

Abstract
Expression of α1antichymotrypsin (ACT) is significantly activated by IL-1 in human astrocytes; however, it is barely affected by IL-1 in hepatocytes. This tissue-specific regulation depends upon an enhancer which contains both NF-κB and AP-1 elements, and is also observed for an NF-κB reporter but not for an AP-1 reporter. We found efficient activation of NF-κB binding in both cell types; however, this binding was persistent in glial cells and only transient in hepatocytes. IL-1-activated NF-κB complexes consisted of p65 and p50, with p65 transiently phosphorylated on serine 536 in glial cells whereas more persistently in hepatic cells. Overexpression of p65 or constitutively active IKKβ resulted in an efficient activation of the ACT reporter in hepatic cells indicating that a specific mechanism exists in these cells terminating IL-1 signaling. IL-1 effectively induced the degradation of IkBα and IkBε in both cell types but IkBβ was not affected. However, IkBα was resynthesized much more rapidly in hepatic cells in comparison to glial cells. In addition, the initial levels of IkBα were much lower in glial cells. We propose that the tissue-specific regulation of the ACT gene expression by IL-1 is determined by different efficiencies of IkBα resynthesis in glial and hepatic cells.
Keywords: astrocytes, α1-antichymotrypsin, IL-1, NF-kB, expression
INTRODUCTION
Activation of brain inflammatory processes and release of proinflammatory cytokines such as IL-1 and tumor necrosis factor (TNF) have been observed in traumatic brain injury, chemical toxicity, multiple sclerosis, AIDS dementia, viral infections, and Alzheimer's disease (AD) (Ghirnikar et al. 1998). In fact, increased expression of IL-1 has been reported in AD, and recently a polymorphism of the IL-1 gene that results in elevated cytokine expression has been shown to correlate with a high risk of developing AD (Grimaldi et al. 2000; Nicoll et al. 2000).
Binding of IL-1 to its cell surface receptors activates several signaling pathways and transcription factors including the NF-κB/Rel family members (p65, RelB, c-Rel, p50, and p52) (Ghosh et al. 1998), which can either homo- or hetero-dimerize. In non-stimulated cells NF-κB proteins reside in the cytoplasm due to the interaction with IkBs (inhibitors of NF-κB) including IkBα, IkBβ, and IkBε(Maniatis 1997). After cell stimulation, NF-κB/IkB complexes are activated by a canonical activation pathway involving the IKKβ subunit of IkB-kinase (IKK) complex, which phosphorylates IkBs targeting them for polyubiqitination, subsequent degradation, and thus liberates dimers containing p65 and c-Rel (Karin and Ben-Neriah 2000). In contrast, RelB/p50 and RelB/p52 complexes are released by a non-canonical activation pathway that requires NF-κB-inducing kinase and the IKKα subunit of IKK complex (Xiao et al. 2001), (Senftleben et al. 2001). The nature of NF-κB activation can be either transient or persistent with several mechanisms proposed to explain this phenomenon that include the exchange of NF-κB dimers (Saccani et al. 2003), differences in resynthesis and phosphorylation kinetics of IkBs (Thompson et al. 1995), (DiDonato et al. 1997), loss of IkBβ (Bourke et al. 2000), occurrence of hypophosphorylated IkBβ(Suyang et al. 1996), formation of the IkBβ:MEKK2 complex (Schmidt et al. 2003), expression protein(s) accelerating export of NF-κB from nucleus (Higashitsuji et al. 2002), SUMO-modification of IkBα(Hay et al. 1999) (Desterro et al. 1998), Pin-1 mediated prolyl isomerization of p65 (Ryo et al. 2003), and the acetylation of p65 (Chen and Greene 2003).
α1-antichymotrypsin (ACT) specifically co-localizes with β-amyloid deposits in the brains of Alzheimer's patients (Abraham et al. 1988), and also enhances the formation of β-amyloid deposits in a double transgenic model of AD (Nilsson et al. 2001). In the brain, astrocytes are the major source of ACT and its expression is regulated by IL-1, TNF, and oncostatin M (OSM) (Kordula et al. 1998), (Kordula et al. 2000). Liver is the major source of ACT found in the blood; however, ACT expression is only slightly activated by IL-1 in hepatic cells although the expression of several other genes including those encoding SAA, CRP, and PAI-1 is strongly activated by IL-1 (Edbrooke et al. 1991) (Cha-Molstad et al. 2000) (Arts et al. 1999).
Since enhanced expression of the ACT gene by astrocytes in vivo is most likely a result of IL-1 (or TNF) stimulation, we analyzed the molecular basis of this sustained cytokine-induced activation, and compared the mechanism that functions in astrocytes to that in hepatic cells.
MATERIALS AND METHODS
Cell culture
Human cortical astrocyte cultures were established using dissociated human cerebral tissue established exactly as described previously (Kordula et al. 1998). Cortical tissue was provide by Advanced Bioscience Resources, and the protocol for obtaining postmortem fetal neural tissue complied with federal guidelines for fetal research and with the Uniformed Anatomical Gift Act. Human astrocytoma U373-MG and human hepatoma HepG2 cells were obtained from American Type Culture Collection (Rockville, MD). Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, antibiotics, sodium pyruvate, and non-essential amino acids.
Cytokines and cell stimulation
Cells were stimulated with 25 ng/ml OSM (R&D, Systems, Inc., Minneapolis, MN), 10 ng/ml IL-1 (a gift from Immunex Corp., Seattle, WA), or 1 μM dexamethasone (DEX) (Sigma Chemical Co., St. Louis, MO).
