

Abstract
Dendritic cells are highly specialized antigen-presenting cells (APC), which may be isolated or generated from human blood mononuclear cells. Although mature blood dendritic cells normally represent ~0.2% of human blood mononuclear cells, their frequency can be greatly increased using the cell enrichment methods described in this unit. More highly purified dendritic cell preparations can be obtained from these populations by sorting of fluorescence-labeled cells. Alternatively, dendritic cells can be generated from monocytes by culture with the appropriate cytokines, as described here. In addition, a negative selection approach is provided that may be employed to generate cell preparations that have been depleted of dendritic cells to be used for comparison in functional studies.
Subject: Immunology, Immunologic Study in Humans, In Vitro Assays for Immune Cell Function, Immune Disease, Innate Immunity, Antigen Presentation
Dendritic cells are highly specialized antigen-presenting cells (APC; see Commentary), which may be isolated or generated from human blood mononuclear cells. Although mature blood dendritic cells normally represent ~0.2% of human blood mononuclear cells, their frequency can be greatly increased by the use of cell enrichment methods (see Basic Protocol 1 and Alternate Protocol). More highly purified dendritic cell preparations can be obtained from these populations by sorting of fluorescence-labeled cells (UNITS 5.3 & 5.4). Alternatively, dendritic cells can be generated from monocytes by culture with the appropriate cytokines (see Basic Protocol 2). In addition, a negative selection approach may be employed to generate cell preparations that have been depleted of dendritic cells (see Basic Protocol 3) to be used for comparison in functional studies.
The ex vivo monocyte-derived DCs are phenotypically and functionally similar to DCs present in blood. Importantly, they can be generated in significant numbers, which is a requisite for clinical studies. This ex vivo DC generation protocol has facilitated clinical vaccine studies using DCs pulsed with antigen; which can be formulated as peptides, proteins, cell lysates, apoptotic tumor cells, DNA and RNA. In addition to the protocol (see Basic Protocol 2) to generate DCs from monocytes, we describe a large-scale version of this procedure that can be used to generate DC-based vaccines from a leukapheresis for clinical studies.
CAUTION: Appropriate caution is advised in working with large volumes of human blood, particularly if from unknown, untested donors. Biosafety practices must be followed (see Chapter 7 introduction). All work with human cells and procedures may need appropriate institutional approval before initiation.
NOTE: All procedures are to be carried out using sterile tissue culture techniques with sterile solutions and equipment.
NOTE: All incubations are performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified.
BASIC PROTOCOL 1: ISOLATION OF DENDRITIC CELLS FROM HUMAN BLOOD MONONUCLEAR CELLS
A peripheral cell population can be enriched for dendritic cells by depletion of T cells and adherent cells. The preparation is then subjected to density gradient centrifugation over metrizamide to isolate low buoyant density cells. The resulting population contains 20% to 80% dendritic cells and is largely free of lymphocytes.
Materials
Leukocyte-enriched leukapheresis packs (leukopaks; i.e., 20- to 50-ml) or buffy coats, ≤ 24 hr old (from blood bank or North American Biologicals; see Critical Parameters)
Complete RPMI-10 (APPENDIX 2), room temperature and 37°C
RPMI 1640
14.5% metrizamide solution (see recipe), room temperature
Dulbecco’s PBS, Ca2+ and Mg2+ free, 4°C
Primary antibody for flow cytometry analysis (see Table 7.32.1; optional)
Fluorescence-labeled secondary antibody for flow cytometry analysis (optional)
15- and 50-ml conical polypropylene centrifuge tubes
9-in. (~23-cm) Pasteur pipets, with and without cotton plugs
100-mm tissue culture plates
Sorvall H1000B rotor (or equivalent)
Additional reagents and equipment for isolating mononuclear cells (UNIT 7.1), counting viable cells (APPENDIX 3B), rosetting with sheep red blood cells (UNIT 7.2), and flow cytometric analysis (UNITS 5.3 & 5.4) or assessment of functional activity by mixed leukocyte reaction (UNIT 7.10; both procedures optional)
Table 7.32.1.
Monoclonal Antibodies Useful in Routine Flow Cytometric Analysis of Human Blood Dendritic Cells
| Antigena | Cell type where primarily expressed | Reactivity with dendritic cells |
|---|---|---|
| MHC class II | Leukocytes | +++++ |
| CD1c | Thymocytes | ++ |
| CD33 | Monocytes | ++ |
| CD40 | B lymphocytes | + |
| CD83 | Dendritic cells | ++ |
| CD80 | Antigen-presenting cells | ++ |
| CD86 | Antigen-presenting cells | ++ |
| CD3 | T lymphocytes | None |
| CD14 | Monocytes | None |
| CD19 | B lymphocytes | None |
| CD56 | NK cells | None |
All of the antigens listed are identified by MAbs that are widely available commercially.
MAb reactivity is indicated on a relative linear scale, with +++++ indicating the highest levels of antigen expression.
Isolate blood mononuclear cells
-
1
Isolate blood mononuclear cells from a leukapheresis pack or buffy coat preparation by Ficoll-Paque density gradient or other suitable method as described in UNIT 7.1. Determine the total number of viable mononuclear cells by trypan blue exclusion (APPENDIX 3B) or other standard procedures.
The usual number of cells obtained at this stage is 6–10 × 108 cells.Prompt processing reduces cellular and platelet aggregation and increases cell yield and purity. If platelet contamination is severe, wash the cells and centrifuge at a lower speed, as excessive platelet contamination will lower the purity of the dendritic cells due to the formation of cellular aggregates. The cell isolation procedures should be carefully monitored by cell counting using a hemacytometer to ensure that there is no significant loss of mononuclear cells. -
2
Adjust the cell concentration to 1–2 × 107 cells/ml in complete RPMI-10.
-
3
Deplete the resuspended cells of T cells by E-rosetting with sheep red blood cells (SRBC) as described in UNIT 7.2, or using other standard methods such as magnetic bead-mediated T cell depletion.
In applying this method, it is advantageous to use 50-ml conical centrifuge tubes because of the large volume of cell suspension. AET-treated SRBC give the best results. Incubate the pelleted mononuclear cells and SRBC for 20 min on ice. -
4
Transfer the interface cells to a 50-ml conical centrifuge tube using a Pasteur pipet. Dilute the density gradient medium four-fold with unsupplemented RPMI 1640 and centrifuge 10 min at 450 × g, room temperature. Remove the supernatant fluid and resuspend the cells in complete RPMI-10. Repeat the wash, then adjust the cell concentration to 1 × 107 cells/ml.
The usual number of E-rosette-negative cells obtained at this stage is 3–6 × 107 cells. The cells consist of B cells, monocytes, macrophages, and dendritic cells.
Deplete monocytes from the mononuclear cell preparation
-
5
Plate the T cell–depleted mononuclear cell suspension into 100-mm tissue culture plates using 10 ml of the cell suspension per plate. Incubate the plates overnight at 37°C to allow monocytes to adhere to the plastic and to allow the dendritic cells to further differentiate into mature dendritic cells.
Plating more densely than ~1 × 108 cells per plate reduces adherence and final yield. During the overnight incubation, initially adherent dendritic cells (and some contaminating monocytes) detach from the plastic surface and float into the overlying medium. -
6
The next morning, swirl the plates gently in a figure 8 to resuspend the nonadherent cells. Collect the nonadherent cells and transfer them to a fresh 100-mm tissue culture plate.
Repeat until all plates have been harvested and the cells replated. It may be helpful to very gently wash each plate with 3 ml fresh medium prewarmed to 37°C to remove additional nonadherent cells. -
7
Incubate the newly plated cells 30 min at 37°C to remove additional monocytes. Swirl the plates again and collect the nonadherent cells in the medium.
Repeat this process again, if necessary, to reduce the frequency of monocytes; however, there is usually little further gain after three adherence steps. The frequency of residual adherent cells can be visualized by examining the plates by phase-contrast microscopy using an inverted microscope. The majority of residual cells in each plate should be adherent, flattened monocytes. If there are a lot of residual nonadherent cells, wash the plates very gently again and recover the wash medium containing the cells. -
8
Harvest the T cell– and monocyte–depleted mononuclear cell preparation and pellet the cells 5 min at 200 × g, room temperature.
-
9
Discard the supernatant fluid and resuspend the cell preparation in complete RPMI-10. Count the number of cells and adjust the cell concentration to ≤1 × 107 cells/ml.
Enrich for dendritic cells by metrizamide density gradient centrifugation
-
10
Add 4 ml sterile 14.5% metrizamide solution to a 15-ml conical centrifuge tube at room temperature. Prepare enough tubes to accommodate the full volume of cell suspension that has to be separated.
