

Abstract
Ovarian cancer (OC) is the leading cause of death from a gynecological malignancy in the United States. By the time a woman is diagnosed with OC, the tumor has usually metastasized. Mouse models that are used to recapitulate different aspects of human OC have been evolving for nearly 40 years. Xenograft studies in immunocompromised and immunocompetent mice have enhanced our knowledge of metastasis and immune cell involvement in cancer. Patient-derived xenografts (PDXs) can accurately reflect metastasis, response to therapy, and diverse genetics found in patients. Additionally, multiple genetically engineered mouse models have increased our understanding of possible tissues of origin for OC and what role individual mutations play in establishing ovarian tumors. Many of these models are used to test novel therapeutics. As no single model perfectly copies the human disease, we can use a variety of OC animal models in hypothesis testing that will lead to novel treatment options. The goal of this review is to provide an overview of the utility of different mouse models in the study of OC and their suitability for cancer research.
Keywords: ovarian cancer, high-grade serous ovarian cancer, xenograft, patient-derived xenograft, genetically engineered mouse models, Adeno-Cre
Introduction
Every year in the United States, over 22,000 women are diagnosed with ovarian cancer (OC) and more than 15,000 die from the disease.1 The poor prognosis for women diagnosed with OC is largely because of the presence of local and distant metastases in 81% of patients at diagnosis, leading to 5-year survival rates of 69% and 30%, respectively.1 These numbers suggest that both early detection and treatment of metastatic disease are crucial in managing OC. These facts also highlight the need to better understand mechanisms driving progression from primary tumor to metastatic disease.
Ovarian tumors can arise in the ovary from germ cells, stromal cells, or epithelial cells. The vast majority (>90%) of OC arises from epithelial tissue. There are several distinct his-totypes of epithelial OC. They are high-grade serous (HGS) and low-grade serous ovarian, mucinous, endometrioid, clear cell, and transitional or undifferentiated.2 HGS is the most common form of OC. The high mortality associated with OC is a direct result of the process of tumor metastasis. OC invasion is a complex process that differs from metastatic programs of other solid tumors, since OC tumor cells shed from the primary site and spread throughout the peritoneal cavity without first entering the blood or lymphatic system.3 OC was originally thought to be derived from the ovarian surface epithelium.4 However, epidemiological, pathological, and molecular studies have made scientists question the origin of OC and hypothesize that different types of ovarian tumors may arise from different precursor cells, including those from the fallopian tube, endometrium, cervix, and gastrointestinal (GI) tract.2,5–7 In addition to the OC subtypes being histologically different, they are also genetically distinct. Ovarian tumors have been divided into two classes. (1) Type 1 cancers progress from tumors with lower malignant activity, are usually low grade, and contain Ras pathway mutations; these include clear cell, mucinous, endometrioid, and low-grade serous tumors. (2) Type 2 tumors arise rapidly de novo and contain TP53 mutations; these include HGS, undifferentiated, and carcinosarcomas.8 Recent work suggests that some HGS tumors may be initiated in the fimbria of the fallopian tube.8,9 Identifying the cell type of origin for OC will change how animal models are generated and evaluated.
The cause of mortality from most solid tumors is metastasis. Frequently, as OC progresses, tumor cells accumulate as single-cell and multi-cell aggregates in the ascites fluid.10 The accumulation of ascites fluid in late stage cancer correlates with poor outcome.11 These shed tumor cells metastasize locally. Preferred sites of metastasis include the omentum, peritoneal wall, the diaphragm, and small bowel mesentery.3 Therefore, design and utilization of mouse models of OC that recapitulate the steps of metastasis are critical. Here, we will review many of the common ways OC is studied in mice. We will first discuss xenografts derived from cell lines and patient samples, followed by genetic models of OC.
Xenograft Models of OC
Xenograft models are a common way of investigating OC growth, metastasis, and treatment response in a live animal. The process of injecting ovarian tumor cells into mice and monitoring tumor growth has been conducted since at least the early 1980s. In these early studies, the thymus of the mice was removed, mice were treated with cytosine arabinoside, given whole-body irradiation, and then engrafted subcutaneously (SC) with either ovarian ascites (n = 5) or a solid peritoneal metastasis from a papillary cystadenocarcinoma ovarian tumor biopsy (n = 8).12 Four of the peritoneal metastases established tumor growth.12 Xenograft studies have been used extensively since the 1980s and offer many advantages. Xenografts are effective at measuring tumor growth in response to mutations or exogenous gene expression. They allow for validation of therapies and studies of drug resistance. Xenografts also can faithfully recapitulate aspects of ovarian tumor progression and metastasis. However, xenografts do not recapitulate the initial transforming events leading to tumorigenesis. Furthermore, many xenograft mouse models are conducted using immunocompromised mice, which will lack the contribution of the full complement of immune cells to the tumor growth process.