RNA preparation and Northern blot analysis
Total RNA was prepared using the phenol extraction method (Rose-John et al. 1988). Briefly, five μg samples of RNA were subjected to formaldehyde gel electrophoresis using standard procedures (Sambrook 1989) and transferred to Hybond-XL membranes (Amersham, Piscataway, NJ) according to the manufacturer's instructions. The filters were prehybridized at 680C for 3 h in 0.5 M sodium phosphate buffer pH 7.2, 7% SDS and 1 mM EDTA, and hybridized in the same solution with cDNA fragments labeled by random priming (Feinberg and Vogelstein 1983). After the hybridization, nonspecifically bound radioactivity was removed by four washes in 40 mM phosphate buffer, 1% SDS and 1 mM EDTA at 680C for 20 min. Intensities of the bands were analyzed by QuantityOne software (BioRad, Hercules, CA)
Synthetic oligonucleotides
The following oligonucleotides were synthesized to amplify the PCI gene promoter; PCITOP (5’-TTTGGGATCCTCTCTCAGGAGTGCCCATG-3’) and PCIBOT (5’-CGGGGGATCCACTCACCTCTGCTGC-3’) (NC_000014, nucleotides 94117275-94117735). The AP-1 and NF–κB double stranded oligonucleotides used both to generate AP-1 and NF–κB reporter constructs, and also in EMSA were described previously (Kordula et al. 2000).
Plasmid construction
Plasmids pΔ5ACTCAT, pStACTCAT, and ptkCATΔEH containing the IL-1-enhancer of the ACT gene linked to its promoter, the ACT promoter (NC_000014, nucleotides 94148122-94148502), and the tk minimal promoter, respectively, were described previously (Kordula et al. 2000). Plasmid pEnhPCICAT contains the IL-1-enhancer of the ACT gene linked to the PCI gene promoter. It was generated as follows; the 466 bp long PCI promoter was amplified by PCR from genomic DNA using the PCITOP and PCIBOT primers. The PCR product was digested with BamHI and inserted into the BamHI/BglII sites of ptkCATΔEH yielding the pPCICAT plasmid. The pΔ5ACTCAT was digested with BamHI, and the DNA fragment containing the IL-1-enhancer of the ACT gene was purified and subsequently cloned into the BamHI site of pPCICAT yielding the pEnhPCICAT. Plasmids p2x(AP-1)CAT and p3x(NFκB)CAT were generated by cloning double stranded oligonucleotides (AP-1 and NF–κB, respectively) into BamHI site of ptkCATΔEH. All constructs were sequenced on both strands. The expression plasmids encoding the pNF–κB(p65) and constitutively active IKKβ were provided by Dr. A. Baldwin (University of North Carolina, Chapel Hill, NC) and Dr. F. Mercurio (Celgene Signal Research Division, San Francisco, CA), respectively.
Transient transfections
Cells were transfected in 12 well clusters using FuGENE6 transfection reagent (Roche, Indianapolis, IN), according to the supplier's instructions. Plasmids (200 ng of the reporter CAT plasmid and 100 ng of pCH110) and 0.6 μl of FuGENE6 diluted in 50 μl of serum free medium were used for each well containing cells growing in 500 μl of culture medium. One day after transfection cells were stimulated, cultured another 24 h, and harvested. Protein extracts were prepared by freeze thawing (Gorman 1985), and protein concentration was determined by the BCA method (Sigma Chemical Co., St. Louis, MO). Chloramphenicol acetyltransferase (CAT) andβ-galactosidase assays were performed as described (Delegeane et al. 1987), (Schrell et al. 1998). CAT activities are normalized to the internal control β-galactosidase activity and are means ± S.E.M. (3-7 determinations).
Nuclear extract preparation and electromobility shift assays (EMSA)
Nuclear and whole cell extracts were prepared as described (Baeuerle and Baltimore 1988). Double stranded DNA fragments were labeled by filling in 5’ protruding ends with Klenow enzyme using [α32P]dCTP (3000 Ci/mmol). EMSA was carried out according to published procedures (Fried and Crothers 1981), (Sawadogo et al. 1988). Five μg of nuclear extracts and approx. 10 fmol (10,000 cpm) of probe were used. Polyclonal anti-c-fos, anti-c-jun, anti-p65, anti-p50, anti-p52, anti-RelB, and anti-c-Rel antisera were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). All oligonucleotides used for EMSA were designed to contain four bases single-stranded 5’ overhangs at each end after annealing.
Western Blotting
Cells growing in 6 cm dishes were lysed in 500 μl of boiling 1% SDS, 10 mM Tris pH 7.4 and 1 mM sodium orthovanadate. Protein concentrations were determined using the BCA kit (Sigma, St. Louis), and 50 μg samples were subjected to SDS-PAGE and electroblotted onto nitrocellulose (Schleicher & Schuell, Keene, NH). C-fos, c-jun, IkBα, IkBβ, and IkBε were detected using following antibodies; SC-52, SC-44, SC-371, SC-946, SC-7156 (Santa Cruz Biotechnology, Santa Cruz, CA). Antigen-antibody complexes were visualized by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ).
RESULTS
IL-1 efficiently activates ACT expression in glial cells
We and others have previously shown that IL-1 and OSM upregulate the expression of the ACT gene in both primary human astrocytes and astrocytoma cells (Das and Potter 1995), (Kordula et al. 1998), (Kordula et al. 2000). This activation is strongly enhanced by glucocorticoids as previously reported for ACT and other acute phase genes expressed in liver and other tissues (Baumann and Gauldie 1994). However, ACT expression is not significantly upregulated in hepatic cells including both HepG2 and Hep3B cell lines which are two of the prominent models used to study hepatic cell functions (Fig. 1 and data not shown). The profound upregulation of ACT expression in glial cells by IL-1 (~30 fold in astrocytes) is very rapid and can be observed as early as one hour after cytokine treatment with maximal induction found after 8-18 hours. In contrast, IL-1 had little effect on the ACT mRNA accumulation in HepG2 cells with a maximal 2-fold increase also observed at 8-18 h (Fig. 1).
Fig. 1. Expression of ACT mRNA in human astrocytes, astrocytoma U373-MG and hepatoma HepG2 cells.