-
11
Gently overlay the metrizamide cushion with 8 ml of the mononuclear cell suspension, forming a sharp interface. Centrifuge the gradient 10 min at 800 × g (1800 rpm in Sorvall H1000B rotor), room temperature. Accelerate the centrifuge slowly and keep the brake turned off.
Alternatively, the metrizamide solution may be layered underneath the mononuclear cell preparation. -
12
After centrifugation, aspirate off the top volume of culture medium until ~0.5 in. (~1 cm) above the interface. Collect the cells that localize at the interface with a cotton-plugged Pasteur pipet. Carefully collect the cells from the interface region, taking the culture medium and the top 1 ml of metrizamide.
Until one becomes proficient at these procedures, one should also collect the cells that pellet through the metrizamide gradient just in case something has gone wrong during the isolation procedures. This avoids the danger of losing everything at this step. The cells can be phenotyped to determine the frequency of dendritic cells and other cell types present in each fraction. -
13
Pool the cells from the metrizamide gradients into a single fresh 50-ml conical tube. Fill the 50-ml tube with cold PBS to dilute the metrizamide at least two-fold and mix the solution by gentle inversion of the tube.
-
14
Pellet the cells by centrifugation for 10 min at 450 × g. Wash the isolated cells twice with complete RPMI-10, count the viable cells by trypan blue exclusion (APPENDIX 3B), and resuspend at 1 × 107 cells/ml in fresh complete RPMI-10.
-
15
Further deplete monocytes from the dendritic cell preparation by plating the cells in 100-mm tissue culture plates, using 10 ml of cell suspension per plate until the entire volume has been plated. Incubate 1 hr at 37°C.
-
16
Gently swirl the plates and remove the supernatant fluid containing nonadherent cells.
This is the dendritic cell–enriched fraction. Monitor the plating process by light microscopy to insure monocyte depletion is occurring without undue loss of non-adherent dendritic cells. -
17
Pellet the cells by centrifugation for 5 min at 450 × g, room temperature.
The usual yield of cells at this stage is 5–10 × 106 cells. Dendritic cells should comprise 20% to 80% of the population. While this purity may suffice for many kinds of experiments, dendritic cells may be further purified by procedures described below. -
18
Determine the purity of dendritic cells by immunofluorescence staining with flow cytometry analysis (UNITS 5.3 & 5.4) using appropriate primary and secondary antibodies; by assessment of cellular morphology (see Fig. 7.32.1C, D); or by assessment of functional activity in mixed leukocyte reactions (UNIT 7.10).
Table 7.32.1 identifies primary antibodies specific for cell-surface antigens present on dendritic cells, and on the major contaminating cell populations obtained by routine purification. MHC class II, CD83, CD3, and CD14 constitute a recommended minimum panel. For the secondary antibody, use fluorescein-conjugated goat anti–mouse Ig antibodies.While the purity of dendritic cells can be estimated using a variety of techniques—including hemacytometer counts and immunostaining after cytocentrifugation onto glass slides—the methods described above are generally superior. Flow cytometry, especially, offers superior quantitation and consistency (Zhou and Tedder, 1995a).
Figure 7.32.1.

Generation and morphology of CD83+ dendritic cells. Monocytes cultured with GM-CSF/IL-4/TNF-α develop a dendritic cell morphology (A–C). Monocytes (CD14+) were isolated and cultured with cytokines for 2 (A) or 5 (B, C) days and examined by phase-contrast microscopy (A–B, 200× magnification; C, 400×). Representative transmission electron micrographs of (D–E) monocyte-derived CD83+ cells isolated by cell sorting from 7-day GM-CSF/IL-4/TNF-α cultures and (F) blood CD83+ dendritic cells (D, 1855× magnification; E-F, 5800×).
This figure, originally published in Zhou and Tedder (1996), is reprinted by permission of Proceedings of the National Academy of Sciences.
Representative phenotype of immature and mature dendritic cells. Phenotype and scatter profiles of monocyte-derived immature DCs (A). Phenotype and scatter profile of monocyte-derived mature DCs (B). Data is depicted as a 2-color analysis using PerCP-labeled class II versus PE-labeled antibodies as indicated in the figure. Peridinin Chlorophyll Protein Complex, PerCP; Phycoerythrin, PE.
ALTERNATE PROTOCOL 1: IMMUNOMAGNETIC ISOLATION OF DENDRITIC CELLS
A preparation of cells enriched for CD83+ dendritic cells as described in Basic Protocol 1 can be isolated using magnetic beads from a peripheral blood cell population depleted of T cells and adherent cells (see UNIT 7.4). Dendritic cells prepared by this method generally exhibit morphological and functional capabilities similar to those of dendritic cells prepared by negative selection (see Basic Protocol 1). It is not possible to isolate CD83+ dendritic cells straight from blood mononuclear cell preparations because their frequency is too low.
Additional Materials (also see Basic Protocol 1)
MAb mix including anti-CD3, -CD14, -CD19, and -CD56 mAbs, each at 10× saturating concentrations (Table 7.32.1)
Goat anti–mouse IgG–coated magnetic beads (Dynabeads), washed (see UNIT 7.4)
Anti-CD83 MAb at a 10× saturating concentration
Clinical rotator
Magnetic separation apparatus
Deplete B cells, T cells, and other mononuclear cells
-
1
Isolate blood mononuclear cells from a leukapheresis pack or buffy coat preparation by density gradient centrifugation (see Basic Protocol 1, step 1). Adjust the cell concentration to 2 × 107 cells/ml of complete RPMI-10 in 15-ml conical polypropylene centrifuge tubes.
The size of the container used for cell separation will depend the magnetic separation apparatus used; adequate times for cell separation must be considered to accommodate larger cell volumes or tube sizes. -
2
Add 1/10 vol of 10× MAb mix at 1× concentration to the mononuclear cell suspension and incubate 30 min with end-over-end rotation at 6 to 10 rpm, 4°C.
-
3
Wash the cells twice by centrifugation and resuspend with fresh medium to remove any unbound MAb, and transfer the cells to a fresh polypropylene centrifuge tube at 2 × 107 cells/ml of complete RPMI-10.
-
4
Deplete the resuspended cells of lymphocytes, monocytes, and NK cells by adding a 2-ml suspension of washed magnetic beads to the cell suspension and rotating 1 hr at 6 to 10 rpm, 4°C.
-
5
Separate the cells labeled with MAb and coated with beads using the magnetic separation apparatus. Allow ~5 min for the beads (and attached cells) to accumulate adjacent to the magnet.
-
6
Transfer the unbound cell suspension to a fresh tube and perform a second magnetic separation. Count the cells and resuspend at 2 × 107 cells/ml of complete RPMI-10.
-
7
Repeat steps 4 and 5 and resuspend the remaining cells at 2 × 107 cells/ml of complete RPMI-10.
This cell population will be greatly enriched for dendritic cells, but they will represent <5% of the mononuclear cell population. -
8
Plate the mononuclear cell suspension into 100-mm tissue culture plates using 10 ml of the cell suspension per plate. Incubate the dishes overnight at 37°C to allow monocytes to adhere to the plastic and to allow the dendritic cells to further differentiate into mature dendritic cells.
Enrich for dendritic cells by positive selection
-
9
The next morning, swirl the plates gently in a figure 8 to resuspend the nonadherent cells, harvest the lymphocyte- and monocyte-depleted mononuclear cell preparation into a polypropylene centrifuge tube, and pellet the cells 5 min at 200 × g, room temperature.
It may be helpful to very gently wash each plate with 3 ml of fresh prewarmed medium to remove additional nonadherent cells. -
10
Discard the supernatant fluid and resuspend the cell preparation in complete RPMI-10. Count the number of cells and adjust the cell concentration to 1 × 107 cells/ml.
-
11
Add 1/10 vol of 10× anti-CD83 MAb to the mononuclear cell suspension and rotate 30 min at 6 to 10 rpm, 4°C.
-
12
Wash the cells twice by centrifugation and resuspend with fresh medium to remove any unbound MAb. Resuspend to a concentration of 1 × 107 cells/ml of complete RPMI-10, and transfer the cells to a fresh polypropylene centrifuge tube.
-
13
Isolate CD83+ cells by adding a 1-ml suspension of washed magnetic beads to the cell suspension and rotating 1 hr at 6 to 10 rpm, 4°C.
-
14
Separate the cells coated with beads using the magnetic apparatus. After 5 min, remove the cells not bound by the magnet using an aspirator tip placed at the bottom of the flask or tube.