The use of xenograft models enables researchers to test factors that influence tumor growth, spread, and drug response in a live animal. These elements cannot be entirely recapitulated in tissue culture. To study tumor growth in mice, murine or human OC cells are injected into mice. Cells from different genetic backgrounds are injected into immunocompromised mice such as Nude (Foxn1, Nu/Nu), SCID, NOD/SCID, or NOD-scid IL2Rcnull (NSG) to enable the cells to engraft without being eliminated by the immune system. Cells are injected SC, intraperitoneally (IP), or intrabursally [(IB) into the bursa that surrounds the mouse ovary]. The SC model is not well suited for ovarian metastasis studies as the tumors do not typically metastasize, and the tumor is not positioned in the right anatomic location or microenvironment. One advantage of the SC model is that it is well suited for investigation with imaging modalities such as two-photon microscopy (Fig. 1). In two-photon microscopy, tumor vasculature (with fluorescent dextrans), collagens (with second harmonic imaging), and fluorescently labeled tumor cells can be measured simultaneously in a live animal. This cannot be done with IP or IB injections because of the depth of the tumors. However, IP and IB tumors can be imaged in live animals with optical imaging approaches using fluorescence or luminescence. In contrast to SC tumors, IP and IB injections of tumor cells into mice can mimic aspects of tumor metastasis, particularly metastatic dissemination. While IP injection cannot mimic the initial steps in metastasis, IP-injected tumor cells such as SKOV3 metastasize to the ovary, peritoneal wall, diaphragm, and form ascites fluid similar to human disease.13 The SKOV3IP cell line is derived from ascites cells that developed in a mouse IP cavity injected with SKOV3 cells.13 Compared to the parental SKOV3 cells, the SKOV3IP cells grow faster, disseminate more, and exhibit overexpressed ERBB2.13 Many cells (including the SKOV3 lines) are frequently transfected or transduced with fluorescence or luminescence-expressing vectors to monitor tumor growth in vivo.14 Similarly, OVCAR3 cells metastasize to the GI tract, omentum, pancreas, kidney, and liver (unpublished data) when injected IP. In some ways, the IB injection mimics the initial steps in metastasis, as the tumor cells exit the bursa to spread throughout the peritoneal cavity. Many cell lines metastasize following IB injection, including A2780, SKOV3, and HEY cells.15,16 Sites of metastasis include diaphragm, mesentery, bowel, and liver.16 In particular, the A2780 cells have been used to study not only metastasis but also chemoresistance. A2780 that are sensitive to cisplatin and a subline of A2780 cells that are resistant to cisplatin cells are also commonly used in xenograft studies of chemoresistance in vivo.17 Many other cell lines not discussed also demonstrate growth in xenograft models.18
Figure 1.
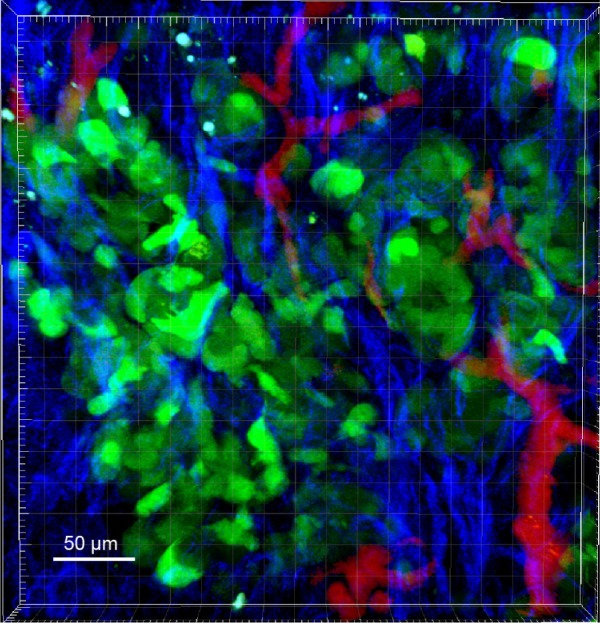
Two-photon microscopy image of SKOV3IP tumor expressing green fluorescent protein (GFP). SKOV3IP labeled with GFP were injected SC into nude mice. After one week, two-photon microscopy was conducted to image tumor cells (green), collagen (blue), and vasculature (red). Therefore, imaging subcutaneous tumor can provide information about how genetic variation affects tumor microenvironment.
Compared to genetic models, xenograft models may require less time to form tumors, from a few weeks to a few months, depending on the cell line and the number of injected tumor cells. Tumor cells can be genetically manipulated in culture, and then implanted in mice (Fig. 2). This allows for the study of how individual genes impact tumor development or metastasis. However, by using xenografts in immunocom-promised mice to study OC, the response of the immune system is ignored.19 A thematically related model of OC is patient-derived xenografts (PDXs). To derive PDXs, ovarian tumors are isolated from patients, and then surgically grafted or injected into mice. This system provides distinct advantages over using traditional xenografts with cell lines. Tumors from diverse genetic backgrounds can be grafted, and the tumor growth can be monitored. Furthermore, the PDXs recapitulate aspects of OC like metastasis and ascites formation. Typically, small fragments of a tumor are isolated and injected SC or orthotopically into immune compromised mice. Tumor engraftment percentages and time to develop tumors vary widely,19 and some xenograft studies result in less than 50% of the mice developing tumors.20 After the tumor reaches a critical size, it can be excised and reimplanted serially into subsequent mice. The original tumor and the serial PDX tumors are characterized for gene expression and mutation profile to assess if the PDXs retain the genetics and behaviors of the original tumor.
Figure 2.