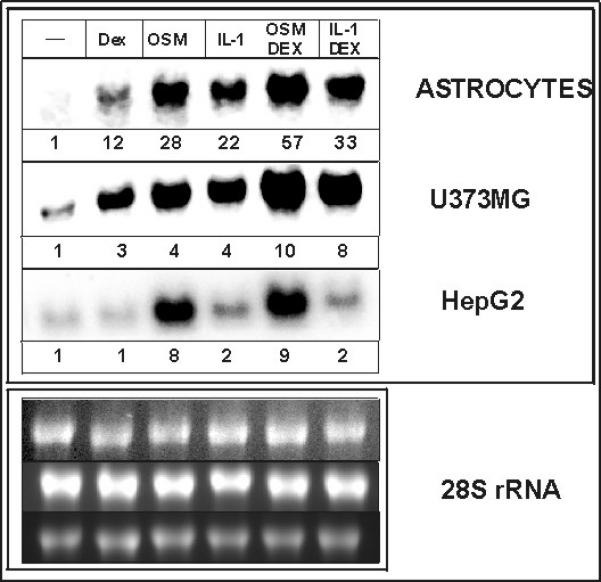
Human astrocytes, human astrocytoma U373-MG and human hepatoma HepG2 cells were treated with IL-1α (10 ng/ml), OSM (25 ng/ml), or 1 μM dexamethasone (DEX) as indicated. RNA was isolated after 18 hours (left panel) or at indicated times (right panel, IL-1 and DEX treatment), and subjected to Northern blot analysis using ACT cDNA as a probe. The bottom panels show 28S rRNA stained with ethidium bromide on the membrane. Numbers correspond to fold-induction.
The –13 kb enhancer of the ACT gene functions in astrocytes but not in hepatic cells
We have previously described the IL-1/TNF-responsive enhancer located 13 kb upstream of the ACT gene (Kordula et al. 2000). Since multiple regulatory mechanisms can account for the observed differences in regulation of the ACT gene by IL-1 in astrocytes and hepatocytes, we analyzed the responsiveness to IL-1 of several reporter constructs containing both the ACT gene promoter and its –13 kb enhancer. Considering that the enhancer is located only 6 kb downstream of the PCI gene, we generated a reporter construct containing the PCI promoter and the enhancer and included this construct in our analysis for comparison. We found that the –13 kb enhancer efficiently mediated the response to IL-1 and TNF in primary astrocytes; however, it was not functional in hepatocytes (Fig. 2). Moreover, the responsiveness mediated by the –13 kb enhancer was independent of the gene promoter used to drive the transcription of the reporter since the enhancer-PCI reporter construct was activated by IL-1 in astrocytes. However, the reporter containing the enhancer linked to the ACT promoter was more efficiently activated than the reporter containing enhancer linked to the PCI promoter. This result suggests that additional yet to be identified elements located within the ACT promoter may contribute to the IL-1 responsiveness. We conclude that the difference in the IL-1-responsiveness of the ACT gene in astrocytes and hepatocytes is mediated by the –13 kb enhancer, which responds to IL-1 in a tissue-specific manner.
Fig.2. The enhancer of the ACT gene is functional in astrocytes but not in hepatoma cells.
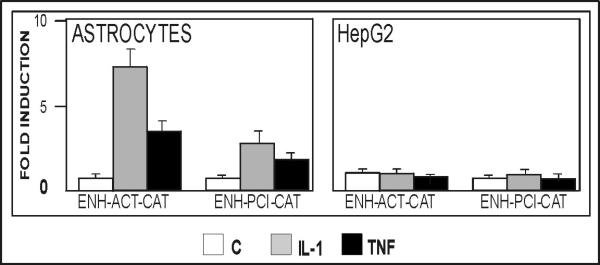
Human astrocytes or HepG2 cells were transfected with plasmids pΔACTCAT (ENH-ACTCAT) or pENHPCICAT and β-galactosidase expression vector as internal control for transfection efficiency. One day after transfection cells were stimulated with IL-1 (10 ng/ml) or TNF (10 ng/ml) in the presence of 1 μM dexamethasone, cultured for another 24 hours, and harvested. CAT activities were normalized to β-galactosidase activities (four separate analysis), and are shown as a fold induction with control cultures equal to 1.
IL-1 induces prolonged activation of NF–κB in glial cells
The –13 kb enhancer contains two NF–κB and one AP-1 binding elements, and both of these transcription factors can be activated by IL-1 in a variety of cell types. We have analyzed the time-dependent activation of NF–κB in astrocytes, astrocytoma and hepatoma cells by EMSA. NF–κB was efficiently activated in all three cell types 20 min after IL-1 treatment (Fig. 3A). This activation was specific to IL-1 since neither OSM nor Dex treatment activated NF–κB in either cell type. Nevertheless, the time course of NF–κB activation was drastically different between glial and hepatoma cells. In all cell types, NF–κB was activated 5 min after cytokine treatment with maximal activity observed between 15-30 min (Fig. 3B). However, glial cells contained substantial levels of activated NF–κB even 21 h after IL-1 stimulation. In contrast, very low levels of activated NF–κB were found in hepatic cells after 2-21 h (Fig. 3B), which were essentially the same as in the untreated cells. This correlated with transient occurrence of p65 in nuclei of HepG2 cells (Fig. 3D).
Fig. 3. Activation of NF–κB in human astrocytes, astrocytoma and hepatoma cells.
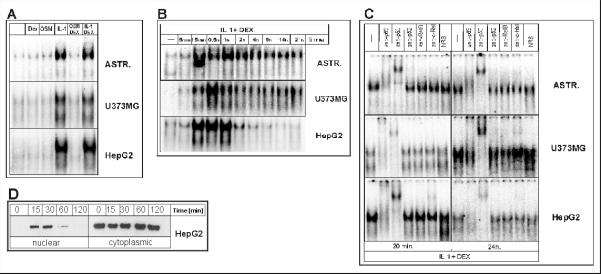
A. Human astrocytes, U373-MG and HepG2 cells were stimulated with IL-1, OSM, or Dex for 20 minutes. Nuclear extracts were prepared and binding was analyzed using the NF–κB oligonucleotide.
