

Abstract
Aims
Diabetes is associated with both dysfunction of the lower urinary tract (LUT) and overactivity of the renin-angiotensin system (RAS). Although it is well known that the RAS affects normal LUT function, very little is known about RAS effects on the diabetic LUT. Accordingly, we investigated the effects of chronic angiotensin II (AngII) treatment on the LUT in a model of type 1 diabetes.
Methods
Ins2(Akita) diabetic mice (20 wk old) and their age-matched background controls underwent conscious cystometric evaluation after 4 wk of chronic AngII treatment (700 ng/kg/min by osmotic pump) or vehicle (saline).
Results
Diabetic mice had compensated LUT function with bladder hypertrophy. Specifically, micturition volume, residual volume and bladder capacity were all increased, while voiding efficiency and pressure generation were unchanged as bladder mass, contraction duration, and phasic urethral function were increased. AngII significantly increased voiding efficiency and peak voiding pressure and decreased phasic frequency irrespective of diabetic state and, in diabetic but not normoglycemic control mice, significantly decreased residual volume and increased contraction duration and nonphasic contraction duration.
Conclusions
The Ins2(Akita) diabetic mice had compensated LUT function at 20 wk of age. Even under these conditions, AngII had beneficial effects on LUT function, resulting in increased voiding efficiency. Future studies should therefore be conducted to determine whether AngII can rescue the decompensated LUT function occurring in end-stage diabetic uropathy.
Keywords: bladder, cystometry, diabetic cystopathy, diabetic uropathy, detrusor, lower urinary tract, renin-angiotensin system, voiding efficiency
Introduction
Diabetes is associated with lower urinary tract dysfunction that seriously compromises quality of life (1). Studies of rats (2) and mice (3) have shown that there are time-dependent changes in bladder function following induction of diabetes with streptozotocin (STZ); initially, enhanced contractility and bladder hypertrophy enable maintained efficient voiding (compensation), but eventually contractility becomes depressed and voiding becomes inefficient (decompensation). It is likely that similar time dependence in humans contributes to variability in bladder function in diabetic patients (1).
Diabetes is also associated with overactivity of the renin-angiotensin system (RAS). Thus, most diabetics develop hypertension and many hypertensives develop diabetes, and treatment with angiotensin converting enzyme inhibitors or type 1 AngII receptor antagonists prevents or delays new onset diabetes (4, 5). This is important because RAS overactivity modifies smooth muscle function. In the short run, AngII binding to AT1 receptors contracts smooth muscle whereas its binding to AT2 receptors relaxes smooth muscle (6). On a longer time scale, AngII stimulates hypertrophy, hyperplasia and extracellular matrix synthesis in smooth muscle via AT1 receptors and inhibits these processes via AT2 receptors (7-9). Moreover, such effects are observed not only in the vasculature but also in the normal lower urinary tract. The rat bladder contains both AT1 receptors (10, 11) and AT2 receptors (12). AngII induces contractions of bladder strips from humans (13-15), mice (16), and rats (17, 18). Apart from these effects on contractility, AngII stimulates proliferation of bladder smooth muscle (19) and neonatal bladder stromal cells (20).
Unfortunately, RAS effects on the lower urinary tract in diabetes have gone unexplored except for a single abstract showing that AT1 receptor expression of rat bladder smooth muscle cells increases as peak bladder pressure drops over an 8-wk period following STZ-induced diabetes (21). It is therefore an open question whether changes in RAS activity are harmful or beneficial to the lower urinary tract and thus whether they might be modulated to improve lower urinary tract function in diabetes.
Accordingly, we assessed lower urinary tract function in mice with and without Ins2(Akita) diabetes and with and without concomitant chronic AngII treatment. Our expectation was that Ins2(Akita) diabetic mice at 20 wk of age would show decompensated bladder function and we hypothesized that chronic AngII treatment would improve that decompensated bladder function.
Materials and methods
Mice
We used 129/SvEv mice, roughly half of them being Ins2(Akita) mice in which one allele of the Ins2 gene for insulin 2 has a substitution of tyrosine for cysteine (22). The resultant diabetes is more prevalent and severe in males (22), which were used in these studies. The normal and Ins2(Akita) 129/SvEv mice were obtained from an institutional colony. Blood glucose was measured without fasting, and diabetes was defined as blood glucose > 300 mg/dL.