Tumor cells can be genetically manipulated in culture, and then implanted in mice to form a tumor. Growth of the tumor can be measured as well as response to chemotherapy. Genetic models of cancer have increased our understanding of how OC originates and what mutations or combinations of mutations may lead to tumor establishment by disruption of tumor suppressor genes, introduction of oncogenes, and tumor origin manipulations. These provide valuable information on metastasis and highlight potential therapeutic targets.
PDX models are used to examine tumor progression and metastasis when the PDXs are implanted into the peritoneal cavity. In one model, human OC biopsies were dissociated and injected in NSG mice.21 The tumor aggregates that were injected contained tumor-associated cells, including lymphocytes and fibroblasts.21 These tumors metastasized to the omentum, ovary, pancreas, uterus, spleen, and liver. Ascites also developed in these mice. Twenty-six percent of mice were found to have lung micrometastases as commonly found in stage IV disease.21 Furthermore, a large study of 241 tumors demonstrated that the PDXs retain the genetics and metastatic spreading pattern of the original tumors.22 Minced tumors were injected IP into female SCID mice, and 74% (168) of these tumors engrafted, mostly in the pelvis.22 Almost 50% of the mice developed metastases in the intestines, mesentery, liver, spleen, diaphragm, and/or omentum, and 17 of these mice developed ascites.22 One intriguing study found that PDXs with ascites or plural effusions were more likely to generate xenograft tumors than solid tumors.23 This raises questions about the metastatic unit in OC. OC stem cells that have been isolated from serous adenocarcinomas based on surface expression of CD177 and CD44 were demonstrated to have increased tumorigenic capacity in xenografts.24 A more recent study used a PDX model of OC to demonstrate that high expression type I receptor tyrosine kinase-like orphan receptor in OC stem cells results in higher tumorigenesis.21,25 More studies on the link between cancer stem cells and metastasis should be conducted using models like the PDXs to discover novel strategies to limit metastatic spread and appropriately target cells with metastatic potential.
One of the major advantages of the PDX models is the ability to investigate the response of tumors to therapeutic agents. Treating PDX tumors with IL-12, FLT-3, and CD40 decreased tumor growth.26–28 Similarly, PDX tumors have been studied for their ability to predict patient response to drug therapy (previously reviewed by Scott et al).19 In one study, HGS tumors were engrafted into NGS mice with an 83% success rate.29 After treatment with cisplatin, it was found that 40% of the mice were platinum sensitive, 30% were refractory, and 30% were platinum resistant.29 These responses correlated with patient outcome,29 which suggests that PDXs retain the genetic/epigenetic program of the original tumor and that the response to standard chemotherapy present in the original tumor is retained. Therefore, PDXs may someday be used to develop personalized patient treatment plans. One disadvantage of PDXs is the variable take rate of the tumors. Additionally, access to patient samples is an impediment to using PDXs as a model system.
Syngeneic mouse models of OC have also been developed. Murine ovarian surface epithelial cells from C57BL/6 mice were transformed (ID8 cells), and then injected back into C57BL/6 mice so that tumor metastasis or growth can be monitored using mice with intact immune systems.30 More recent syngeneic mouse models use cells with spontaneously generated increased aggressiveness to study resistance to anoikis.31 The tumors generated in this syngeneic model develop extensive metastasis in the peritoneal cavity reminiscent of advanced OC. McCloskey et al also described a strategy for creating spontaneously transformed ovarian surface epithelial cell lines by maintaining cells for many passages. These transformed cells exhibited aberrant Wnt and NFKB signaling and resulted in tumors resembling HGS cancer when injected into the IP cavity of FVB/N mice.32 The syngeneic experimental design more accurately reflects the response of the immune system during tumor progression than immunocompromised models. However, the cells are of murine origin, and thus may not resemble human cancer as well as models using human cells.
Genetically Engineered Mouse Models of OC
Genetically engineered mouse models (GEMMs) have yielded many important advances in understanding cancer. Mouse models of leukemia, breast cancer, and colon cancer can faithfully recapitulate many aspects of human disease. The OC genetic models have been more complicated to generate, partially because of the lack of understanding of OC biology and the heterogeneity of OC. Infertility resulting from ovarian tumors has also hampered generation of transgenic models of OC. The controversy about the cell type of origin for OC and identification of transgenic promoters that reflect the proper cell type of origin are further complications. GEMMs are especially important for studying the beginning stages of OC that cannot be mimicked in xenografts. Some OCs may originate in the fallopian tube, while others may arise from ovarian epithelium.5 Different GEMMs allow us to evaluate how tumors arising from mutations at different anatomic locations compare to human disease. Thus, GEMMs provide an invaluable tool for studying the development of OC with mutations observed in patients. For example, women with familial Brca1 mutations have increased the risk of developing OC and develop cancer at a younger age than those with sporadic disease.33 Thus, mouse models with alterations to Brca1 and other genes commonly mutated in OC, such as Tp53, c-Myc, Kras, Akt, and Brca2,34–36 are important to increasing our understanding regarding how genetic predisposition leads to OC (summarized in Table 1).
Table 1.