B. Cells were stimulated with IL-1 and DEX for the indicated time periods, nuclear extracts were prepared, and binding analyzed using the NF–κB oligonucleotide. C. Cells were stimulated with IL-1 and DEX for either 20 min or 24 hours, nuclear cell extracts were prepared, incubated with anti-p65, anti-p50, anti-p52, anti-RelB, anti-c-Rel or normal rabbit serum (NRS) and binding was analyzed using the NF–κB oligonucleotide. D. HepG2 cells were stimulated with IL-1 and DEX for the indicated time periods, nuclear and cellular extracts were prepared and analyzed by Western blotting using anti-p65 antibodies. Representative results of three separate experiments are shown.
Since NF–κB is a complex of several proteins, we analyzed the composition of activated NF–κB complex after short and long term IL-1 treatment in astrocytes, astrocytoma and hepatoma cells. We found that the composition of activated complex is identical in all cell types analyzed after both short and long time cytokine exposure (Fig. 3C). We detected p65 and p50 as the major components of the complex with very low amounts of c-Rel found after long cytokine treatment. We conclude that IL-1 treatment of glial and hepatic cells results in rapid activation of NF–κB containing the same components; however, in contrast to hepatic cells, glial cells are characterized by prolonged activation of this transcription factor.
IL-1 activates NF–κB reporter in astrocytes but not in hepatocytes
To test whether activation of NF–κB or AP-1 manifest as an increased responsiveness to IL-1 in glial cells, we generated reporter constructs containing several copies of either NF–κB or AP-1 elements (derived from the enhancer) linked to the minimal tk promoter. We tested these constructs in transient transfections of human primary astrocytes and hepatoma cells. The AP-1 reporter was not activated in either astrocytes or hepatocytes in response to IL-1 (Fig. 4). In contrast, the NF–κB reporter was activated in astrocytes but not in hepatocytes indicating that this element from the enhancer mimics the response profile of the endogenous ACT gene. We conclude that NF-κB is the key regulator controlling IL-1-induced transcription of the ACT gene in a tissue-specific manner.
Fig. 4. IL-1 activates the NF–κB reporter construct in human astrocytes but not in HepG2 cells.
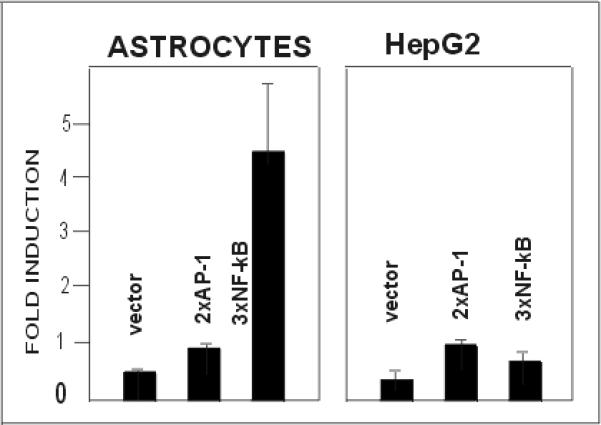
Human astrocytes or HepG2 cells were transfected with reporter plasmids ptkCATDEH (vector), p2xAP-1CAT (2xAP-1), or p3x(NF–κB) (3xNF–κB), and β-galactosidase expression vector as internal control for transfection efficiency. Cells were stimulated with IL-1 and DEX, cultured for another 24 hours, and harvested. CAT activities were normalized to β-galactosidase activities (three separate analysis), and are shown as a fold induction with control cultures equal to 1.
Overexpression of p65 or constitutively active mutant of IKKβ activates the ACT reporter in hepatocytes
Since the initial activation of NF–κB in hepatocytes was essentially identical to that found in glial cells we hypothesized that there is a cell-type specific mechanism that terminates NF–κB signaling in hepatocytes. To test this hypothesis we overexpressed p65 together with the ACT reporter to override this mechanism. In fact, we found that overexpression of NF–κB resulted in a significant activation of the reporter containing the –13 kb enhancer linked to the ACT promoter but not the control reporter containing only the ACT promoter (Fig. 5A). This result proves that a continuous supply of NF–κB can activate the ACT gene in hepatic cells.
Fig. 5. Overexpression of p65 or constitutively active IKKβactivates NF–κB reporter in human hepatoma cells.

HepG2 cells were cotransfected with reporter plasmids pΔ5ACTCAT (ENH-ACT-CAT) or pStACTCAT (ACT-CAT), and plasmids encoding NF–κB(p65) (A) or constitutively active IKKβ (B), and β-galactosidase expression vector as internal control for transfection efficiency. Cells were stimulated with IL-1 and OSM (in the presence of DEX) as indicated, cultured for another 24 hours, and harvested. CAT activities were normalized to β-galactosidase activities (3-5 separate experiments).
To test if hepatic cells contain sufficient amounts of endogenous NF–κB to activate the ACT gene, which cannot be continuously activated after cytokine treatment, we overexpressed constitutively active IKKβ together with our reporters. Once again the reporter containing the enhancer was efficiently activated but not the control construct containing only the ACT promoter (Fig. 5B). These results suggest that signaling to NF–κB is quickly terminated after an initial activation by IL-1 in hepatocytes. This inhibition results in a very low level of activated NF–κB found several hours after cytokine treatment.