-
15
Resuspend the bound CD83+ cells in fresh complete RPMI-10 to the volume used in step 10. Resuspend the cells thoroughly and perform a second magnetic separation. Resuspend the recovered cells in fresh medium and count the cells.
Additional cycles of washing and magnetic isolation can be used to increase cell purity, but more than three cycles does not usually increase purity significantly. Dendritic cell populations isolated using these procedures are usually homogenous in morphologic appearance and display the morphologic features of dendritic cells. Procedures for removing magnetic beads from cells are outlined in UNIT 7.5. However, some of the beads attached to dendritic cells through CD83 are endocytosed with overnight culture and other methods produce only modest success at removing the beads. Flow cytometry analysis of isolated dendritic cells is not usually practical because of the attached beads.
BASIC PROTOCOL 2: GENERATION OF HUMAN DENDRITIC CELLS FROM MONOCYTES
A relatively homogenous population of functionally mature dendritic cells can be generated from CD14+ blood monocytes by incubating them with the proper cytokines (Zhou and Tedder, 1996). Under the conditions described, monocytes differentiate into dendritic cells without cell proliferation, so that the number of monocytes used is the determining factor for dendritic cell recovery. Because monocytes are much more plentiful than dendritic cells, this method can result in higher yields than those obtained using Basic Protocol 1.
Materials
Leukocyte-enriched leukapheresis packs (leukopaks; i.e., 20- to 50-ml) or buffy coats, ≤24 hr old (from blood bank or North American Biologicals; see Critical Parameters)
Complete RPMI-10 (APPENDIX 2), 37°C
0.2 mM EDTA in PBS, Ca2+ and Mg2+ free (APPENDIX 2)
14.5% metrizamide solution (see recipe), room temperature
Recombinant human granulocyte/macrophage colony stimulating factor (GM-CSF; see recipe)
Recombinant human IL-4 (see recipe)
Recombinant human TNF-α (see recipe)
Inverted phase-contrast microscope
Sorvall H1000B rotor (or equivalent)
15-ml conical polypropylene centrifuge tube
Tissue culture flask
Additional reagents and equipment for isolation of peripheral blood monocytes (UNIT 7.1) followed by adherence to plastic (UNIT 7.6)
Isolate blood monocytes by adherence to plastic
-
1
Isolate peripheral blood monocytes from a leukapheresis pack or buffy coat preparation using Ficoll-Paque density gradient centrifugation and plastic adherence (UNITS 7.1 & 7.6).
-
2
Gently aspirate the medium that contains the nonadherent cells from the plate and wash each plate very gently with warm complete RPMI-10 to remove the nonadherent cells. Examine each plate using an inverted phase-contrast microscope to determine the level of contamination with small round lymphocytes.
It will take a little practice to efficiently remove the nonadherent cells without removing dendritic cell precursors (CD14+ monocytes). A balance between lymphocyte contamination and optimal yields of dendritic cells is achieved by proper washing techniques. Trial and error is unfortunately the best guide until an investigator develops a feel for how gently to wash the plates. If lymphocytes are present, rewash the plates more vigorously, yet not so vigorously as to dislodge adherent monocytes. -
3
Remove adherent monocytes by gently scraping with a plastic cell scraper (UNIT 7.6). Alternatively, incubate the cells 10 min at 4°C in ice-cold 0.2 mM EDTA/PBS. Use a cotton-plugged Pasteur pipet with a pipet bulb to vigorously wash the adherent cells off the plate.
-
4
Pellet the harvested monocytes by centrifugation for 5 min at 450 × g, room temperature.
-
5
Adjust the cell density to <1 × 107 cells/ml in PBS containing 0.2 mM EDTA.
-
6
Overlay 8 ml of cell suspension on 4 ml of 14.5% metrizamide solution in a 15-ml conical centrifuge tube.
-
7
Centrifuge the metrizamide gradients 10 min at 800 × g, room temperature, to pellet contaminating lymphocytes.
-
8
Recover the interface cells and wash the recovered cells twice with 0.2 mM EDTA in PBS.
The harvested interface cells can be subjected to a second metrizamide gradient to remove residual lymphocytes if these represent a significant proportion of the cell population. -
9
Resuspend the cells in a small volume of complete RPMI-10 and determine the number of viable cells recovered.
Monocytes usually comprise 5% to 10% of the mononuclear cell population isolated directly from blood and are recovered proportionally at this stage given the loss in overall cell number with each isolation step. The level of cell recovery will vary between laboratories and with the degree of vigor used to wash the plates. Few monocytes should pellet in the metrizamide gradient and most of the pelleted cells should be lymphocytes.
Culture monocytes with cytokines to induce differentiation
-
10
Resuspend the monocytes to a density of 1 × 106 cells/ml in complete RPMI-10 containing 800 U/ml GM-CSF and 500 U/ml IL-4.
-
11
Incubate the monocytes in an appropriately sized tissue culture flask or other suitable vessel. Add fresh complete RPMI-10 and cytokines to the cultures on day 3.
Usually ~75% of spent medium is exchanged for fresh medium and cytokines. The monocytes will sediment to the bottom of the culture flasks (unless they are resuspended during movement of the tissue culture plates); thus, most of the culture medium can be aspirated without disturbing the cells. Cell differentiation within the cultures can be monitored by light microscopy.Monocytes cultured with GM-CSF and IL-4 generate small, distinctive clusters of cells that are primarily semi-adhered to the tissue culture plates (Fig. 7.32.1A, B). The cells are tightly clustered together by day 5, with some cells located between clusters (Fig. 7.32.1C). Some cells with a dendritic morphology can be observed. -
12
On day 5 of incubation, add fresh complete RPMI-10 containing 100 U/ml TNF-α as well as 800 U/ml GM-CSF and 500 U/ml IL-4.
-
13
On day 8 or 9 of incubation, resuspend the cells by vigorous pipetting to break up the cellular aggregates and to wash the semiadherent cells from the culture wells.
At this point the cells should be predominantly CD83+ dendritic cells, forming large clusters. The monocyte-derived dendritic cells should be morphologically homogenous and exhibit the characteristic cellular projections and motility of dendritic cells. While in culture, the cells will continually extend, retract, and reorient their cellular processes and veils.Recoveries vary, but typically range between 60% and 90% of the input monocytes. The cellular aggregates should be thoroughly disrupted before counting to obtain accurate counts. The cell preparations should remain >95% viable, as determined by trypan blue exclusion until day 14 to 15, when the cells increase in size and the cell clusters begin to deteriorate. -
14
Determine the frequency of dendritic cells (see Basic Protocol 1, step 18).
ALTERNATE PROTOCOL 2: GENERATION OF MONOCYTE-DERIVED DENDRITIC CELLS FROM A LEUKAPHERESIS PACK
A large number of autologous DCs are required for clinical studies using DC-based vaccines. Since DCs are found in trace amounts in human blood, isolating DCs from blood for clinical studies is cumbersome. Generating a homogenous population of DCs ex vivo that are functionally equivalent to DCs isolated from blood addresses this problem (Romani et al., 1996; Nair et al., 1998; Nair et al., 1999; Feuerstein et al., 2000; Spisek et al., 2001; Gilboa, 2007).
To generate DC-based vaccines, immature DCs are loaded with antigen followed by overnight maturation of the antigen-loaded DCs (Steinman and Banchereau, 2007; Gilboa, 2007; Ueno et al., 2010). The entire protocol to generate DCs from monocytes is a 7 day procedure followed by an additional 16–20 hours during which the antigen-loaded immature DCs are matured using a cocktail containing proinflammatory cytokines (IL-1β, IL-6, and TNFα) and prostaglandin E2 (PGE2) (Jonuleit et al., 1997; Lee et al., 2002). This cytokine mix is also referred to as the maturation cytokine cocktail.
The protocol below describes the ex vivo generation of monocyte-derived DCs from a leukopak. DC generation and maturation is also described in, “Differentiation of peripheral blood monocytes into dendritic cells”, in UNIT 22F.4.