Summary of GEMMs.
| REFERENCE | TRANSFORMING GENES | METHOD OF INDUCTION | TISSUE ORIGIN | HISTOLOGY |
|---|---|---|---|---|
| Flesken-Nikitin et al34 | Tp53, Rb1 | Injected Cre adenovirus-induced knockout | Ovary | Serous epithelial ovarian cancer |
| Szbova et al35 | Tp53, Rb “pocket proteins”, Brca1/2 | SV40 T antigen expression, Injected Cre adenovirus-induced knockout | Ovary | Serous epithelial ovarian cancer |
| Orulic et al36 | C-myc, K-ras, Akt1 | Syngeneic cells transduced with RCAS viral vectors injected into p53-null mice | Ovary | Undifferentiated carcinoma |
| Connolly et al40 | Tp53, Rb1 | Chimeric expression of SV40A under MISIIR promoter | Ovary | Poorly-differentiated neoplasia |
| Xing et al44 | C-myc, Tp53, Brca1 | Syngeneic cells transduced with RCAS viral vectors, Cre-induced knockout | Ovary | Serous epithelial ovarian cancer |
| Perets et al45 | Brca1, Brca2, Tp53, Pten | Cre-recombinase driven by Pax8 promoter | Fallopian tube | Serous tubal intraepithelial carcinomas |
| Sherman-Baust et al46 | Tp53, Top2A | Expression of SV40A under Ovp-1 promoter | Fallopian tube | Serous tubal intraepithelial carcinomas |
| Tirodkar et al47 | Pten, K-ras, Muc1 | Injected Cre adenovirus-induced knockout | Ovary, oviduct, and uterus | Endometrioid adenocarcinoma |
| Wu et al50 | Pten, Apc | Injected Cre adenovirus-induced knockout | Uterus | Endometrioid adenocarcinoma |
The most common mutation in HGS OC occurs in Tp53,37 which is mutated in about 48% of sporadic OCs and 96% of HGS cancers.37,38 In addition, Rb1 is dysregulated in about 67% of HGS OCs.38 GEMMs can be used to study these tumor suppressors as well as oncogenes such as c-Myc, Kras, and Akt1. An early method for inhibiting both of Tp53 and Rb1 is to express Simian virus 40 (SV40) large T antigen that binds to and inactivates both p53 and pRB.39 Transgenic mice expressing SV40 under control of the promoter for Mullerian inhibiting substance type II receptor (MISIIR) develop poorly differentiated ovarian carcinomas.40 MISIIR is expressed in 92% of OC, but most nongynecological tissues do not express this receptor.41 These carcinomas express cyto-keratins 8 and 19 and lack alpha-inhibin, suggesting that they are derived from epithelium and not derived from granulosa cells.40 These tumors result in ascites formation and metastatic spread to omentum.40 One mouse demonstrated metastasis to the kidney and one had metastasis to the uterus, but metastasis to other organs was not observed.40 This was the first published model of a transgenic model of epithelial OC. A major disadvantage to this approach is that the mice were infertile because of the ovarian tumors. Therefore, other approaches to inactivate tumor suppressors that may be more indicative of human cancer needed to be developed.
Conditional knockouts of Tp53 and Rb1 have also been used to generate mouse models of OC. As reported by Flesken-Nikitin, mice with Tp53 and Rb1 flanked by loxP sites (floxed) were injected into the bursa ovary with an adenovirus expressing Cre recombinase under control of a CMV promoter.34 This double knockout resulted in a metastatic OC resembling human ovarian epithelial cancer, which spread to the lung, liver, and noninjected ovary.34 A total of 97% of the mice with knockout of Tp53 and Rb1 in the ovary died within eight months.34 Single knockouts of either Tp53 or Rb1 were much less efficient at inducing metastatic cancer: 13% of Tp53 knockout mice and 3% of Rb1 knockout mice developed tumors.34 Importantly, these studies reveal that Tp53 mutations alone are sufficient for ovarian tumor initiation, but mutation in both Tp53 and Rb1 lead to metastatic OC.34
In a more recent study, combinations of Tp53 mutation or loss, loss of pocket protein function, and loss of Brca1/2 were generated to thoroughly examine the contributions of these genes to OC. Expression of T121 (a domain of the SV40 large T antigen) in the surface epithelium is induced by injecting Cre-expressing adenovirus (Adeno-Cre). T121 is in a Cre-conditional loxP-GFP-stop-loxP (LSL) cassette driven by the cytokeratin 18 promoter. Injection of Adeno-Cre resulted in inhibition of Rb1, p107, and p130, also known as the “pocket proteins.” In this study, antigen T121 is used in concert with floxed alleles for Tp53, a point mutation in Tp53 (p53R172H), and inactivation of either Brca1 or Brca2.35 Inactivation of the pocket proteins was sufficient for tumorigenesis, resulting in stage I serous epithelial OC.35 However, loss of pRb, p53, and Brca1 or Brca2 in mice more closely mimics serous OC.35 When Brca1 or Brca2 were deleted in this model (in addition to inhibition of Tp53 and the RB pathway), the tumors metastasized to the liver and demonstrated a gene expression profile resembling serous OC.35 A later version of this GEMM was used to study drug response in Brca1-deficient mice, showing that tumors in these mice respond to a combination chemotherapy of cisplatin and olaparib.42 This provides an example of how GEMMs can be used to test effective therapies. Through these varying combinations of genetic alterations, different stages of OC progression were modeled, from early stage I epithelial OC (Rb pathway disruption alone) to an advanced metastatic cancer (RB loss/Tp53/Brca1/Brca2 alterations).
