henikoff S. 2009. Major evolutionary transitions in centromere complexity. Cell 138:1067–1082. [DOI] [PubMed] [Google Scholar]
- Melters DP, et al. 2013. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 14:R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misof B, et al. 2014. Phylogenomics resolves the timing and pattern of insect evolution. Science 346:763–767. [DOI] [PubMed] [Google Scholar]
- O’Leary MA, et al. 2013. The placental mammal ancestor and the Post-K-Pg radiation of placentals. Science 339:662–667. [DOI] [PubMed] [Google Scholar]
- Oliver PL, et al. 2009. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 5:e1000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overlack K, et al. 2015. A molecular basis for the differential roles of Bub1 and BubR1 in the spindle assembly checkpoint. eLife 4:e05269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panhuis TM, Clark NL, Swanson WJ. 2006. Rapid evolution of reproductive proteins in abalone and Drosophila. Philos Trans R Soc Lond B Biol Sci. 361:261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic A, et al. 2014. Modular assembly of RWD domains on the Mis12 complex underlies outer kinetochore organization. Mol Cell. 53:591–605. [DOI] [PubMed] [Google Scholar]
- Primorac I, et al. 2013. Bub3 reads phosphorylated MELT repeats to promote spindle assembly checkpoint signaling. eLife 2:e01030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacristan C, Kops GJPL. 2015. Joined at the hip: kinetochores, microtubules, and spindle assembly checkpoint signaling. Trends Cell Biol. 25:21–28. [DOI] [PubMed] [Google Scholar]
- Santaguida S, Musacchio A. 2009. The life and miracles of kinetochores. Embo J. 28:2511–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasao T, et al. 2004. The protein encoded by cancer/testis gene D40/AF15q14 is localized in spermatocytes, acrosomes of spermatids and ejaculated spermatozoa. Reproduction 128:709–716. [DOI] [PubMed] [Google Scholar]
- Shepperd LA, et al. 2012. Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr Biol. 22:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Tzvetkova A, Morgenstern B. 2006. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 7:S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Chan GK, Yen TJ. 2001. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 154:925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suijkerbuijk SJE, et al. 2012. The vertebrate mitotic checkpoint protein BUBR1 is an unusual pseudokinase. Dev Cell. 22:1321–1329. [DOI] [PubMed] [Google Scholar]
- Tang Z, Bharadwaj R, Li B, Yu H. 2001. Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell. 1:227–237. [DOI] [PubMed] [Google Scholar]
- Vleugel M, et al. 2013. Arrayed BUB recruitment modules in the kinetochore scaffold KNL1 promote accurate chromosome segregation. J Cell Biol. 203:943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vleugel M, et al. 2015. Sequential multisite phospho-regulation of KNL1-BUB3 interfaces at mitotic kinetochores. Mol Cell. 57:824–835. [DOI] [PubMed] [Google Scholar]
- Vleugel M, Hoogendoorn E, Snel B, Kops GJPL. 2012. Evolution and function of the mitotic checkpoint. Dev Cell. 23:239–250. [DOI] [PubMed] [Google Scholar]
- Welburn JPI, et al. 2011. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Curr Biol. 21:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH. 2011. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc Natl Acad Sci U S A. 108:10863–10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Raetz EA, Kitagawa M, Virshup DM, Lee SH. 2013. BUBR1 recruits PP2A via the B56 family of targeting subunits to promote chromosome congression. Biol Open. 2:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi Y, Honda T, Tanno Y, Watanabe Y. 2010. Two histone marks establish the inner centromere and chromosome bi-orientation. Science 330:239–243. [DOI] [PubMed] [Google Scholar]
- Yang C, Stiller JW. 2014. Evolutionary diversity and taxon-specific modifications of the RNA polymerase II C-terminal domain. Proc Natl Acad Sci U S A. 111:5920–5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.