Low levels of IkBαin glial cells
In untreated cells NF–κB components are complexed with members of the IkB family and reside in the cytoplasm. After cytokine treatment the IkB proteins are phosphorylated, ubiqitinated, and degraded by the proteasome, which results in the subsequent unmasking of an NLS within NF–κB, its nuclear translocation and activation of target genes including the gene encoding IkBα (Sun et al. 1993). To test if differing levels of IkB proteins or differing kinetics of their degradation and/or resynthesis can explain the drastically different responsiveness of glial and hepatic cells to IL-1, we analyzed the time-dependent effect of IL-1 treatment on the levels of IkBα, IkBβ, and IkBε. We found that IkBα and IkBε are rapidly degraded after cytokine treatment in both glial and hepatic cells, whereas levels of IkBβ were only slightly diminished in astrocytes (Fig. 6A). However, the kinetics of IkBα degradation and resynthesis was drastically different between glial and hepatic cells. In hepatocytes, the amount of IkBα was back to initial levels within 1 h, while it almost completely depleted from glial cells at this time. In addition, we compared the initial levels of IkBs in all three cell types, and found that the levels of IkBα and IkBε are very low in astrocytes with low levels of IkBα also found in astrocytoma cell (Fig. 6B). We conclude that responsiveness to IL-1 can be inversely proportionally correlated with the levels of IkBα. The drastic attenuation of IL-1-induced accumulation of activated NF–κB may result from rapid resynthesis of IkBα in hepatic cells.
Fig. 6. Analysis of IkB isoforms in astrocytes, astrocytoma and hepatoma cells.

(A). Human astrocytes, U373-MG or HepG2 cells were stimulated with IL-1 and DEX for the indicated time periods and cell lysates were prepared. IkBα, IkBβ, and IkBε were detected by Western blotting. (B). Protein concentrations were determined in the lysates and equal amounts of total cellular protein were analyzed as in A.
Representative results of two experiments are shown.
Low activation of IL-1-induced IkBα synthesis in glial cells
To analyze if relatively slow resynthesis of IkBα in glial cells (in comparison to hepatic cells) results from low activity of the IkBα gene in these cells, we measured levels of IkBα mRNA after IL-1 treatment. The kinetics and magnitude of IkBα mRNA accumulation were drastically different between glial and hepatic cells (Fig. 7). In glial cells, the levels of IkBα mRNA were significantly upregulated 1-2 hours after cytokine treatment with maximal levels found at 1 hour (7 and 3 fold increase for astrocytes and astrocytoma, respectively) and increased levels were still found hours later. In hepatocytes, the activation was very dramatic (25 fold increase); however transient, with amounts of IkBα mRNA only slightly increased at 2 hours after cytokine treatment. This transient and dramatic activation correlates with the kinetics of IL-1 signaling termination in these cells.
Fig. 7. Expression of IkBα mRNA in human astrocytes, astrocytoma and hepatoma cells.
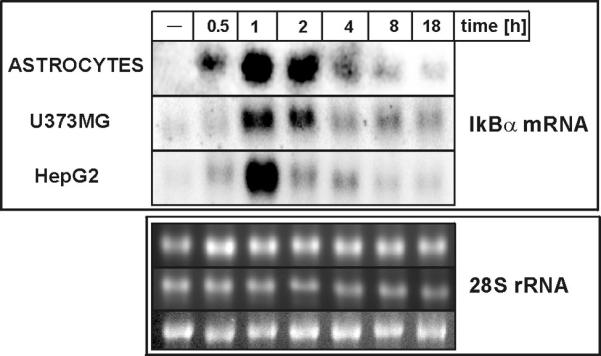
Human astrocytes, astrocytoma U373-MG and hepatoma HepG2 cells were treated with IL-1α (10 ng/ml) and 1 μM DEX for indicated time periods. RNA was isolated after 18 h, and subjected to Northern blot analysis using IkBα cDNA as a probe. Bottom panels show 28S rRNA stained with ethidium bromide on the membrane.
Transient phosphorylation of p65 on serine 536 in glial cells
Recently, phosphorylation of serine 536 of p65 by IKK complex has been shown to regulate recruitment of p300/CBP to the nuclear pool of p65 (Sakurai et al. 2003) (Zhong et al. 2002). Subsequently, these coactivators acetylate p65 on several residues, which leads to the increased transactivation potential of p65 and modifies its association rate with IkBα (Chen and Greene 2003). We compared the kinetics of p65 phosphorylation on serine 536 in both glial and hepatic cells. The phosphorylation of p65 on serine 536 was very rapid and transient in astrocytes with very low levels of phosphorylated p65 found after 2 hours of cytokine treatment (Fig. 8). Very rapid phosphorylation was also evident in hepatic cells; however, maximal levels of phosphorylated p65 were found 1 hour after IL-1 treatment, and these levels were still elevated after 2 hours (Fig. 8). It should be noted that in hepatic cells p65 is not present in the nucleus already 2 hours after IL-1 stimulation (Fig. 3B).
Fig. 8. Transient phosphorylation of p65 on serine 536.
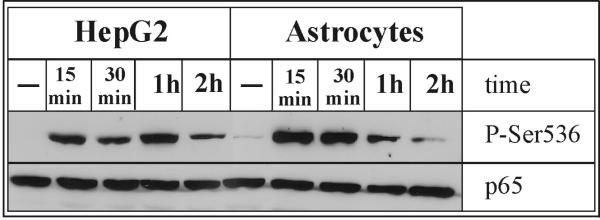
Human astrocytes and hepatoma HepG2 cells were treated with IL-1α (10 ng/ml) and 1 μM DEX for indicated time periods, and cell lysates were prepared. Protein concentrations were determined in the lysates and equal amounts of total cellular protein were analyzed by Western blotting using anti-Phospho-NF-κB p65 (Ser536) or anti-p65 antibodies. Representative results of two experiments are shown.
DISCUSSION
The ACT found in the brain is identical to that produced in the liver and found in the blood; however, in the brain it is produced by astrocytes in response to inflammatory cytokines (Hwang et al. 1999). The mechanisms that regulate the expression of ACT in the brain are of primary interest since its increased levels amplify formation of β-amyloid plaques in a double transgenic mouse model of Alzheimer's disease (Nilsson et al. 2001). Here, we investigated the molecular mechanism that allows astrocytes to continuously synthesize ACT in response to IL-1. This mechanism is non functional in hepatic cells since IL-1 has little, if any, effect on ACT expression in these cells (Fig. 1). Our results clearly demonstrate that the same pattern of tissue-specific regulation is found for the –13 kb enhancer of the ACT gene (Fig. 2) suggesting that this enhancer is the sole mediator of the gene's tissue-specific responsiveness. Furthermore, the specificity of the enhancer correlates with the specificity of the reporter containing κB elements derived from the enhancer (Fig. 4). These data suggest that astrocyte-specific activation of ACT gene expression by IL-1 is solely determined by persistently activated NF–κB in glial cells while its activation is transient in hepatic cells (Fig. 3B).