In addition to disrupting tumor suppressor genes, introduction of oncogenes has also been employed to generate mouse models of OC. Transgenic mice expressing the avian retroviral receptor TVA (with a-actin or keratin 5 promoter) were generated.36 These mice were then crossed with p53 null mice.36 Ovarian surface epithelial cells were isolated from the mice and transduced in vitro with replication-competent avian-leukemia-virus splice-acceptor (RCAS)43 viral vectors. These viral vectors expressed common OC oncogenes: c-Myc, Kras, or Akt1. Subcutaneous tumor formation resulted when any two of these oncogenes were expressed in ovarian cells that were injected SC into nude mice.36 IB injection of the β-actin-TVA/p53−/− cells with the RCAS viruses containing c-Myc or Kras oncogenes resulted in metastastic OC.36 Metastasis to the liver, spleen, intestine, kidney, and malignant ascites was observed.36 This result mirrors that of later mouse models from the same laboratory, showing that TVA/RCAS-induced expression of c-Myc is sufficient to induce transformation in ovarian cells from mice with floxed Tp53 and Brca1.44 These transformed cells displayed chromosomal defects and an unusually high centromere count; injection of these modified cells into the ovary resulted in metastatic tumors that resemble serous ovarian carcinomas and spread to the peritoneum, pancreas, and intestines.44 Collectively, these models demonstrate that Tp53 mutation in concert with oncogene expression or loss of other tumor suppressors results in OC generation and metastasis.
As our understanding of the genetics and epigenetics of OC evolves, so must the ways of modeling the disease. It has been hypothesized that different cell types of origin and different genetic mutations are responsible for the distinct histological subtypes of OC. Therefore, establishing models of fallopian tube and uterine-derived OC is essential. To determine if high-grade serous ovarian cancer (HGSOC) can originate from fallopian tube, Pax8-driven Cre recombinase was used to excise Brca1, Brca2, Tp53, and/or Pten.45 Loss of Brca1/2, Tp53 mutation, and Pten loss resulted in fallopian tube serous tubal intraepithelial carcinomas (STICs), ovarian metastases, and peritoneal metastases.45 The histology, metastatic profile, and transcriptome of these tumors closely mimics HGS OC.45
Another method to transform the fallopian tube used SV40 large T antigen expressed under control of the Mullerian Ovgp-1 promoter.46 This model results in STICs and invasive adenocarcinomas that spread to the ovaries.46 Therefore, this model is capable of recapitulating early development of OC46 in the fallopian tube. One additional advantage of this model is that the Ovgp-1 promoter ensures tissue-specific expression of SV40 in mice that have reached sexual maturity.
Since the ovary, uterus, and fallopian tube may be sites of origin for different types of OC, a comparison of transformation at each of these locations was conducted. To compare tumorigenesis in the ovary, oviduct, and endometrium using the same genetic backgrounds, a triple transgenic mouse model was created.47 In these mice, KrasG12D (using a LSL cassette) is induced, and floxed Pten is deleted using Adeno-Cre in different locations of the female reproductive tract.48 Additionally, these transgenic mice also express the human glycoprotein MUC1 that is often overexpressed in OC.48 Injection of Adeno-Cre into the ovary, oviduct, or uterus resulted in the formation of epithelial tumors capable of metastasis, spreading to the pancreas and diaphragm.47 However, the nuclear grade of the tumors and survival of the mice varied depending on the anatomical site of injection.47 Tumors arising from ovaries, the oviduct, and uterus generated endometrioid histology and metastasis to spleen, diaphragm, and liver were detected.47 The oviduct tumors developed into poorly differentiated high-grade tumors, while ovarian and uterine tumors were more differentiated.47 Mice with ovarian and oviduct tumors exhibited the shortest mean survival times compared to mice with uterine tumors (89, 82, and 132.5 days, respectively). A key feature of this model is that overexpression of MUC1 results in a similar immune response as that seen in human patients.47 This enables testing of immunotherapy treatments that target OC cells over-expressing MUC1.47
While many of the recent GEMMs are aimed at understanding the origin of OC or recapitulating HGSOC, mouse models of other OC histotypes have also emerged. Ovarian endometrioid adenocarcinoma is a subtype of OC originating from malignant uterine cells created during endometriosis.6 Some models of endometrioid OC resulted from oncogenic Kras and a conditional Pten deletion.47,49 However, Kras mutations are uncommon in human endometrioid cancer. To address this issue, a mouse model of endometrioid OC was created in which Apc and Pten are deleted, since perturbations in Wnt and phosphoinositide 3-kinase (PI3K) signaling are linked in some endometrioid OC.50 Mice with floxed Apc and Pten were IB injected with Adeno-Cre.50 Resultant adenocarcinomas were similar to metastatic human ovarian endome-trioid adenocarcinoma and50 were generally low grade, unlike those resulting from Tp53 mutations.50 The tumors resulting from Pten/Apc knockout showed increased nuclear β-catenin and overactive AKT signaling.50 These GEMMs have been used to test therapeutics against endometrioid cancer; a combination of cisplatin and paclitaxel together with inhibitors of mTOR and AKT signaling has proven effective in mouse models.51 This work highlights the possibility of using treatments that target the Wnt and PI3K pathways to combat endometrioid cancer.