Experimental design
The experimental protocol was approved by the Animal Care and Use Committee of the Durham Veterans Affairs Medical Center where this work was carried out. Control and diabetic mice were implanted with osmotic pumps at 16 wk of age to deliver either vehicle (saline) or AngII for 4 wk, thereby defining 4 groups of mice (Table I). We chose 700 ng/kg/min (1.008 mg/kg/day) as the AngII dose to ensure adequate stimulation while avoiding undue mortality (23, 24) and 4 weeks as the duration of treatment after consulting local experts and Alzet's references on AngII treatment with Alzet pumps (http://www.alzet.com/research_applications/AII.html). In a prior study of C57BL/6 mice with STZ-induced diabetes (3), blood glucose levels were 100-120 mg/dL in controls rising to 356 mg/dL at 3 wk post-STZ and 548-591 at 9-20 wk post-STZ. In a study of C57BL/6 Ins2(Akita) diabetic mice blood glucose levels rose from about 110 mg/dL at 3 wk of age to 270 at 4 wk, 325 at 5 wk, 450 at 6 wk, and 540-600 at 9-20 wk (22), and blood glucose levels around 500 mg/dL were maintained from 2-6 months of age for Ins2(Akita) diabetic mice on C57BL/6 and 129/SvEv-Ins2(Akita) backgrounds (25). Given that STZ-treated mice change from compensated to decompensated bladder function at 9-12 wk post-STZ (3), we therefore expected that our 20 wk 129/SvEv-Ins2(Akita) mice would have decompensated bladder function. We had planned separate study of Ins2(Akita) mice with compensated bladder function, which turned out to be unnecessary.
Table 1. Group Characteristics (Mean ± SEM).
| Variable | Control Saline | Control AngII | DM Saline | DM AngII |
|---|---|---|---|---|
| Number of mice | 12 | 10 | 8 | 7 |
| Blood glucose (mg/dl) | 122 ± 7 | 103 ± 9 | 381 ± 13 | 381 ± 24 |
| Body mass (g) | 24.4 ± 0.6 | 23.3 ± 0.6 | 23.0 ± 0.8 | 21.2 ± 0.9 |
| Bladder mass (mg) | 64 ± 3 | 59 ± 3 | 99 ± 7 | 85 ± 10 |
| Cardiac mass (mg) | 152 ± 11 | 185 ± 8 | 154 ± 15 | 174 ± 15 |
| Filling time (min) | 11.6 ± 0.6 | 9.8 ± 0.5 | 13.8 ± 1.3 | 14.3 ± 0.9 |
AngII, angiotensin II; DM, Ins2(Akita) model of diabetes mellitus.
Osmotic pump implantation
At 16 wk of age, mice were anesthetized with isoflurane, an incision was made between the shoulder blades, and a pocket was created in which Alzet model 2004 osmotic pumps (Durect, Cupertino, CA, USA) were inserted. Pumps contained either saline or AngII in saline for delivery at 700 ng/kg/min. Following skin closure and application of local anesthetic cream, mice were treated with 0.05-0.1 mg/kg s.c. buprenorphine 2×/day for 3 days and 5-10 mg/kg s.c. enrofloxacin 2×/day for 5 days.
Chronic bladder catheter implantation
Three weeks after pump implantation, a suprapubic catheter was surgically implanted under isoflurane anesthesia for cystometric study 1 wk later. This interval was chosen because rat bladder is hyperreflexic at 2 but not 6 days after catheterization (26, 27), presumably because cystitis present at 2 days after catheterization is largely repaired after 7 days (27). The catheter was formed of PE10 polyethylene tubing distally heated to create a flared end. The bladder was exposed via a midline abdominal incision, the flared catheter end was passed through an incision in the bladder dome, and the catheter was tied in place in the bladder using a purse-string suture. The catheter end was heat-sealed and tunneled subcutaneously to the back of the neck where it was anchored subcutaneously. Mice were then treated with buprenorphine and enrofloxacin as above.
Restraint training
Cystometry was performed in awake restrained mice. To reduce restraint stress, mice spent 1-2 hr/day during the week before cystometry to accommodate to a Ballman restraint cage (KN-326 Type 4, Natsume Seisakusho, Tokyo, Japan). The cage is formed of axial metal rods forming the border of a cylindrical cage through which the mouse can freely void.
Cystometry
One week after catheter implantation, the free end of the catheter was released under isoflurane anesthesia after which the incision was closed and treated with local anesthetic cream. The catheter tip was cut off and connected via PE50 tubing to a syringe pump (Model 975, Harvard Apparatus, South Natick, MA, USA) for infusion of saline and to a pressure transducer (BLPR, World Precision Instruments, Sarasota, FL, USA) for monitoring of intravesical pressure. Voids were measured by collection in a beaker mounted on a force transducer (FORT100, World Precision Instruments). Pressure and force transducer signals were amplified by a Transbridge TBM4M transducer amplifier (World Precision Instruments) whose outputs were sent to a PowerLab 8/30 data acquisition system running LabChart v7 software (ADInstruments, Colorado Springs, CO, USA), and continuously sampled at 1000 Hz.