Materials
Leukopak containing blood product
Sorvall RT-6000D centrifuge (or equivalent)
Ficoll-Paque Premium™ (GE Healthcare #17-5442-02)
PBS (Invitrogen #14190-144)
AIM V® cell culture media (Invitrogen #870112DK)
Cellstripper, enzyme-free cell dissociation buffer (Cellgro #25-056-CI)
Recombinant human GM-CSF (Berlex Laboratories, Inc, Leukine® (sargramostim), recombinant granulocyte-macrophage colony-stimulating factor [GM-CSF])
Recombinant human IL-4 (R&D Systems # 204-IL/CF)
Recombinant human TNF-α (R&D Systems, # 210-TA/CF)
Recombinant human IL-1β (R&D Systems, # 201-LB/CF)
Recombinant human IL-6 (R&D Systems, # 208-IL/CF)
Prostaglandin E2 (PGE2) (γ-irradiated powder, BioXtra from Sigma-Aldrich #P6532)
Human AB serum (Valley Biomedical #HP1022)
Cryoserv dimethyl sulfoxide (DMSO) (Baxter #88-850)
50% Dextrose (Abbott Laboratories NDC 0074-6648-02)
2 ml cryogenic vials (Corning #25704-2)
Trypan blue (Sigma #T-8154)
Hemocytometer (Hausser #1492)
T-75 and T-150 cell culture flasks
50 ml sterile polypropylene conical tubes
500 ml and 1000 ml sterile bottles
Isolate peripheral blood mononuclear cells (PBMCs) from a leukopak
-
1
Wipe the leukopak tubing with alcohol swab, cut with sterile scalpel and transfer the contents of the leukapheresis bag (leukopak) into an appropriately sized sterile Corning® bottle (usually 500 ml to 1000 ml).
Appropriate precautions must be taken when handling human cells at all times. Biosafety practices must be followed. All procedures use sterile tissue culture techniques with sterile solutions and equipment and are conducted in biosafety containment hoods.A detailed protocol for the isolation of peripheral blood mononuclear cells is described in the chapter on “Preparation of human mononuclear cell populations from peripheral blood”, in UNIT 7.1. -
2
Measure the volume of the leukapheresis product.
-
3
Add an equal volume of sterile PBS to the leukapheresis product.
Always use PBS that is maintained at room temperature, unless specified. -
4
Add 20 ml of Ficoll-Paque Premium™ (density 1.077 g/ml) to 50 ml sterile polypropylene conical tubes.
-
5
Gently layer 30 ml of the PBS-diluted leukapheresis product on top of the Ficoll.
Ensure that the interphase between Ficoll and the leukapheresis product remains distinct and is not disturbed. It helps to hold the tube at a slight angle when layering on top of the Ficoll layer. Alternatively, add 30 ml of the PBS-diluted leukapheresis product to the 50 ml tubes and underlay the Ficoll by inserting the pipette to the bottom of the tube and slowly releasing 20 ml of the Ficoll-Paque solution. -
6
Spin at 900 × g for 25 minutes.
The centrifuge brakes should be in off position and the centrifuge should be maintained at room temperature. -
7
Carefully harvest the interface containing peripheral blood mononuclear cells (PBMCs) into a new 50 ml sterile polypropylene conical tube. Use one 50 ml conical tube for each interface.
-
8
Add room temperature PBS to each tube and bring the total volume to 50 ml per tube.
-
9
Centrifuge the tubes for 10 minutes at 300 × g at room temperature.
-
10
Decant the supernatant and resuspend each pellet in 50 ml PBS and centrifuge as above.
-
11
Repeat the washes with PBS as described above for a total of 3 washes.
-
12
Combine all the cells into one 50 ml conical tube and resuspend the cells in 50 ml of PBS.
-
13
Determine the number of live and dead cells via trypan blue exclusion with the aid of a hemacytometer. Record that number.
Recovery of Pumps in patients can vary widely and range from 80–140 × 108 cells per pheresis.
Isolate monocytes by adherence to plastic
-
14
Resuspend the PBMCs in AIM V media at 2 × 108 per ml.
AIM V® medium CTS™ is an FDA 510(k) cleared device which is intended for human ex vivo cell culture. -
15
Add 1 ml of PBMCs to each T-150 sterile cell culture flask.
Please be cognizant that at this point you will be working with a large number of flasks (ranges from 40 to 70 flasks). It might help to handle the flasks in batches of 10. All subsequent steps should be handled in batches so as to minimize process-related variations in protocol. -
16
Add 29 ml of AIM V media to each flask.
-
17
Place all cell culture flasks in a humidified incubator maintained at 37°C and 5% CO2. Incubate the flasks for 1 hour to allow the monocyte precursors to adhere.
Time of incubation can vary from 1–2 hours. The CD14+ monocytes (adherent cells) will adhere to the tissue culture flasks and T and B lymphocytes will remain non-adherent (non-adherent cells).
Generate dendritic cells from adherent monocytes
-
18
After 1 hour, remove the non-adherent cells by firmly tapping the flask and harvest the non-adherent cells into 50 ml conical tubes. Ensure that all the medium is completely removed from the T-150 cell culture flasks by aspirating off traces of media left behind in the flask.
Maintain non-adherent cells on ice. Centrifuge the cells at 300 × g for 10 min. The non-adherent cells can be frozen at 5 × 107 cells/ml, 2 ml per cryovial in 90% human AB serum or autologous plasma and 10% dimethyl sulfoxide (DMSO). Freezing mix should be chilled on ice prior to adding to cells.If autologous plasma is available, freeze human cells in autologous plasma. Transfer the plasma (usually collected in bags similar to the leukapheresis product) into 50 ml sterile tubes. Centrifuge the tubes containing plasma at 1600 × g for 20 min at room temperature. Collect the supernatant fluid and filter through a 0.45 micron filter. Store plasma at 4°C in the refrigerator. -
19
Immediately add 30 ml of AIM-V medium supplemented with 800 U/ml GM-CSF and 500 U/ml IL-4 to each T-150 cell culture flask containing the adherent monocytes.
-
20
Incubate the cells in a humidified incubator at 37°C and 5% CO2 for 7 days.
-
21
After the 7-day culture period, harvest DCs by firmly tapping the flasks with the palm of your hand. This will dislodge the loosely adherent DC clusters. Harvest the DCs into a 50 ml conical tube and maintain the cells on ice.
-
22
Wash the adherent DCs with cold PBS and collect the PBS wash in 50 ml conical tubes.
-
23
Add 20 ml of enzyme free dissociation buffer to each T-150 tissue culture flask and incubate at 37°C for 15–20 minutes.
-
24
Harvest all DCs by firmly tapping the flasks with the palm of your hand followed by vigorous pipetting. Harvest all cells and combine them with previously harvested DCs.
-
25
Centrifuge all tubes at 300 × g for 10 minutes at 4°C.
-
26
Resuspend the DCs in the desired medium and count. These are immature DCs.
If the cells are to be loaded with antigen to generate antigen-loaded DCs for vaccines, perform this procedure on these freshly harvested immature DCs. Alternatively, DCs may be matured and then loaded with antigens (Michiels et al., 2005).Immature DC recovery should be 5–12% of the initial PBMC count with a purity of 90% or above in the final population (by morphology and phenotype). The wide variation in cell yield is due to donor-to-donor variation. DCs can be frozen at 5 × 106 cells/ml, 2 ml per cryovial in 90% autologous plasma (if available) or human AB serum and 10% DMSO and stored in a liquid nitrogen freezer.
Dendritic cell maturation
-
27
Culture immature DCs loaded with antigen (using the protocol of choice) at 106/ml in AIM-V medium containing GM-CSF and IL-4 and the maturation cytokine cocktail in tissue culture flasks. Maintain DCs in incubator at 37°C and 5% CO2 for 16–20 hours.
DCs are maintained in cell culture flasks for overnight maturation. A T-75 flask will hold 15–16 ml of medium, a T-150 flask will hold 30–35 ml of medium and a T-225 flask will hold 50 ml of medium.For DC maturation the two most commonly used combinations are: 1] AIM-V media containing GM-CSF (800 U/ml), IL-4 (500 U/ml), TNF-α (5 ng/ml), IL-1β (5 ng/ml), IL-6 (150 ng/ml) and prostaglandin E2 (PGE2; 1 μg/ml) (Lee et al., 2002) and 2] AIM-V media containing GM-CSF (800 U/ml), IL-4 (500 U/ml), TNF-α (10 ng/ml), IL-1β (10 ng/ml), IL-6 (15 ng/ml) and prostaglandin E2 (PGE2,1 μg/ml) (Jonuleit et al., 1997).
Dendritic cell harvest and cryopreservation
-
28
Harvest DCs after overnight incubation by firmly tapping the flask and collecting the cells into a 50 ml conical tube. Maintain the DCs on ice.
-
29
Wash the flask containing the adherent DCs with ice cold PBS and combine the wash PBS with the previously harvested DCs.
-
30
Add the appropriate amount of enzyme free dissociation buffer to each tissue culture flask (10–30 ml) and incubate at 37°C for 5–15 minutes.
Matured DCs tend to come of the flask very easily and it is important to check the cells after 5 minutes. -
31
Tap the flask against the palm of the hand, gently but firmly, to dislodge the adherent DCs. Harvest the DCs into 50 ml tubes.