Summary
The survival rates and treatment for OC have remained relatively unchanged for the past two decades. Recent advances in understanding the genetic landscape of the histological subtypes of OC generate hope that targeted therapies can be devised that will increase patient survival. In order to see these targeted therapies come into practice, preclinical mouse models that reflect the genetics, metastatic potential, and different responses to therapy need to be used. In the last 30 years, mouse models have progressed from early xenografts to xenografts with immunocompetant mice, and PDXs that reflect individual patient’s genetics and therapeutic response. Moreover, genetic models of cancer have increased our understanding of how OC originates and what mutations or combinations of mutations may lead to tumor establishment. A recent mouse model with mutations in c-Kit further allows the study of ovarian tumors in the context of a postmenopausal environment, potentially allowing for the above-discussed genetic mutations to be further improved by incorporating hormone responsiveness seen in the older population most likely to be affected by OC.52 Now that we have a variety of mouse models (summarized in Table 2) (as well as other animal models, such as the laying hen)53 at our disposal that reflect a wide range of steps of OC progression, we can develop diagnostic, prognostic, and therapeutic approaches that will increase OC survival.
Table 2.
Ovarian mouse model systems.
| MODEL TYPE | ADVANTAGES | DISADVANTAGES |
|---|---|---|
| Xenograft | Effective at measuring tumor growth in response to mutations or exogenous gene expression. They allow for validation of therapies and studies of drug resistance. Xenografts also can faithfully recapitulate aspects of ovarian tumor progression and metastasis. | Does not recapitulate the initial transforming events leading to tumorigenesis. Many xenograft mouse models are conducted using immunocompromised mice, which will lack the contribution of the full complement of immune cells to the tumor growth process. |
| Subcutaneous xenografts | It is well suited for investigating imaging modalities such as 2-photon microscopy. | It is not well suited for ovarian metastasis studies as the tumors do not typically metastasize and the tumor is not positioned in the right anatomic location or microenvironment. |
| Intraperitoneal xenografts | Can mimic aspects of tumor metastasis, particularly metastatic dissemination; IP injected tumor cells, such as SKOV3IP cells, metastasize to the ovary, peritoneal wall, diaphragm, and form ascites fluid similarly to human disease. Tumor growth can be monitored using in vivo fluorescence or luminescence. | Tumor initiation and initial tranforming events cannot be studied. Cannot image in vivo using 2-photon microscopy due to the depth of the tumors. |
| Intrabursal xenografts | IB injection mimics the initial steps in metastasis, as the tumor cells exit the bursa to spread throughout the peritoneal cavity. | Tumor initiation and initial tranforming events cannot be studied. Cannot image in vivo using 2-photon microscopy due to the depth of the tumors. |
| Patient Derived Xenografts | Tumors from diverse genetic backgrounds can be grafted, and the tumor growth can be monitored. PDXs recapitulate aspects of OC like metastasis and ascites formation. PDX models have the ability to investigate the response of the tumors to therapeutic agents and could predict patient response to drug therapy. | PDXs have variable take rate and access to patient samples is a limitation for many investigators. |
| Syngeneic xenografts | The experimental design more accurately reflects the response of the immune system during tumor progression than immunocompromised models. | The cells are of murine origin, and thus may not resemble human cancer as well as models using human cells. |
| Genetically engineered mouse models | GEMMS are especially important for studying the beginning stages of ovarian cancer that cannot be mimicked in xenografts. Different genetically engineered mouse models allow us to compare evaluate how OC tumors arising from mutations at different anatomic locations compare to human disease. | OC genetic models have been more complicated to generate, partially due to the lack of understanding of OC biology and the heterogeneity of ovarian cancer. Infertility resulting from ovarian tumors has also hampered generation of transgenic models of ovarian cancer. |
Note: This is an overview of the different mouse models used in the study of OC, summarizing advantages and disadvantages of each.
Abbreviations
- OC
ovarian cancer
- GEMM
genetically engineered mouse model
- PDX
patient-derived xenograft
- HGS
high-grade serous
- GI
gastrointestinal
- HGSOC
high-grade serous ovarian cancer
- IP
intraperitoneally
- IB
intrabursally
- SC
subcutaneously
- STOSE
spontaneously transformed ovarian surface epithelial
- LSL
loxP-GFP-stop-loxP
- MISIIR
Mullerian inhibiting substance type II receptor
- RCAS
replication competent avian-leukemia-virus splice-acceptor
- STIC
serous tubal intraepithelial carcinoma
- SV40
Simian virus 40
Footnotes
ACADEMIC EDITOR: Marc D. Basson, Editor in Chief
PEER REVIEW: Eight peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1,449 words, excluding any confidential comments to the academic editor.