After an hour for recovery from anesthesia, infusion of room-temperature physiological saline through the bladder catheter was begun after the bladder was emptied via the catheter and ended when voiding began. The volume remaining in the bladder after voiding (residual volume) was measured by withdrawal into a syringe. Expecting that mice in different groups would have different bladder capacities, we chose to keep the time required to fill an initially empty bladder until the voiding contraction begins (filling time) constant rather than infusion rate. (Varying infusion rate to keep filling time constant for bladders of markedly different sizes may affect cystometric outcomes, but this is equally true for keeping infusion rate constant and thereby allowing marked differences in filling time.) We targeted 10 minutes as a filling time to enable acquisition of ten cystometrograms without keeping the mice restrained too long. Note that bladder capacity varies somewhat among cystometrograms in a single animal; thus, it is not possible to precisely regulate filling time. The final infusion rate varied from 0.45 to 5 ml/hr (0.0075 to 0.0833 ml/min) to achieve a mean filling time of 12.1 min. Filling time was often higher in diabetic mice with larger bladders (Table I) because filling rate is limited by increasing backpressure generated in the narrow catheters at higher filling rates. The greater backpressure at higher filling rates was also manifest by a slight elevation in recorded pressure when the infusion pump was turned on (compare the beginning of panels A1 and B1 in Figure 1).
Figure 1.
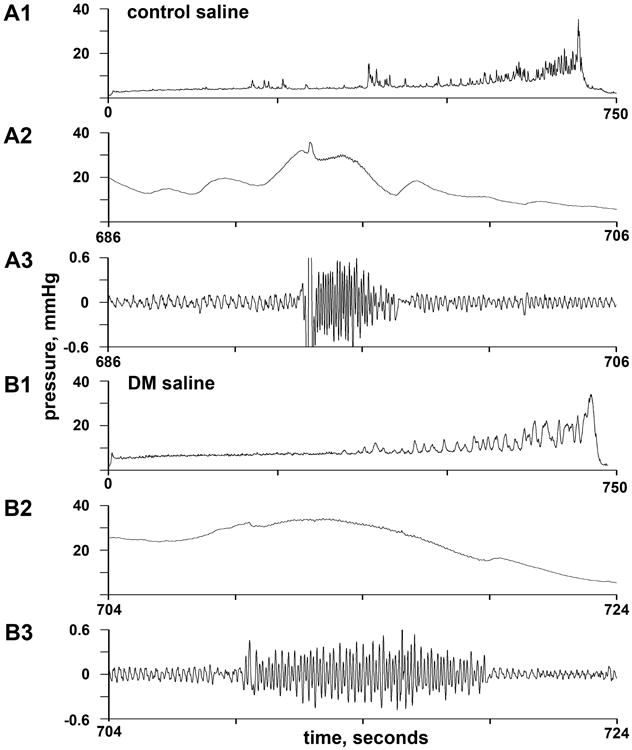
Representative cystometrograms of saline-treated control (A) and diabetic (B) mice. Panels A1 and B1 show complete cystometrograms begun after emptying the bladder; although filling time was similar, the infusion rate for A was about 3 times higher than that for B. Panels A2 and B2 show the 20 seconds surrounding voiding, and panels A3 and B3 show the same data after bandpass filtering used to emphasize high-frequency oscillations attending phasic EUS contraction. DM = diabetes mellitus.
Cystometric measurements
Data were obtained from 10 cystometrograms per mouse. Measured cystometric variables were micturition volume (voided volume), residual volume, and intravesical pressure; derived cystometric variables were bladder capacity (micturition volume + residual volume) and percent voiding efficiency as a measure of the effectiveness of bladder emptying by voiding (100 × micturition volume / bladder capacity). Intravesical pressure was measured at six points: Baseline pressure was taken as pressure before the pump was turned on and filling started, threshold pressure was taken as pressure at the take-off of the voiding contraction, opening pressure was taken as peak pressure before the onset of phasic external urethral sphincter (EUS) activity and voiding, peak voiding pressure was taken as maximal pressure during phasic EUS activity and voiding, closing pressure was taken as peak pressure after phasic EUS activity and voiding, and post-contraction pressure was taken as pressure after the micturition contraction was complete and the pump was turned off. Phasic activity of the EUS was assessed by measurement of “high-frequency oscillations” (28) in the intravesical pressure record. The number of EUS contractions (phasic number, Pn) was counted as the number of high-frequency oscillations and the duration over which they occurred (phasic duration, Pd) were measured as the aggregate duration of high-frequency oscillations per void. Discernment of high-frequency oscillations was made easier by bandpass filtering intravesical pressure with high- and low-frequency cut-offs of 25 and 3 Hz (see Figure 1). The frequency of EUS contractions (phasic frequency, Pf) was taken as Pn/Pd. Finally, contraction duration was estimated as the time between take-off of the voiding contraction and the attainment of post-contraction pressure, and nonphasic contraction duration was taken as contraction duration minus phasic duration.