Sometimes it is preferable to use 60 mm or 100 mm tissue culture dishes during the overnight maturation step. If that is the case, do not tap the dishes; instead use a 5 ml pipette to harvest the cells by gently pipetting the cells up and down. -
32
Centrifuge all tubes containing DCs at 300 × g for 10 minutes.
-
33
Combine all the cell pellets into a single 50 ml tube and resuspend in 50 ml PBS. Count the mature, antigen-loaded DCs. Record the number.
It is important to work swiftly yet gently with DCs. The recovery of DCs post-antigen loading and post-maturation is between 50–70% of the starting DC number. -
34
Centrifuge at 300 × g for 10 minutes.
-
35
Resuspend DCs at 5–10 × 106 per ml in 90% human AB serum and 10% DMSO. Add 1 ml to each cryovial and maintain cryovials in a liquid nitrogen freezer.
Alternatively, DCs may be frozen in 80% human AB serum, 10% DMSO and 10% dextrose.
BASIC PROTOCOL 3: DEPLETION OF DENDRITIC CELLS BY NEGATIVE SELECTION
Dendritic (i.e., CD83+) cells may also be removed from mononuclear cell populations using immunomagnetic depletion. It is often useful to examine the function of cell populations in the absence of dendritic cells. Depletion of CD83+ cells from mixed leukocyte populations using this protocol significantly reduces the stimulator activity of cell populations in allogeneic mixed lymphocyte reactions (Zhou and Tedder, 1995a). Because CD83+ cells usually represent only a very small fraction of the cell population being examined (0.1% to 0.2% of mononuclear cells) and are such potent stimulators in mixed lymphocyte reactions, it is recommended that the cell population being depleted of CD83+ cells goes through two rounds of negative selection, after which they are essentially undetectable. However, significant reductions in stimulator activity of starting cell populations are achieved with only one round. Because the CD83+ cells make up such a small proportion of the initial population, there is usually no discernible loss in overall cell numbers following this procedure.
Materials
Complete RPMI-10 (APPENDIX 2)
CD83 MAb at a 10× saturating concentration
Goat anti–mouse IgG–coated magnetic beads (Dynabeads), washed (see UNIT 7.4)
Tissue culture flask or Petri plate
15-ml conical polypropylene centrifuge tubes
Clinical rotator
Magnetic separation apparatus
Additional reagents and equipment for isolating mononuclear cells (UNIT 7.1) and counting viable cells (APPENDIX 3B)
Isolate peripheral blood mononuclear cells from a leukopak or buffy coat preparation by density gradient centrifugation (see Basic Protocol 1, step 1). Culture the cells overnight at 37°C in complete RPMI-10 at 1 × 107 cells/ml in a tissue culture flask or Petri plate.
Harvest and wash the isolated blood mononuclear cells once in complete RPMI-10 and adjust the cell concentration to 2 × 107 cells/ml of complete RPMI-10 in 15-ml conical polypropylene centrifuge tubes.
Add 1/10 vol of 10× CD83 MAb to the mononuclear cell suspension and rotate 30 min at 6 to 10 rpm, 4°C.
Wash the cells twice with fresh medium to remove any unbound MAb and transfer the cells to a fresh polypropylene centrifuge tube at 2 × 107 cells/ml of complete RPMI-10.
Deplete the resuspended cells of CD83+ cells by adding a 2-ml suspension of washed magnetic beads to the cell suspension and rotate 1 hr at 6 to 10 rpm, 4°C.
Separate the bead-coated cells using the magnetic separation apparatus for ~5 min. Transfer the unbound cell suspension to a fresh tube and repeat the magnetic separation. Harvest the unbound cells.
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2; for suppliers, see APPENDIX 5.
Metrizamide, 14.5% (w/v)
Place 25 g metrizamide (Sigma 99% pure, grade 1) in a sterile 250-ml graduated cylinder. Rinse the bottle with 150 ml complete RPMI-10 (APPENDIX 2) and add this to the graduated cylinder. Dissolve the powder and then bring the total volume to 172 ml with more medium. Filter sterilize the solution into a sterile container and store at 4°C protected from light.
The density of metrizamide is critical for optimal cell separation. Each newly prepared batch should be evaluated to determine that it optimally separates the appropriate high-density and low-density populations of cells before it is used.
Bring the solution to room temperature prior to use.
Recombinant human GM-CSF, 100×
Dilute (by ≤1:5) a 1 μg/ml vial of granulocyte/macrophage colony-stimulating factor (GM-CSF) in complete RPMI-10 (APPENDIX 2) to generate a solution containing 80,000 U/ml GM-CSF. Divide into appropriately sized aliquots and store at −70°C as recommended by manufacturer. Use or store the solution immediately after reconstitution and dilution.
Recombinant human IL-4, 100×
Resuspend a 10-μg vial of recombinant human IL-4 in complete RPMI-10 (APPENDIX 2) at 50,000 U/ml. Divide into appropriately sized aliquots and store at −70°C as recommended by manufacturer. Use or store the solution immediately after reconstitution and dilution.
Recombinant human TNF-α, 100×
Dilute a 5-μg vial of recombinant human tumor necrosis factor α (TNF-α) in complete RPMI-10 (APPENDIX 2) to generate 10,000 U/ml. Divide into appropriately sized aliquots and store at −70°C as recommended by manufacturer. Use or store the solution immediately after reconstitution and dilution.
Cell culture medium for human DC generation (clinical use)
AIM-V medium supplemented with 800 U/ml human GM-CSF, 500 U/ml human IL-4.
Cell culture medium for human DC maturation (clinical use)
AIM-V media supplemented with GM-CSF (800 U/ml), IL-4 (500 U/ml), TNF-α (5 ng/ml), IL-1β (5 ng/ml), IL-6 (150 ng/ml) and prostaglandin E2 (PGE2) (1 μg/ml).
GM-CSF (clinical use)
Leukine® (sargramostim) is available in a liquid formulation and can be directly added to AIM-V medium. The liquid Leukine® presentation is formulated as a sterile, injectable solution in a vial and contains 500 μg (2.8 × 106 IU/ml) sargramostim, 1.9 mg/ml edetate disodium, and 1.1% benzyl alcohol in a 1 ml solution. Liquid Leukine® should be stored in the refrigerator.
IL-4 (clinical use) 1000×
Reconstitute lyophilized IL-4 (R&D Systems, 1 μg = 2.9 × 104 international units [IU]) in AIMV at 5 × 105 IU/ml (1000x stock solution). 100–200 μl aliquots of the 1000x stock solution can be stored frozen at −20°C to −80°C.
TNF-α and IL-1β 1000×
Reconstitute lyophilized cytokines in AIM-V at 5 μg/ml and add to DC maturation medium. 100–200 μl aliquots of this 1000x stock solution can be stored frozen at −20°C to −80°C.
IL-6 1000×
Reconstitute lyophilized IL-6 in AIM-V at 150 μg/ml and add to DC maturation medium. 100–200 μl aliquots of this 1000x stock solution can be stored frozen at −20°C to −80°C.
PGE2 1000×
Dissolve PGE2 powder at a concentration of 1 mg/ml in 100% ethanol (1000x). 100–200 μl aliquots of this 1000x stock solution can be stored frozen at 20°C, protected from light.
COMMENTARY
Background Information
Dendritic cells represent a small subpopulation of bone marrow–derived leukocytes that develop within the monocyte lineage (Steinman, 1991; Zhou and Tedder, 1996). Dendritic cells are classified primarily based on their tissue location; thus, they are classified as interdigitating reticulum cells when present in lymphoid organs, as veiled cells when present in afferent lymph, as blood dendritic cells when in the circulation, as Langerhans cells when present in the epidermis, and as dermal dendritic cells when found in the dermis of the skin (Steinman and Cohn, 1973; Steinman, 1991). Human dendritic cells have been isolated from many of these tissues (Table 7.32.2). Although dendritic cells are widely dispersed throughout the body they exhibit many common features: an irregular shape with extensive and elongated dendritic processes (Fig. 7.32.1D, E, F), a distinct cell-surface phenotype (including very high levels of MHC class II antigens), low buoyant density, active motility, and the ability to stimulate the vigorous proliferation of unprimed T cells.
Table 7.32.2.