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Wrote the first draft of the manuscript: ASB and KDCD. Contributed to the writing of the manuscript: ASB, JMC, and KDCD. Jointly developed the structure and arguments for the paper: ASB, JMC, and KDCD. Made critical revisions and approved final version: ASB, JMC, and KDCD. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.American Cancer Society . Cancer Facts and Figures. New York: American Cancer Society; 2015. [Google Scholar]
- 2.Rescigno P, Cerillo I, Ruocco R, Condello C, De Placido S, Pensabene M. New hypothesis on pathogenesis of ovarian cancer lead to future tailored approaches. Biomed Res Int. 2013;2013:852839. doi: 10.1155/2013/852839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177(3):1053–1064. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev. 2001;22(2):255–288. doi: 10.1210/edrv.22.2.0422. [DOI] [PubMed] [Google Scholar]
- 5.Piek JM, van Diest PJ, Verheijen RH. Ovarian carcinogenesis: an alternative hypothesis. Adv Exp Med Biol. 2008;622:79–87. doi: 10.1007/978-0-387-68969-2_7. [DOI] [PubMed] [Google Scholar]
- 6.Campbell IG, Thomas EJ. Endometriosis: candidate genes. Hum Reprod Update. 2001;7(1):15–20. doi: 10.1093/humupd/7.1.15. [DOI] [PubMed] [Google Scholar]
- 7.Yang-Hartwich Y, Gurrea-Soteras M, Sumi N, et al. Ovulation and extraovarian origin of ovarian cancer. Sci Rep. 2014;(4):6116. doi: 10.1038/srep06116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer. 2010;10(11):803–808. doi: 10.1038/nrc2946. [DOI] [PubMed] [Google Scholar]
- 9.Koshiyama M, Matsumura N, Konishi I. Recent concepts of ovarian carcinogenesis: type I and type II. Biomed Res Int. 2014;2014:934261. doi: 10.1155/2014/934261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmed N, Stenvers KL. Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Front Oncol. 2013;(3):256. doi: 10.3389/fonc.2013.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puls LE, Duniho T, Hunter JE, Kryscio R, Blackhurst D, Gallion H. The prognostic implication of ascites in advanced-stage ovarian cancer. Gynecol Oncol. 1996;61(1):109–112. doi: 10.1006/gyno.1996.0106. [DOI] [PubMed] [Google Scholar]
- 12.Selby PJ, Thomas JM, Monaghan P, Sloane J, Peckham MJ. Human tumour xenografts established and serially transplanted in mice immunologically deprived by thymectomy, cytosine arabinoside and whole-body irradiation. Br J Cancer. 1980;41(1):52–61. doi: 10.1038/bjc.1980.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu D, Wolf JK, Scanlon M, Price JE, Hung MC. Enhanced c-erbB-2/neu expression in human ovarian cancer cells correlates with more severe malignancy that can be suppressed by E1A. Cancer Res. 1993;53(4):891–898. [PubMed] [Google Scholar]
- 14.Mikuła-Pietrasik J, Sosińska P, Kucińska M, et al. Peritoneal mesothelium promotes the progression of ovarian cancer cells in vitro and in a mice xenograft model in vivo. Cancer Lett. 2014;355(2):310–315. doi: 10.1016/j.canlet.2014.09.041. [DOI] [PubMed] [Google Scholar]
- 15.Bao R, Connolly DC, Murphy M, et al. Activation of cancer-specific gene expression by the survivin promoter. J Natl Cancer Inst. 2002;94(7):522–528. doi: 10.1093/jnci/94.7.522. [DOI] [PubMed] [Google Scholar]
- 16.Sengupta S, Kim KS, Berk MP, et al. Lysophosphatidic acid downregulates tissue inhibitor of metalloproteinases, which are negatively involved in lysophos-phatidic acid-induced cell invasion. Oncogene. 2007;26(20):2894–2901. doi: 10.1038/sj.onc.1210093. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Cardenas H, Fang F, et al. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014;74(17):4922–4936. doi: 10.1158/0008-5472.CAN-14-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther. 2004;10(6):1032–1042. doi: 10.1016/j.ymthe.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 19.Scott CL, Becker MA, Haluska P, Samimi G. Patient-derived xenograft models to improve targeted therapy in epithelial ovarian cancer treatment. Front Oncol. 2013;(3):295. doi: 10.3389/fonc.2013.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luca G, Cameron DF, Arato I, et al. Xenograft of microencapsulated Sertoli cells for the cell therapy of type 2 diabetes mellitus in spontaneously diabetic nonhuman primates: preliminary data. Transplant Proc. 2014;46(6):1999–2001. doi: 10.1016/j.transproceed.2014.06.053. [DOI] [PubMed] [Google Scholar]
- 21.Bankert RB, Balu-Iyer SV, Odunsi K, et al. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLoS One. 2011;6(9):e24420. doi: 10.1371/journal.pone.0024420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weroha SJ, Becker MA, Enderica-Gonzalez S, et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin Cancer Res. 2014;20(5):1288–1297. doi: 10.1158/1078-0432.CCR-13-2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verschraegen CF, Hu W, Du Y, et al. Establishment and characterization of cancer cell cultures and xenografts derived from primary or metastatic Mullerian cancers. Clin Cancer Res. 2003;9(2):845–852. [PubMed] [Google Scholar]
- 24.Zhang S, Balch C, Chan MW, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68(11):4311–4320. doi: 10.1158/0008-5472.CAN-08-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Cui B, Lai H, et al. Ovarian cancer stem cells express ROR1, which can be targeted for anti-cancer-stem-cell therapy. Proc Natl Acad Sci U S A. 2014;111(48):17266–17271. doi: 10.1073/pnas.1419599111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghamande S, Hylander BL, Oflazoglu E, Lele S, Fanslow W, Repasky EA. Recombinant CD40 ligand therapy has significant antitumor effects on CD40-positive ovarian tumor xenografts grown in SCID mice and demonstrates an augmented effect with cisplatin. Cancer Res. 2001;61(20):7556–7562. [PubMed] [Google Scholar]