Statistical methods
Linear mixed effects models (29), with mouse as a random intercept, were used to determine the significance of differences among the means of the four groups. The 10 cystometrograms performed on each mouse were nested within that mouse in the analysis. Comparisons among groups were carried out only if the omnibus F test was significant (P ≤ 0.05). Data transformations were used as necessary to satisfy the statistical assumptions of constant variance and normality; we used the Box-Cox power transformations as obtained from the boxcox() function in the R package MASS (30). The R programming language was used for all statistical analyses. Linear mixed models were fit using R package NLME (29, 31); group comparisons were carried out using the R package gmodels (32).
Results of significance testing are presented in Table II. Comparisons of phenotypes without respect to treatment (column 1) and of treatments without respect to phenotype (column 2) were performed when the phenotype-by-treatment interaction was not significant. Individual group comparisons (last 4 columns of Table II) were only performed when the phenotype-by-treatment interaction was significant.
Table 2. Results of Significance Testing, Separated According to the Significance of the Interaction Between Phenotype (Control or DM) and Treatment (Saline or AngII).
| Interaction Insignificant | Interaction Significant | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| DM vs. Control | AngII vs. Saline | |||||
|
|
|
|||||
| DM vs. Control | AngII vs. Saline | Saline | AngII | Control | DM | |
| MV | <0.001 | 0.71 | -- | -- | -- | -- |
| RV | -- | -- | <0.001 | 0.22 | 0.94 | <0.001 |
| Capacity | -- | -- | <0.001 | 0.003 | 0.15 | 0.11 |
| VE | 0.49 | 0.009 | -- | -- | -- | -- |
| conDur | -- | -- | <0.001 | <0.001 | 0.73 | 0.005 |
| Peak void P | 0.23 | 0.008 | -- | -- | -- | -- |
| Pn | <0.001 | 0.85 | -- | -- | -- | -- |
| Pd | <0.001 | 0.21 | -- | -- | -- | -- |
| Pf | <0.001 | 0.04 | -- | -- | -- | -- |
| conDur-Pd | -- | -- | <0.001 | <0.001 | 0.70 | 0.002 |
Significant differences are shown in bold italic type. DM, diabetic; AngII, angiotensinII; MV, micturition volume; RV, residual volume; VE, voiding efficiency; conDur, contraction duration; peak void P, peak voiding pressure; Pn, phasic number; Pd, phasic duration; and Pf, phasic frequency.
Results
Demographic variables (Table I)
Blood glucose levels are shown in Table I, and ranged from 68-159 mg/dL in control and 303-502 mg/dL in diabetic mice. Body mass. Phenotype by treatment interaction was not significant (P = 0.69). Body mass was 9% lower in Ins2(Akita) mice than control mice (P = 0.017), and was 6% lower in AngII-treated than vehicle-treated mice (P = 0.048). Bladder mass. Phenotype by treatment interaction was not significant (P = 0.48). Bladder mass was lower in control mice than in Akita mice (P < 0.001). There was no statistically significant difference between saline and AngII treatments (P = 0.10). Because bladder mass may depend in part on body mass, we adjusted for the effect of body mass by refitting the model using body mass as a covariable; this did not change the significance of the results (for phenotype, P < 0.001; for treatment, P = 0.24). Cardiac mass. Phenotype by treatment interaction was not significant (P = 0.55). Cardiac mass was not significantly different between controls and Akita mice (P = 0.58). However, 4 weeks of AngII treatment significantly increased cardiac mass ((P = 0.007), as expected with the dose rate employed. Because cardiac mass may depend in part on body mass, we refitted the model with body mass as a covariable; this did not change the conclusions (for phenotype, P = 0.95; for treatment, P < 0.001).
Cystometric variables (Figures 1 and 2 and Table II)
Figure 2.