Our data raises the question as to whether the AP-1 element located within the enhancer, and the very high levels of AP-1 found in glial cells (Kasza et al. 2002), are important for IL-1 mediated activation of the ACT gene. We found neither an activation of the AP-1 reporter in response to IL-1 (Fig. 4) nor could we see any substantial activation of AP-1 binding in response to IL-1 (Kasza et al. 2002). These results suggest that AP-1 itself does not likely mediate the activation of the ACT gene in response to IL-1 (at least activation of the episomal reporter). However, the binding of AP-1 to its element within the –13 kb enhancer may serve as a recruiting signal for remodeling complexes and/or coactivators. Our data show that in contrast to glial cells, rapid termination of IL-1 signaling to the ACT gene occurs in hepatocytes. However, this inhibitory mechanism can be overcome by overexpression of p65 or a constitutively active mutant of IKKβ (Fig. 5). These data suggest that sufficient amounts of NF–κB are present in hepatic cells, which can be effectively activated if active kinase is present. The IkB proteins are the obvious candidates that may be responsible for the observed termination of signaling. Our analysis of IkBs expression levels in hepatic cells and the kinetics of their resynthesis clearly points to IkBα as the protein that likely terminates IL-1 signaling. The dramatic upregulation of IkBα mRNA expression one hour after cytokine treatment of hepatic cells results in rapid reappearance of the IkBα protein (Fig. 6 and 7). This pool of newly synthesized protein is no longer degraded, and likely is responsible for the export of NF–κB from nucleus back to the cytoplasm. It was proposed by others that this pool of IkBα can be modified by SUMO and, therefore, not accessible for degradation (Desterro et al. 1998). However, we could not detect SUMO-modified IkBα prior to or after IL-1 stimulation of hepatic or glial cells (data not shown). It is possible that after initial activation by IL-1, IKKα is no longer available in the cytoplasm since it translocates to the nucleus (Yamamoto et al. 2003), (Anest et al. 2003).
The question why IkBα is so efficiently induced in hepatic cells in comparison to glial cells remains? Our in silico analysis of IkBα promoter (http://www.genomatix.de/cgibin/matinspector_prof) produced several putative binding sites for hepatocyte nuclear factor–1 (HNF-1), HNF-4 and CCAAT enhancer binding protein (C/EBP) all of which are highly expressed in hepatic cells but only some isoforms of C/EBP are expressed in glial cells (Cardinaux et al. 2000). It is likely that these factors constitutively occupy the promoter of the IkBα gene and once NF–κB appears in the nucleus and binds to its three binding elements within the IkBα promoter (Ito et al. 1994), it can very efficiently activate transcription. However, other mechanisms including regulation by CBF-1 and Notch may also apply (Higashitsuji et al. 2002).
The persistent activation of NF–κB in glial cells correlates with the transcriptional activity of the ACT gene in these cells. Over the last few years multiple mechanisms have been proposed to explain the persistent activation of NF–κB, and we have analyzed which of these proposed mechanisms could apply to glial cells. Clearly, the exchange of NF–κB dimers (Saccani et al. 2003) does not occur in astrocytes since p65 and p50 are the major components of the NF–κB complex after both short- and long-term exposure to IL-1 (Fig. 3C). Changes in the kinetics of degradation or resynthesis of IkBβ or presence of hypophosphorylated IkBβ also do not apply since we did not observe degradation of IkBβ in astrocytes or hepatocytes (Fig. 6). These results suggest that it is likely that newly synthesized IkBα is modified, and this prevents its association with NF–κB, or in turn NF–κB is modified and cannot interact with IkBα. To date the sumoylation of IkBα has been shown; however, we could not detect any SUMO-modified IkBα in glial or hepatic cells. Recently, NF–κB has been shown to undergo cytokineinducible acetylation on at lest five different lysine residues (Chen et al. 2002), (Kiernan et al. 2003). Although published papers propose two contradictory functional models (acetylation inhibited or enhanced interaction with IkBα), acetylation of the NF–κB occurs after cytokine treatment and results in prolonged nuclear localization of NF–κB. Acetylation of p65 is preceded by phosphorylation of p65 on serine 536 which generates a signal for recruitment of coactivators (Zhong et al. 2002). In fact, we observed rapid phosphorylation of p65 in both cell types; however, it was transient in astrocytes and more persistent in hepatic cells (Fig. 8). Nuclear phosphatases were implicated in the dephosphorylation process of p65 (Sakurai et al. 2003). Since p65 is rapidly exported from the nuclei of hepatic cells (Fig. 3B) its prolonged phosphorylation in these cells (Fig. 8) supports the finding that nuclear phosphatases are the key players responsible for the dephosphorylation of p65. This rapid export of p65 in hepatic cells will also likely prevent any efficient acetylation of p65 by coactivators.
We propose the following model that explains differences in responsiveness of the ACT gene to IL-1 in glial versus hepatic cells. In hepatic cells IL-1 induces a rapid degradation of IkBα that results in the release of NF–κB, its translocation to the nucleus and activation of target genes including IkBα gene. The resynthesis of IkBα is rapid due to the elements binding hepatocyte-specific factors within the promoter of IkBα gene. Since newly synthesized IkBα quickly accumulates in the nucleus there is little time for NF–κB to become acetylated by nuclear acetyltransferases. Therefore, NF–κB binds to IkBα, it is efficiently exported back to the cytoplasm, and the signal is terminated. Recently, hepatoma subtracted-cDNA library clone one (HSCO) has been shown to enhance the nuclear export of NF–κB. This protein may be an important component of the export mechanism in hepatocytes.