- 27.Silver DF, Hempling RE, Piver MS, Repasky EA. Effects of IL-12 on human ovarian tumors engrafted into SCID mice. Gynecol Oncol. 1999;72(2):154–160. doi: 10.1006/gyno.1998.5239. [DOI] [PubMed] [Google Scholar]
- 28.Silver DF, Hempling RE, Piver MS, Repasky EA. Flt-3 ligand inhibits growth of human ovarian tumors engrafted in severe combined immunodeficient mice. Gynecol Oncol. 2000;77(3):377–382. doi: 10.1006/gyno.2000.5782. [DOI] [PubMed] [Google Scholar]
- 29.Topp MD, Hartley L, Cook M, Australian Ovarian Cancer Study et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol Oncol. 2014;8(3):656–668. doi: 10.1016/j.molonc.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roby KF, Taylor CC, Sweetwood JP, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21(4):585–591. doi: 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- 31.Cai Q, Yan L, Xu Y. Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene. 2014;34(25):3315–3324. doi: 10.1038/onc.2014.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCloskey CW, Goldberg RL, Carter LE, et al. A new spontaneously transformed syngeneic model of high-grade serous ovarian cancer with a tumor-initiating cell population. Front Oncol. 2014;(4):53. doi: 10.3389/fonc.2014.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer. 2003;97(9):2187–2195. doi: 10.1002/cncr.11310. [DOI] [PubMed] [Google Scholar]
- 34.Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res. 2003;63(13):3459–3463. [PubMed] [Google Scholar]
- 35.Szabova L, Yin C, Bupp S, et al. Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res. 2012;72(16):4141–4153. doi: 10.1158/0008-5472.CAN-11-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell. 2002;1(1):53–62. doi: 10.1016/s1535-6108(01)00002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pipas JM, Levine AJ. Role of T antigen interactions with p53 in tumorigenesis. Semin Cancer Biol. 2001;11(1):23–30. doi: 10.1006/scbi.2000.0343. [DOI] [PubMed] [Google Scholar]
- 40.Connolly DC, Bao R, Nikitin AY, et al. Female mice chimeric for expression of the Simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 2003;63(6):1389–1397. [PubMed] [Google Scholar]
- 41.Bakkum-Gamez JN, Aletti G, Lewis KA, et al. Mullerian inhibiting substance type II receptor (MISIIR): a novel, tissue-specific target expressed by gynecologic cancers. Gynecol Oncol. 2008;108(1):141–148. doi: 10.1016/j.ygyno.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Szabova L, Bupp S, Kamal M, et al. Pathway-specific engineered mouse allograft models functionally recapitulate human serous epithelial ovarian cancer. PLoS One. 2014;9(4):e95649. doi: 10.1371/journal.pone.0095649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hughes SH, Greenhouse JJ, Petropoulos CJ, Sutrave P. Adaptor plasmids simplify the insertion of foreign DNA into helper-independent retroviral vectors. J Virol. 1987;61(10):3004–3012. doi: 10.1128/jvi.61.10.3004-3012.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xing D, Orsulic S. A mouse model for the molecular characterization of brca1-associated ovarian carcinoma. Cancer Res. 2006;66(18):8949–8953. doi: 10.1158/0008-5472.CAN-06-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perets R, Wyant GA, Muto KW, et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell. 2013;24(6):751–765. doi: 10.1016/j.ccr.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sherman-Baust CA, Kuhn E, Valle BL, et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. J Pathol. 2014;233(3):228–237. doi: 10.1002/path.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tirodkar TS, Budiu RA, Elishaev E, et al. MUC1 positive, Kras and Pten driven mouse gynecologic tumors replicate human tumors and vary in survival and nuclear grade based on anatomical location. PLoS One. 2014;9(7):e102409. doi: 10.1371/journal.pone.0102409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Budiu RA, Elishaev E, Brozick J, et al. Immunobiology of human mucin 1 in a preclinical ovarian tumor model. Oncogene. 2013;32(32):3664–3675. doi: 10.1038/onc.2012.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endo-metrioid ovarian cancer. Nat Med. 2005;11(1):63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 50.Wu R, Hendrix-Lucas N, Kuick R, et al. Mouse model of human ovarian endo-metrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11(4):321–333. doi: 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 51.Wu R, Hu TC, Rehemtulla A, Fearon ER, Cho KR. Preclinical testing of PI3K/AKT/mTOR signaling inhibitors in a mouse model of ovarian endometrioid adenocarcinoma. Clin Cancer Res. 2011;17(23):7359–7372. doi: 10.1158/1078-0432.CCR-11-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith ER, Wang Y, Xu XX. Development of a mouse model of menopausal ovarian cancer. Front Oncol. 2014;(4):36. doi: 10.3389/fonc.2014.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barua A, Bitterman P, Abramowicz JS, et al. Histopathology of ovarian tumors in laying hens: a preclinical model of human ovarian cancer. Int J Gynecol Cancer. 2009;19(4):531–539. doi: 10.1111/IGC.0b013e3181a41613. [DOI] [PMC free article] [PubMed] [Google Scholar]