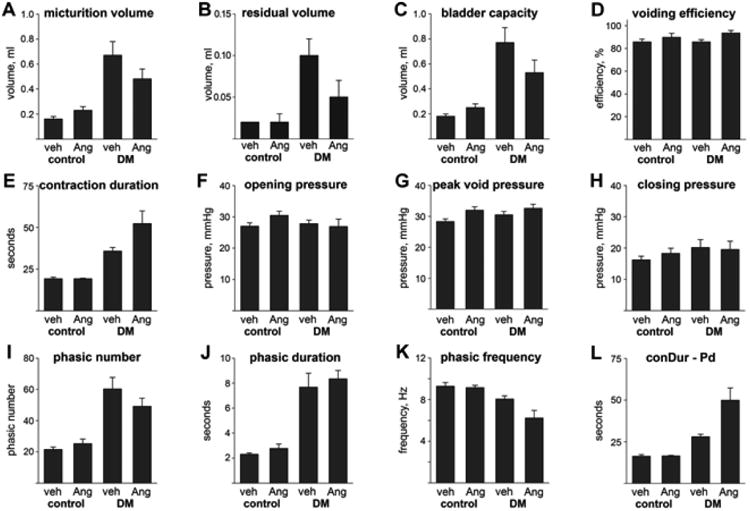
Cystometric variables in the four groups of mice (mean ± SEM). Abbreviations: veh = vehicle, Ang = AngII, DM = diabetes mellitus, conDur = contraction duration, and Pd = phasic duration. Significant differences are indicated in Table II.
Typical cystometrograms are shown in Figure 1 (control in A1, diabetic in B1, both saline-treated). The filling rate was roughly three-fold higher for the diabetic mouse, so the capacity was roughly three-fold higher despite the similar filling time. Both cystometrograms show large fluctuations in pressure during filling which are often called nonvoiding contractions; they were often broader in diabetic mice as they are here. (Some of these pressure fluctuations may be the consequence of abdominal contractions rather than bladder contractions.) Panels A2 and B2 show intravesical pressure in a 20-second period including the time of voiding. Panels A3 and B3 show the same 20-second period but with bandpass filtering to make the high-frequency oscillations of pressure corresponding to phasic opening and closing of the external urethral sphincter more obvious. In this instance, the control mouse has a higher phasic frequency (shorter individual high-frequency oscillations) and a shorter phasic duration.
Cystometric volumes and voiding efficiency (Fig 2A-D)
Micturition volume (Fig. 2A) was significantly greater in diabetic mice (P < 0.001) and not significantly altered by treatment (P = 0.71). Residual volume (Fig. 2B) depended on both phenotype and treatment, with a significant interaction (P = 0.01). Residual volume was greater in saline-treated than in AngII-treated diabetic mice (P = 0.001) but not control mice (P = 0.94), and was greater in diabetic than in control mice following treatment with saline (P < 0.001) but not AngII (P = 0.22). Bladder capacity (Fig. 2C) was significantly elevated in diabetic mice whether they were treated with saline (P < 0.001) or AngII (P = 0.003), with the magnitude of the elevation depending significantly on treatment (being lower with AngII treatment; P = 0.036). Within phenotypes, bladder capacity was not significantly affected by treatment (P = 0.15 for control and 0.11 for diabetic mice). Voiding efficiency was quite high in all four groups (Fig. 2D). Nonetheless, AngII treatment resulted in significantly higher voiding efficiency than saline treatment (P = 0.009), independent of phenotype (P = 0.49).
Contraction duration and intravesical pressures (Fig. 2E-H)
Contraction duration (Fig. 2E) depended on both phenotype and treatment, with a significant interaction (P < 0.036). Thus, contraction duration was longer in diabetic than in control mice whether saline- or AngII-treated (both P < 0.001), and contraction duration was longer in diabetic mice treated with AngII rather than saline (P = 0.005) though not in control mice treated with AngII rather than saline (P = 0.73). Intravesical pressure was significantly changed among groups at only one of the six times measured: peak voiding pressure was significantly higher with AngII than with saline treatment (P = 0.008), and not affected by phenotype (P = 0.23).
Phasic variables (Fig. 2I-L)
Phasic number (Fig. 2I) was significantly higher in diabetic than in control mice (P < 0.001) and was not significantly affected by treatment (P = 0.85). Phasic duration (Fig. 2J) was also significantly higher in diabetic than in control mice (P < 0.001), and not significantly affected by treatment (P = 0.21). Finally, phasic frequency (Fig. 2K) was significantly lower in diabetic than control mice (P < 0.001) and in AngII-treated than saline-treated mice (P = 0.04).