In contrast to hepatic cells, glial cells express much lower levels of IkBα, and its resynthesis is much slower. NF–κB released from IkBα complexes is present in the nucleus for longer times before a new wave of IkBα reaches the nucleus, this allows NF–κB to be efficiently acetylated, thus preventing its association with IkBα and results in persistent activation. The difference in the initial levels of IkBα between glial and hepatic cells is likely determined by a CBF-1 repressor that recently was shown to bind to the IkBα gene promoter (Oakley et al. 2003).
The final question remains how IL-1 in hepatic cells regulates other IL-1-responsive genes including those coding for CRP, PAI, and SAA? The mechanisms governing the IL-1 induced activation of the PAI-1 gene are not known but may involve transcription factors other than NF–κB. Expression of the CRP gene is controlled by C/EBP and p50 that both interact at the CRP promoter (Cha-Molstad et al. 2000). Two NF–κB binding elements were identified in the promoter of the SAA gene; however, identity of the dimers or interacting proteins is not known (Edbrooke et al. 1991).
We propose that the low levels of IkBα in glial cells coupled with its slow resynthesis after IL-1 stimulation allow NF–κB to reside within the nucleus for a longer duration and therefore undergo efficient modification (likely acetylation) preventing its interaction with newly synthesized IkBα. This leads to the persistent activation of NF–κB and, in turn, efficient activation of glial NF–κB-responsive genes. Acetylation of NF–κB or mechanisms allowing faster expression of IkBα therefore may be the future targets in order to lower ACT expression in the brain.
Acknowledgments
We would also like to thank Dr. Joseph Fontes for helpful discussions and critical reading of the manuscript. This work was supported by an NINDS grant NS044118-01 (to T.K.)
Abbreviations
- ACT
α1-antichymotrypsin
- AD
Alzheimer's disease
- AP-1
activating protein 1
- CAT
chloramphenicol acetyl transferase
- CRP
C reactive protein
- DEX
dexamethasone
- EMSA
electromobility shift assay
- IkB
inhibitor of NF–κB
- IKK
IkB kinase
- IL
interleukin
- NF–κB
nuclear factor κB
- OSM
oncostatin M
- PCI
protein C inhibitor
- SAA
serum amyloid A
- serpin
serine proteinase inhibitor
- TNF
tumor necrosis factor
REFERENCES
- Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine protease inhibitor alpha 1-antichymotrypsin in the brain amyloid deposits of Alzheimer's disease. Cell. 1988;52:487–501. doi: 10.1016/0092-8674(88)90462-x. [DOI] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- Arts J, Grimbergen J, Toet K, Kooistra T. On the role of c-Jun in the induction of PAI-1 gene expression by phorbol ester, serum, and IL-1alpha in HepG2 cells. Arterioscler Thromb Vasc Biol. 1999;19:39–46. doi: 10.1161/01.atv.19.1.39. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- Baumann H, Gauldie J. The acute phase response [see comments]. Immunol Today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- Bourke E, Kennedy EJ, Moynagh PN. Loss of Ikappa B-beta is associated with prolonged NF-kappa B activity in human glial cells. J Biol Chem. 2000;275:39996–40002. doi: 10.1074/jbc.M007693200. [DOI] [PubMed] [Google Scholar]
- Cardinaux JR, Allaman I, Magistretti PJ. Pro-inflammatory cytokines induce the transcription factors C/EBPbeta and C/EBPdelta in astrocytes. Glia. 2000;29:91–97. [PubMed] [Google Scholar]
- Cha-Molstad H, Agrawal A, Zhang D, Samols D, Kushner I. The Rel family member P50 mediates cytokine-induced C-reactive protein expression by a novel mechanism. J Immunol. 2000;165:4592–4597. doi: 10.4049/jimmunol.165.8.4592. [DOI] [PubMed] [Google Scholar]
- Chen LF, Greene WC. Regulation of distinct biological activities of the NF-kappaB transcription factor complex by acetylation. J Mol Med. 2003;81:549–557. doi: 10.1007/s00109-003-0469-0. [DOI] [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. Embo J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Potter H. Expression of the Alzheimer amyloid-promoting factor antichymotrypsin is induced in human astrocytes by IL-1. Neuron. 1995;14:447–456. doi: 10.1016/0896-6273(95)90300-3. [DOI] [PubMed] [Google Scholar]
- Delegeane AM, Ferland LH, Mellon PL. Tissue-specific enhancer of the human glycoprotein hormone alpha-subunit gene: dependence on cyclic AMP-inducible elements. Mol Cell Biol. 1987;7:3994–4002. doi: 10.1128/mcb.7.11.3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell. 1998;2:233–239. doi: 10.1016/s1097-2765(00)80133-1. [DOI] [PubMed] [Google Scholar]
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Edbrooke MR, Foldi J, Cheshire JK, Li F, Faulkes DJ, Woo P. Constitutive and NF-kappa B-like proteins in the regulation of the serum amyloid A gene by interleukin 1. Cytokine. 1991;3:380–388. doi: 10.1016/1043-4666(91)90041-b. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- Fried M, Crothers DM. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 1981;9:6505–6525. doi: 10.1093/nar/9.23.6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirnikar RS, Lee YL, Eng LF. Inflammation in traumatic brain injury: role of cytokines and chemokines. Neurochem Res. 1998;23:329–340. doi: 10.1023/a:1022453332560. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Gorman C. In: DNA Cloning: A Practical Approach. Glover D, editor. IRL Press; 1985. pp. 143–190. [Google Scholar]