Procedures for the Isolation of Human Dendritic Cells from Tissues
| Tissue | Nomenclature | References |
|---|---|---|
| Tonsil | Interdigitating reticulum cell | Hart and McKenzie (1988) King and Katz (1989) Prickett et al. (1992) |
| Thymus | Thymic dendritic cell | Landry et al. (1990); Lafontaine et al. (1990) |
| Skin | Dermal dendritic cell, Langerhans cell | Romani et al. (1989); Lenz et al. (1993); Nestle et al. (1993) |
| Synovial exudate | Synovial dendritic cell | Zvaifler (1985); Thomas et al. (1994) |
| Lung | Lung dendritic cell | Nicod et al. (1987) |
| Bone marrow | Dendritic cell precursors | Egner et al. (1993b); Egner and Hart (1995) |
Dendritic cells are of central importance for antigen presentation during the initiation of primary immune responses such as the sensitization of MHC-restricted T cells and the formation of T cell–dependent antibodies (Steinman, 1991). They are also the principal stimulator cells of primary mixed leukocyte reactions and are actively involved in autoimmune diseases, graft rejection, and HIV infection. Therefore, the ability to isolate and further characterize dendritic cells is of considerable importance and practical utility.
Isolating adequate numbers of pure dendritic cells from humans has always proven difficult because of their very low frequency in blood (~0.2%) and tissues (Zhou and Tedder, 1995a) Nonetheless, populations of human blood mononuclear cells that contain dendritic cells have been isolated by numerous laboratories and studied using a variety of methods (Van Voorhis et al., 1982; Knight et al., 1986; Young and Steinman, 1988; Freudenthal and Steinman, 1990; Egner et al., 1992; Wood and Freudenthal, 1992; Xu et al., 1992; Egner et al., 1993a; Thomas et al., 1993). Mononuclear cell preparations enriched for dendritic cells are commonly isolated by negative selection, because dendritic cells in the blood lack surface antigens found on T cells, B cells, monocytes, and NK cells (Table 7.32.1). The numbers of dendritic cells in cell preparations have been quantified primarily by determining the frequency of cells with a dendritic morphology, the frequency of cells with a limited phenotypic profile, or the ability of the cells to stimulate in mixed leukocyte reactions. However, the recent identification of CD83 as a marker preferentially expressed by human dendritic cells has greatly facilitated their isolation, identification, and characterization (Zhou et al., 1992; Engel et al., 1995; Weissman et al., 1995; Zhou and Tedder, 1995a,b, 1996).
CD83 is predominantly expressed on the surface of dendritic cells, including blood dendritic cells, skin Langerhans cells, and interdigitating reticulum cells present in the T cell zones of lymphoid organs (Zhou et al., 1992; Engel et al., 1995). CD83+ cells isolated from blood are a homogenous and unique population of cells with the characteristic cellular morphology and functional activities of mature dendritic cells. CD83+ cells express the highest levels of MHC class II molecules compared with other leukocyte lineages and are the most potent stimulator cells in allogeneic mixed leukocyte reactions. Other circulating leukocytes do not express detectable levels of CD83 ex vivo, but this antigen is weakly expressed by some germinal center lymphocytes in vivo (Zhou et al., 1992).
Human CD83+ cells have a unique phenotype, expressing several lineage-associated antigens such as the T cell markers CD2 and CD5, the B cell markers CD40 and CD78, and the myeloid cell markers CD13, CD32 (FcγR II), CD33, CD36, and CD63, as well as a large number of leukocyte-associated antigens (Zhou and Tedder, 1995a). It is therefore possible to define mature dendritic cells as being CD83+ cells, rather than relying principally on the cells having a dendritic cell–like morphology or having the greatest potency in a mixed leukocyte reaction.
The use of CD83 MAbs provides a convenient and efficient way to rapidly separate dendritic cells from bulk mononuclear cells without apparent functional perturbation. Basic Protocol 1 uses CD83 MAbs as a phenotypic marker for assessing dendritic cell purity. The Alternate Protocol uses CD83 MAbs to positively select dendritic cells from populations of mononuclear cells using immunomagnetic selection. CD83 MAbs can also be used in panning procedures to isolate pure dendritic cells from dendritic-enriched populations of cells.
The CD2 and CD4 cell-surface antigens on dendritic cells are also useful. Using immunomagnetic selection protocols, CD2 and CD4 have each been used to isolate dendritic cell populations from mononuclear cell populations depleted of lymphocytes, NK cells, and monocytes. However, CD2 is expressed only at low levels by mature dendritic cells while it is expressed at high levels by T cells, and CD4 is expressed by “immature” blood dendritic cells but not mature CD83+ dendritic cells (Zhou and Tedder, 1995a, 1996). A new nomenclature for blood monocytes and dendritic cells has been recently proposed (Ziegler-Heitbrock, et. al., 2010). Because dendritic cells are the only cells in the blood that express CD83, CD83 MAbs can also be used to deplete dendritic cells from mononuclear cell preparations using magnetic bead isolation procedures (Zhou and Tedder, 1995a).
Three populations of cells in blood that have or can develop a dendritic cell morphology have been identified: one small subpopulation that is already CD83+, one population that becomes CD83+ following very brief periods of culture, and one population that derives from myelomonocytic cells but does not express CD83 even following culture or activation (Weissman et al., 1995; Zhou and Tedder, 1995a). The population of cells with a dendritic morphology that derives from myelomonocytic cells may represent a portion of precursor cells that become CD83+ when cultured in the presence of GM-CSF, IL-4, and TNF-1 (Zhou and Tedder, 1996).
The above findings are consistent with other observations that many blood dendritic cells circulate as precursor cells, which undergo phenotypic and morphological changes during their differentiation into functional antigen-presenting cells after a period of maturation (Knight et al., 1986; Markowicz and Engleman, 1990; O’Doherty et al., 1993; Romani et al., 1994; Thomas and Lipsky, 1994). Mixed populations of myeloid lineage cells that include cells with a dendritic cell morphology, phenotype, and function can also be generated from human bone marrow or blood progenitor cells after culture with combinations of granulocyte/monocyte colony stimulating factor (GM-CSF), and either tumor necrosis factor α (TNF-α) or interleukin 4 (IL-4; Caux et al., 1992; Reid et al., 1990, 1992; Santiago-Schwarz et al., 1992, 1993; Romani et al., 1994; Szabolcs et al., 1995). This strategy has been used to show that dendritic cells can actually be derived directly from human blood CD14+ monocytes (Zhou and Tedder, 1996). Therefore, cells with a dendritic cell morphology within human blood are heterogeneous, but likely represent a common lineage of cells at different stages of differentiation.
Blood monocyte differentiation into a fairly homogenous population of functionally mature CD83+ dendritic cells under the influence of a specific cascade of cytokines can be divided into several stages (Zhou and Tedder, 1996). In the first stage, CD14+ CD1a+ cells with a dendritic morphology are induced by GM-CSF/IL-4. In the second stage, the cells are induced by TNF-α to differentiate into CD83+ CD14– CD4+ cells. Finally, in a third stage involving continued culture, the monocyte-derived CD83+ cells develop into cells with decreased CD1a expression that have a dermal dendritic cell phenotype. Depletion of CD14+ cells from mononuclear cell preparations depletes most dendritic cell precursors (Zhou and Tedder, unpub. observ.). This finding supports and extends earlier findings in which blood monocyte subpopulations or adherent cells with dendritic cell morphology, size, cell surface phenotype, or function have been identified (Knight et al., 1986; Kabel et al., 1989; Najar et al., 1990; Peters et al., 1991; Ruppert et al., 1991; Grage-Griebenow et al., 1993). The ability to generate dendritic cells from monocytes provides a new and simple method for generating functionally mature dendritic cells from a population of cells that are relatively easy to isolate to homogeneity, as opposed to CD34+ stem cells or other precursor cells.
In the alternate protocol 2, we describe DC generation from a leukapheresis product: this protocol is used in many DC-based clinical studies to generate a large number of DCs ex vivo (Steinman and Banchereau, 2007; Gilboa, 2007; Ueno et al., 2010). The DCs are matured with a cocktail of three proinflammatory cytokines (IL-1β, IL-6, and TNF-α) and prostaglandin E2 (Jonuleit et al., 1997; Lee et al., 2002). Because the impact of PGE2 on IL-12 production by DCs is questionable (Kalinski et al., 2001; Landi et al., 2011), some studies use the above cocktail without PGE2. Many labs are also evaluating other DC maturation cocktails including toll-like receptor (TLR) agonists, CD40-ligand, and various combinations thereof and the generation of optimal DCs for vaccination remains an active topic of research (Macagno et al., 2007; Ueno et al., 2010; Schreibelt et al., 2010; Skalova et al., 2010; Castiello et al., 2011). It is critical to evaluate all DC generation and maturation strategies by phenotype, function (presentation of antigen to T and/or B lymphocytes), migration (in vitro migration towards the chemokine CCL19 or macrophage inflammatory protein 3 beta, MIP-3β) and cytokine secretion upon activation.