The difference between contraction duration and phasic duration, i.e., nonphasic contraction duration, was examined to assess whether the change in phasic duration was alone responsible for changes in contraction duration. Nonphasic contraction duration depended on both phenotype and treatment, with a significant interaction (P = 0.029). Thus, nonphasic contraction duration was significantly greater in AngII-treated than in saline-treated diabetic mice (P = 0.002) but not control mice (P = 0.70), and significantly greater in diabetic than in control mice following treatment with either saline (P < 0.001) or AngII (P < 0.001).
Discussion
Effects of Ins2(Akita) diabetes
Ins2(Akita) diabetes was associated with significant increases in bladder mass, micturition volume, bladder capacity, contraction duration, nonphasic contraction duration, phasic number, and phasic duration, and with a significant decrease in phasic frequency regardless of treatment. Residual volumes significantly increased in saline- but not AngII-treated mice. Voiding efficiency and a variety of measured pressures were unchanged across treatments. Overall, the observed changes are consistent with compensated bladder function, where the increased diuresis attending diabetes induces bladder hypertrophy (2, 3) but a variety of compensatory mechanisms preserve effective function as manifest by the unchanged voiding efficiency in the face of increased bladder capacity. Thus, increased contraction duration (and phasic duration in particular) and phasic number with maintained pressure generation ensured that essentially complete voiding was still obtained despite the increased workload. Electromyographic study of EUS electromyographic activity would be required to determine whether the decrease in phasic frequency was the result of an increase in duration of bursts, pauses, or both. Interestingly, a direct relationship between pause duration and higher voiding efficiency has been shown (33, 34).
We expected that 20 wk 129/SvEv-Ins2(Akita) mice would have decompensated bladder function secondary to prolonged hyperglycemia comparable to that seen in the mouse STZ model with decompensation. Such was clearly not the case. This may be because blood glucose levels at 20 weeks in our 129/SvEv-Ins2(Akita) mice (381 ± 24 mg/dL) were lower than those reported in other chronic studies of Ins2(Akita) mice (around 500-600 in C57BL/6-Ins2(Akita) mice (3, 22, 25) and around 500 mg/dL in 129/SvEv-Ins2(Akita) mice (25)). Additionally, the rate of development of hyperglycemia may have been slower in our 129/SvEv-Ins2(Akita) mice than in mice of other studies. Alternatively, the difference in bladder function between the two models may be the consequence of a strain difference in the consequences of hyperglycemia (e.g., in degree of diabetic neuropathy) an important possibility warranting further investigation. It is noteworthy that some other models of diabetes also show considerably compensated LUT function (35).
Effects of chronic AngII treatment
Chronic AngII treatment induced significant cardiac hypertrophy as expected, confirming 700 ng/kg/min to be an effective dose. The absence of such a difference in untreated control vs. diabetic mice suggests that any diabetes-induced change in AngII was small. Independent of glycemic state, chronic AngII treatment slightly but significantly increased voiding efficiency and peak voiding pressure and decreased phasic frequency. AngII effects on peak intravesical pressure could arise from augmented contraction of either the bladder or the outlet tract. In diabetic but not control mice, chronic AngII treatment also significantly decreased residual volume and increased contraction duration and nonphasic contraction duration. Except for the increase in nonphasic contraction duration, these results are all in the direction of compensatory mechanisms to produce more efficient voiding, particularly in diabetic mice, and are in line with the contractility-promoting actions of AngII. For this reason, and because AT1 receptors probably greatly outnumber AT2 receptors in rat bladder (12), the AngII actions probably reflect binding to AT1 receptors. Expression of AT1 and AT2 receptors may change with chronic AngII exposure (36) or with chronic diabetes (10) and is an important avenue for investigation.
Apart from direct effects on lower urinary tract AT1 and AT2 receptors, chronic AngII likely increased urine output as recently shown in C57BL/6 mice (23) and may have thereby altered lower urinary tract function. On the other hand, increased urine output induced with dietary sucrose causes bladder hypertrophy (3) whereas AngII treatment in this study had no such effect in either control or Ins2(Akita) mice (Table 1). Further study of this question is warranted.
Conclusions
Against our expectations, the Ins2(Akita) diabetic mice had compensated LUT function at 20 wk of age. It is very interesting that even with compensated LUT function, AngII had beneficial effects on LUT function which resulted in increased voiding efficiency. Future studies should be conducted to determine whether AngII can rescue the decompensated LUT function found in other models of diabetic uropathy such as the STZ-treated rodent (2, 3). Use of AT1 receptor agonists would likely be contraindicated in hypertensive diabetic patients. Nonetheless, reduced doses of AngII-lowering drugs in combination with other antihypertensive treatments could potentially allow for blood pressure control with reduced LUT dysfunction.