- Grimaldi LM, Casadei VM, Ferri C, Veglia F, Licastro F, Annoni G, Biunno I, Bellis G, Sorbi S, Mariani C, Canal N, Griffin WS, Franceschi M. Association of early-onset Alzheimer's disease with an interleukin-1alpha gene polymorphism [see comments]. Ann Neurol. 2000;47:361–365. [PubMed] [Google Scholar]
- Hay RT, Vuillard L, Desterro JM, Rodriguez MS. Control of NF-kappa B transcriptional activation by signal induced proteolysis of I kappa B alpha. Philos Trans R Soc Lond B Biol Sci. 1999;354:1601–1609. doi: 10.1098/rstb.1999.0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashitsuji H, Nagao T, Nonoguchi K, Fujii S, Itoh K, Fujita J. A novel protein overexpressed in hepatoma accelerates export of NF-kappa B from the nucleus and inhibits p53-dependent apoptosis. Cancer Cell. 2002;2:335–346. doi: 10.1016/s1535-6108(02)00152-6. [DOI] [PubMed] [Google Scholar]
- Hwang SR, Steineckert B, Kohn A, Palkovits M, Hook VY. Molecular studies define the primary structure of alpha1-antichymotrypsin (ACT) protease inhibitor in Alzheimer's disease brains. Comparison of act in hippocampus and liver. J Biol Chem. 1999;274:1821–1827. doi: 10.1074/jbc.274.3.1821. [DOI] [PubMed] [Google Scholar]
- Ito CY, Kazantsev AG, Baldwin AS., Jr Three NF-kappa B sites in the I kappa B-alpha promoter are required for induction of gene expression by TNF alpha. Nucleic Acids Res. 1994;22:3787–3792. doi: 10.1093/nar/22.18.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Kasza A, Kiss DL, Gopalan S, Xu W, Rydel RE, Koj A, Kordula T. Mechanism of plasminogen activator inhibitor-1 regulation by oncostatin M and interleukin-1 in human astrocytes. J Neurochem. 2002;83:696–703. doi: 10.1046/j.1471-4159.2002.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan R, Bres V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S, Benkirane M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003;278:2758–2766. doi: 10.1074/jbc.M209572200. [DOI] [PubMed] [Google Scholar]
- Kordula T, Bugno M, Rydel RE, Travis J. Mechanism of interleukin-1-and tumor necrosis factor alpha-dependent regulation of the alpha 1-antichymotrypsin gene in human astrocytes. J Neurosci. 2000;20:7510–7516. doi: 10.1523/JNEUROSCI.20-20-07510.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordula T, Rydel RE, Brigham EF, Horn F, Heinrich PC, Travis J. Oncostatin M and the interleukin-6 and soluble interleukin-6 receptor complex regulate alpha1-antichymotrypsin expression in human cortical astrocytes. J Biol Chem. 1998;273:4112–4118. doi: 10.1074/jbc.273.7.4112. [DOI] [PubMed] [Google Scholar]
- Maniatis T. Catalysis by a multiprotein IkappaB kinase complex. Science. 1997;278:818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Mrak RE, Graham DI, Stewart J, Wilcock G, MacGowan S, Esiri MM, Murray LS, Dewar D, Love S, Moss T, Griffin WS. Association of interleukin-1 gene polymorphisms with Alzheimer's disease [see comments]. Ann Neurol. 2000;47:365–368. [PMC free article] [PubMed] [Google Scholar]
- Nilsson LN, Bales KR, DiCarlo G, Gordon MN, Morgan D, Paul SM, Potter H. Alpha-1-antichymotrypsin promotes beta-sheet amyloid plaque deposition in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:1444–1451. doi: 10.1523/JNEUROSCI.21-05-01444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley F, Mann J, Ruddell RG, Pickford J, Weinmaster G, Mann DA. Basal expression of IkappaBalpha is controlled by the mammalian transcriptional repressor RBP-J (CBF1) and its activator Notch1. J Biol Chem. 2003;278:24359–24370. doi: 10.1074/jbc.M211051200. [DOI] [PubMed] [Google Scholar]
- Rose-John S, Dietrich A, Marks F. Molecular cloning of mouse protein kinase C (PKC) cDNA from Swiss 3T3 fibroblasts. Gene. 1988;74:465–471. doi: 10.1016/0378-1119(88)90179-5. [DOI] [PubMed] [Google Scholar]
- Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12:1413–1426. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G. Modulation of NF-kappaB activity by exchange of dimers. Mol Cell. 2003;11:1563–1574. doi: 10.1016/s1097-2765(03)00227-2. [DOI] [PubMed] [Google Scholar]
- Sakurai H, Suzuki S, Kawasaki N, Nakano H, Okazaki T, Chino A, Doi T, Saiki I. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278:36916–36923. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Sawadogo M, Dyke MW, Gregor PD, Roeder RG. Multiple forms of the human gene-specific transcription factor USF. I. Complete purification and identification of USF from HeLa cell nuclei. J Biol Chem. 1988;263:11985–11993. [PubMed] [Google Scholar]
- Schmidt C, Peng B, Li Z, Sclabas GM, Fujioka S, Niu J, Schmidt-Supprian M, Evans DB, Abbruzzese JL, Chiao PJ. Mechanisms of proinflammatory cytokine-induced biphasic NF-kappaB activation. Mol Cell. 2003;12:1287–1300. doi: 10.1016/s1097-2765(03)00390-3. [DOI] [PubMed] [Google Scholar]
- Schrell UM, Koch HU, Marschalek R, Schrauzer T, Anders M, Adams E, Fahlbusch R. Formation of autocrine loops in human cerebral meningioma tissue by leukemia inhibitor factor, interleukin-6, and oncostatin M: inhibition of meningioma cell growth in vitro by recombinant oncostatin M. J Neurosurg. 1998;88:541–548. doi: 10.3171/jns.1998.88.3.0541. [DOI] [PubMed] [Google Scholar]
- Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- Suyang H, Phillips R, Douglas I, Ghosh S. Role of unphosphorylated, newly synthesized I kappa B beta in persistent activation of NF-kappa B. Mol Cell Biol. 1996;16:5444–5449. doi: 10.1128/mcb.16.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, Ghosh S. I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappa B. Cell. 1995;80:573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9:625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]