The ability to generate dendritic cells in vitro from their precursors has considerable utility. Given the capacity of dendritic cells to elicit strong antigen-specific helper and cytotoxic T cell responses, the ability to isolate or generate large numbers of homogeneous preparations of CD83+ dendritic cells will facilitate the manipulation of this cell lineage for vaccine development, organ grafting, and ex vivo therapy for a broad range of human diseases. For purposes of comparison, it is also useful to generate cell preparations depleted of dendritic cells, as detailed in Basic Protocol 3. The ability to deplete CD83+ dendritic cells may prove useful for the prevention of graft-versus-host disease (Wilson et al., 2009).
As outlined above, there are several ways to isolate or generate human dendritic cells. Caution should be used when dendritic cell properties or function are assessed using dendritic cell–enriched preparations of cells that are not homogeneous, as considerable variability can be obtained. Because there is tremendous variability in the way individual laboratories assess dendritic cell purity and function, there remains considerable disagreement in the literature. This heterogeneity is further magnified by the fact that dendritic cells can be at differing stages of “maturity,” which may also affect their function. Although CD83+ dendritic cells isolated by each of these techniques are phenotypically, functionally, and morphologically homogenous in most assays, it remains possible that there is additional heterogeneity that is not currently appreciated.
Critical Parameters
The single most important factor in isolating adequate numbers of dendritic cells from human blood mononuclear cells is the availability of large numbers of starting cells. Small volumes of blood do not work well for the isolation of dendritic cells and in these cases it is advisable to generate dendritic cells from monocytes (see Basic Protocol 2). Freshly isolated mononuclear cells are best, but more than adequate results can be obtained with day-old mononuclear cell preparations that have been maintained on ice. Leukocyte-enriched “leukopaks” that have been isolated within the last 24 hr can be obtained from large blood banking facilities or from North American Biologicals. Leukopaks are usually obtained by total leukapheresis of donors, so it is possible to isolate 2–12 × 108 mononuclear cells from each leukopak. However, the number of mononuclear cells in a leukopak will vary considerably based on the source and blood donor. Alternatively, leukocyte-enriched “buffy coats” prepared from donated units of peripheral blood or from plasmapheresis procedures can be utilized. Differences in isolation procedures can affect neutrophils within leukopaks, thereby making it difficult to remove the neutrophils from the mononuclear cell preparation by Ficoll-Paque density gradient centrifugation. In these cases, the purity of the final dendritic cell preparation can be significantly diminished since neutrophils share many of the buoyant properties of dendritic cells. Units of whole blood can also be used as a starting population of cells, but the final yield of dendritic cells is closely linked with the number of starting cells, so sufficient numbers of cells are required to obtain reasonable yields. This is particularly important for experiments where dendritic cells need to be purified to homogeneity by cell sorting. Typical yields after cell sorting represent ~10% of the starting population of labeled cells that are being sorted when the frequency of CD83+ cells is >50%, but vary greatly with the level of dendritic cell purity and the sort rate.
The technique-sensitive steps are those involving cell manipulation, because these can lead to large cell losses. The single most critical step is washing the tissue culture dishes free of nonadherent dendritic cells after plastic adherence (see Basic Protocol 1). Care must be taken to clear the entire surface of nonadherent cells, pipetting with sufficient force to dislodge the nonadherent cells, while at the same time avoiding excessive force that can strip off adherent cells. It is essential to use warm medium in these steps, because cold medium detaches adherent cells from plastic.
The cells that are obtained using Basic Protocol 1 are enriched for dendritic cells. The relative number of dendritic cells can be roughly assessed by phase-contrast microscopy of the cells in a hemacytometer. Dendritic cells are large and have irregular, long membrane processes (Fig. 7.32.1C), while lymphocytes are small and round. If a higher purity of dendritic cells is desired, a second metrizamide density gradient centrifugation can be performed to remove contaminating lymphocytes. Contaminating B lymphocytes and monocytes, which bind EA via their surface Fc receptors, can be further depleted from the dendritic cell preparations by EA rosetting as described in UNIT 3.7 for mouse dendritic cells.
Alternatively, monocytes can be depleted by adherence to human Ig–coated plates and B lymphocytes depleted on plates coated with goat anti–human Ig. Depletion of contaminating cells using lineage-associated MAbs that do not bind to dendritic cells (Table 7.32.1) can also be achieved using magnetic bead–mediated depletion techniques (see Alternate Protocol) or panning. If total purity is required, cell sorting should be performed. Due to protocol variations in each laboratory, these procedures may need some optimization in order to isolate maximal numbers of dendritic cells. It is important to always start with a sufficient number of mononuclear cells, such as are present in leukapheresis packs or buffy coats from leukapheresis donors, to ensure optimal cell isolation and sufficient cell yield.
Depleting contaminating cells from the dendritic cell preparations by Fc receptor–mediated procedures is highly effective. However, the authors have found that dendritic cells express the CD32 and CD64 Fc receptors (Fanger et al., 1996), so these isolation procedures may limit the heterogeneity of the dendritic cell preparations obtained. Additionally, mature dendritic cells express the sheep erythrocyte receptor, CD2 (Zhou and Tedder, 1995a). While it is possible that some dendritic cells may be depleted from the E-rosette-negative fraction of blood mononuclear cells by rosetting with sheep red blood cells, the levels of CD2 expression are low and may not be sufficient to result in effective rosette formation.
Troubleshooting
Because of the many manipulations that are required in these protocols, there are many things that can go wrong. Until these protocols become standard to the investigator, all cell fractions should be retained until the end of the procedure. In this way, it is possible to determine where things went wrong and, in many cases, to recover the dendritic cell–containing fraction.
Anticipated Results
The density centrifugation and immunomagnetic isolation procedures (Basic Protocol 1 and Alternate Protocol) yield cell populations consisting of 20% to 80% dendritic cells and are largely free of lymphocytes. Incubation of CD14+ monocytes with cytokines (Basic Protocol 2) yields a relatively homogeneous population of functionally mature dendritic cells, with typical recoveries of 60% to 90% of input monocytes.
Immature DC recovery should be 5–12% of the initial PBMC count with a purity of 90% or above in the final population (by morphology and phenotype). The wide variation in cell yield is due to donor-to-donor variation. The cells are typically large cells and form loosely adherent clusters or may be firmly attached to the cell culture flasks on day 7. Once again this is due to donor-to-donor variation. Post-maturation the cells remain adherent but they are easily harvested after a short incubation with enzyme-free cell dissociation buffer (sometimes within 5 minutes). When counting mature DCs one can easily identify DCs as large cells. Mature DCs are typically large cells with dendrites. Very low yield could indicate problems with cytokines, but often this may be due to donor variation and the treatments the patient has undergone prior to being pheresed.
A representative phenotype of immature and mature DCs derived from healthy donor PBMCs is presented in Figure 7.32.2. DCs express high levels of MHC class II, CD11c, CD86 and CD40. Upon maturation there is a clear increase in the expression of CD80, CD83 and CCR7 (marker associated with DC migration). DCs do not express the monocyte marker, CD14. Less than 10–20% of the cells should be lymphocytes, which are the small cell population at the bottom, visible in the DC scatter profile (Figure 7.32.2).
Figure 7.32.2.
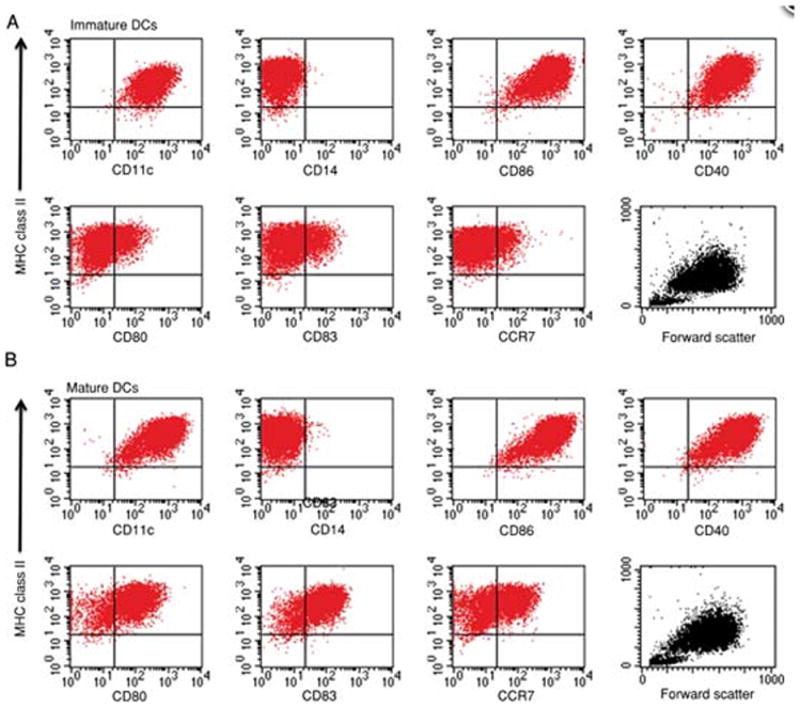
Representative phenotype of immature and mature dendritic cells. Phenotype and scatter profiles of monocyte-derived immature DCs (A). Phenotype and scatter profile of monocyte-derived mature DCs (B). For the color version of this figure go to http://www.currentprotocols.com/protocol/im0732.