Acknowledgments
Funding: This work was funded by Diabetic Complications Consortium grants DK076169 and DK076136 and by VA Merit Review BX000334. The work was additionally supported with resources and the use of facilities at the Durham Veterans Affairs Medical Center, Durham, NC.
Literature cited
- 1.Daneshgari F, Liu G, Birder L, Hanna-Mitchell AT, Chacko S. Diabetic bladder dysfunction: current translational knowledge. J Urol. 2009;182:S18–26. doi: 10.1016/j.juro.2009.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daneshgari F, Liu G, Imrey PB. Time dependent changes in diabetic cystopathy in rats include compensated and decompensated bladder function. J Urol. 2006;176:380–6. doi: 10.1016/S0022-5347(06)00582-9. [DOI] [PubMed] [Google Scholar]
- 3.Daneshgari F, Huang X, Liu G, Bena J, Saffore L, Powell CT. Temporal differences in bladder dysfunction caused by diabetes, diuresis, and treated diabetes in mice. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1728–35. doi: 10.1152/ajpregu.00654.2005. [DOI] [PubMed] [Google Scholar]
- 4.Aguilar D, Solomon SD. ACE inhibitors and angiotensin receptor antagonists and the incidence of new-onset diabetes mellitus: an emerging theme. 2006;66:1169–77. doi: 10.2165/00003495-200666090-00001. [DOI] [PubMed] [Google Scholar]
- 5.Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. 2007;112:375–84. doi: 10.1042/CS20060247. [DOI] [PubMed] [Google Scholar]
- 6.Lee JH, Xia S, Ragolia L. Upregulation of AT2 receptor and iNOS impairs angiotensin II-induced contraction without endothelium influence in young normotensive diabetic rats. 2008;295:R144–54. doi: 10.1152/ajpregu.00191.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. 2000;52:11–34. [PubMed] [Google Scholar]
- 8.Perret-Guillaume C, Joly L, Jankowski P, Benetos A. Benefits of the RAS blockade: clinical evidence before the ONTARGET study. 2009;27(Suppl 2):S3–7. doi: 10.1097/01.hjh.0000354511.14086.f1. [DOI] [PubMed] [Google Scholar]
- 9.Matsubara H. Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. 1998;83:1182–91. doi: 10.1161/01.res.83.12.1182. [DOI] [PubMed] [Google Scholar]
- 10.Hatada T, Noguchi M, Tobu S, Mori K, Matsuo M, Kanetaka H. Differential expression of angiotensin II type-1 receptor in the rat detrusor muscle with partial bladder outlet obstruction and diabetes mellitus model. 2007 [Google Scholar]
- 11.Yamada S, Takeuchi C, Oyunzul L, Ito Y. Bladder angiotensin-II receptors: characterization and alteration in bladder outlet obstruction. 2009;55:482–9. doi: 10.1016/j.eururo.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 12.Nakazawa R, Tanaka M, Takahashi T, Kobayashi S, Iwamoto T. Effects of castration and testosterone administration on angiotensin II receptor mRNA expression and apoptosis-related proteins in rat urinary bladder. 2007;54:211–9. doi: 10.1507/endocrj.k06-134. [DOI] [PubMed] [Google Scholar]
- 13.Andersson KE, Hedlund H, Stahl M. Contractions induced by angiotensin I, angiotensin II and bradykinin in isolated smooth muscle from the human detrusor. 1992;145:253–9. doi: 10.1111/j.1748-1716.1992.tb09362.x. [DOI] [PubMed] [Google Scholar]
- 14.Lam DS, Dias LS, Moore KH, Burcher E. Angiotensin II in child urinary bladder: functional and autoradiographic studies. 2000;86:494–501. doi: 10.1046/j.1464-410x.2000.00771.x. [DOI] [PubMed] [Google Scholar]
- 15.Waldeck K, Lindberg BF, Persson K, Andersson KE. Characterization of angiotensin II formation in human isolated bladder by selective inhibitors of ACE and human chymase: a functional and biochemical study. 1997;121:1081–6. doi: 10.1038/sj.bjp.0701240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acevedo CG, Contreras E. Possible involvement of adenine nucleotides in the neurotransmission of the mouse urinary bladder. 1985;82:357–61. doi: 10.1016/0742-8413(85)90176-8. [DOI] [PubMed] [Google Scholar]
- 17.Tanabe N, Ueno A, Tsujimoto G. Angiotensin II receptors in the rat urinary bladder smooth muscle: type 1 subtype receptors mediate contractile responses. 1993;150:1056–9. doi: 10.1016/s0022-5347(17)35685-9. [DOI] [PubMed] [Google Scholar]
- 18.Cristofaro V, Peters CA, Yalla SV, Sullivan MP. Smooth muscle caveolae differentially regulate specific agonist induced bladder contractions. 2007;26:71–80. doi: 10.1002/nau.20361. [DOI] [PubMed] [Google Scholar]
- 19.Lin HK, Cowan R, Moore P, Zhang Y, Yang Q, Peterson JA, Jr, et al. Characterization of neuropathic bladder smooth muscle cells in culture. 2004;171:1348–52. doi: 10.1097/01.ju.0000108800.47594.8b. [DOI] [PubMed] [Google Scholar]
- 20.Cheng EY, Decker RS, Lee C. Role of angiotensin II in bladder smooth muscle growth and function. In: Baskin LS, Hayward SW, editors. Advances in bladder research. New York: Plenum; 1999. pp. 183–91. [DOI] [PubMed] [Google Scholar]
- 21.Hatada T, Noguchi M, Tobu S, Mori K, Matsuo M, Kanetaka H. Differential expression of angiotensin II type-1 receptor in the rat detrusor muscle with partial bladder outlet obstruction and diabetes mellitus model (Abstract 390) 37th Annual Meeting of the International Continence Society. 2007 [Google Scholar]
- 22.Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. 1997;46:887–94. doi: 10.2337/diab.46.5.887. [DOI] [PubMed] [Google Scholar]
- 23.Jennings BL, Anderson LJ, Estes AM, Yaghini FA, Fang XR, Porter J, et al. Cytochrome P450 1B1 contributes to renal dysfunction and damage caused by angiotensin II in mice. Hypertension. 2012;59:348–54. doi: 10.1161/HYPERTENSIONAHA.111.183301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang MZ, Yao B, Wang S, Fan X, Wu G, Yang H, et al. Intrarenal dopamine deficiency leads to hypertension and decreased longevity in mice. J Clin Invest. 2011;121:2845–54. doi: 10.1172/JCI57324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurley SB, Mach CL, Stegbauer J, Yang J, Snow KP, Hu A, et al. Influence of genetic background on albuminuria and kidney injury in Ins2(+/C96Y) (Akita) mice. 2010;298:F788–95. doi: 10.1152/ajprenal.90515.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yaksh TL, Durant PA, Brent CR. Micturition in rats: a chronic model for study of bladder function and effect of anesthetics. Am J Physiol Regul Integr Comp Physiol. 1986;251:R1177–85. doi: 10.1152/ajpregu.1986.251.6.R1177. [DOI] [PubMed] [Google Scholar]
- 27.Morikawa K, Ichihashi M, Kakiuchi M, Yamauchi T, Kato H, Ito Y, et al. Effects of various drugs on bladder function in conscious rats. Jpn J Pharmacol. 1989;50:369–76. doi: 10.1254/jjp.50.369. [DOI] [PubMed] [Google Scholar]
- 28.Conte B, Maggi CA, Parlani M, Lopez G, Manzini S, Giachetti A. Simultaneous recording of vesical and urethral pressure in urethane-anesthetized rats: effect of neuromuscular blocking agents on the activity of the external urethral sphincter. J Pharmacol Methods. 1991;26:161–71. doi: 10.1016/0160-5402(91)90041-3. [DOI] [PubMed] [Google Scholar]
- 29.Pinheiro JC, Bates DM. Mixed-effects models in S and S-plus. New York: Springer-Verlag; 2000. [Google Scholar]
- 30.Venables WN, Ripley BD. Modern Applied Statistics with S. Fourth. New York: Springer; 2002. [Google Scholar]
- 31.Pinheiro J, Bates D, DebRoy S, Sarkar D. nlme: Linear and Nonlinear Mixed Effects Models, R package version 3.1-87. 2008 [Google Scholar]
- 32.Warnes GR. gmodels: Various R programming tools for model fitting. 2007 Available from: http://cran.r-project.org/web/packages/gmodels/index.html.
- 33.Cheng CL, de Groat WC. Effect of ovariectomy on external urethral sphincter activity in anesthetized female rats. J Urol. 2011;186:334–40. doi: 10.1016/j.juro.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng CL, de Groat WC. Role of 5-HT1A receptors in control of lower urinary tract function in anesthetized rats. Am J Physiol Renal Physiol. 2010;298:F771–8. doi: 10.1152/ajprenal.00266.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aizawa N, Homma Y, Igawa Y. Characteristics of lower urinary tract dysfunction and bladder afferent nerve properties in type 2 diabetic Goto-Kakizaki rats. J Urol. 2013;189:1580–7. doi: 10.1016/j.juro.2012.10.060. [DOI] [PubMed] [Google Scholar]
- 36.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]