Time Considerations
In Basic Protocol 1, 5 to 6 hr of work are required on the first day of the procedure, from the isolation of blood mononuclear cells to the overnight incubation of mononuclear cells in culture plates. On the second day, the enrichment of dendritic cells by metrizamide gradients followed by additional adherence and panning procedures requires 4 to 6 hr. It is possible to institute many shortcuts, but these usually result in decreased yields or purity of the final cell preparation, so it is best to carefully follow the protocols as outlined. The use of magnetic beads can speed up the negative and positive isolation procedures, as indicated in the Alternate Protocol.
Isolation of dendritic cells from GM-CSF/IL-4/TNF-α–induced monocytes or total mononuclear cell preparations (see Basic Protocol 2) is less time-consuming. This procedure does require an 8-day period of cell culture in addition to the time spent preparing the cells, and it is a more expensive approach, because of the cost of recombinant cytokines. Nonetheless, the generation of dendritic cells from monocytes, can yield significant numbers of cells and a fairly homogenous population of dendritic cells, which has considerable advantages for some applications.
As described above, the generation of DC from adherent monocytes spans 8 days from start to finish, when one starts with a leukapheresis. Day 1 tends to be an all-day procedure, starting with the processing of leukapheresis sample (3 to 4 hours) and ending with DC culture (1 hour for adherence and then adding medium to DC flasks in batches of 10). Recovery of PBMCs in patients can vary widely and range from 80–140 × 108 cells per pheresis. The amount of time increases because of the number of cells being processed, which can range from 40–70 T-150 tissue culture flasks for DC generation).
Harvesting DCs on day 7 and plating them for maturing is also time-consuming because of the large volume of cells being handled. This work should be limited to 10 T-150 tissue culture flasks at one time in the biosafety hood. It is best to allow up to 1 hour for each batch of 10 T-150 tissue culture flasks, from the time of DC harvest (including 15–20 minutes for cell dissociation buffer), washing the cells, counting them and then culturing DCs in maturation medium (this does not include the time required for loading DCs with antigen). Harvesting DCs and counting and freezing antigen-pulsed or -unpulsed DCs will also take approximately 40–60 minutes for 10 T-150 cell culture flasks.
Acknowledgments
These studies were supported by grants from the Department of Defense (PCRP Award W81XWH-10-1-0339; to S.N.) and the NIH [AI56363 and Southeastern Regional Center of Excellence for Emerging Infections and Biodefense (U54 AI057157)] and the Lymphoma Research Foundation (to T.F.T.).
Footnotes
Contributed by Smita Nair, Gerald E. Archer and Thomas F. Tedder, Duke University Medical Center, Durham, North Carolina
Key References
Steinman, R.M. 1991. See above.
Presents a general review of dendritic cell biology in mouse and man.
Zhou, L.-J. and Tedder, T.F. 1995a. See above.
Provides an extensive flow cytometric study of the antigenic phenotype of human dendritic cells.
Zhou, L.-J. and Tedder, T.F. 1995b. See above.
Surveys the cytokines and chemokines produced by human blood dendritic cells.
Zhou, L.-J. and Tedder, T.F. 1996. See above.
Details procedures for generating human dendritic cells from monocytes.
Literature Cited
- Castiello L, Sabatino M, Jin P, Clayberger C, Marincola FM, Krensky AM, Stroncek DF. Monocyte-derived DC maturation strategies and related pathways: a transcriptional view. Cancer Immunol Immunother. 2011;60:457–466. doi: 10.1007/s00262-010-0954-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-α cooperate in the generation of dendritic Langerhans cells. Nature. 1992;360:258–261. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- Egner W, Hart DNJ. The phenotype of freshly isolated and cultured human bone marrow allostimulatory cells: Possible heterogeneity in bone marrow dendritic cell populations. Immunology. 1995;85:611–620. [PMC free article] [PubMed] [Google Scholar]
- Egner W, Prickett TCR, Hart DNJ. Adhesion molecule expression: A comparison of human blood dendritic cells, monocytes, and macrophages. Transplant Proc. 1992;24:2318. [PubMed] [Google Scholar]
- Egner W, Andreesen R, Hart DNJ. Allostimulatory cells in fresh human blood: Heterogeneity in antigen-presenting cell populations. Transplantation. 1993a;56:945–950. doi: 10.1097/00007890-199310000-00032. [DOI] [PubMed] [Google Scholar]
- Egner W, McKenzie JL, Smith SM, Beard MEJ, Hart DNJ. Identification of potent mixed leukocyte reaction–stimulatory cells in human bone marrow. J Immunol. 1993b;150:3043–3053. [PubMed] [Google Scholar]
- Engel P, Wagner N, Tedder TF. CD83 workshop report. In: Schlossman SF, Boumsell L, Gilks W, Harlan JM, Kishimoto T, Morimoto C, Ritz J, Shaw S, Silverstein R, Springer T, Tedder TF, Todd RF, editors. Leukocyte Typing V. White Cell Differentiation Antigens. Oxford University Press; Oxford: 1995. pp. 693–695. [Google Scholar]
- Fanger NA, Wardwell K, Shen L, Tedder TF, Guyre PM. Type I (CD64) and type II (CD32) Fcγ receptor–mediated phagocytosis by human blood dendritic cells. J Immunol. 1996;156:541–548. [PubMed] [Google Scholar]
- Feuerstein B, Berger TG, Maczek C, Roder C, Schreiner D, Hirsch U, Haendle I, Leisgang W, Glaser A, Kuss O, Diepgen TL, Schuler G, Schuler-Thurner B. A method for the production of cryopreserved aliquots of antigen-preloaded, mature dendritic cells ready for clinical use. J Immunol Methods. 2000;245:15–29. doi: 10.1016/s0022-1759(00)00269-6. [DOI] [PubMed] [Google Scholar]
- Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117:1195–1203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal PS, Steinman RM. The distinct surface of human blood dendritic cells, as observed after an improved isolation method. Proc Natl Acad Sci USA. 1990;87:7689–7702. doi: 10.1073/pnas.87.19.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grage-Griebenow E, Lorenzen D, Fetting R, Flad HD, Ernst M. Phenotypical and functional characterization of Fcγ receptor I (CD64)–negative monocytes, a minor human monocyte subpopulation with a high accessory and antiviral activity. Eur J Immunol. 1993;23:3126–3135. doi: 10.1002/eji.1830231213. [DOI] [PubMed] [Google Scholar]
- Hart DNJ, McKenzie JL. Isolation and characterization of human tonsil dendritic cells. J Exp Med. 1988;168:157–170. doi: 10.1084/jem.168.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk AH. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27:3135–3142. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- Kabel PJ, De Haan-Meulman M, Voorbij HAM, Kleingeld M, Knol EF, Drexhage HA. Accessory cells with the morphology and marker pattern of dendritic cells can be obtained from elutriator-purified blood monocyte fractions. An enhancing effect of metrizamide in this differentiation. Immunobiology. 1989;179:395–411. doi: 10.1016/S0171-2985(89)80044-0. [DOI] [PubMed] [Google Scholar]
- King PD, Katz DR. Human tonsillar dendritic cell–induced T cell responses: Analysis of molecular mechanisms using monoclonal antibodies. Eur J Immunol. 1989;19:581–587. doi: 10.1002/eji.1830190402. [DOI] [PubMed] [Google Scholar]
- Knight SC, Farrant J, Bryant A, Edwards AJ, Burman S, Lever A, Clarke J, Webster ADB. Non-adherent, low density cells from human peripheral blood contain dendritic cells and monocytes, both with veiled morphology. Immunology. 1986;57:595–603. [PMC free article] [PubMed] [Google Scholar]
- Lafontaine M, Landry D, Montplaisir S. The human thymic dendritic cell phenotype and its modification in culture. Cell Immunol. 1992;142:238–251. doi: 10.1016/0008-8749(92)90286-x. [DOI] [PubMed] [Google Scholar]
- Landry D, Doyon L, Poudrier J, Lafontaine M, Pelletier M, Montplaisir S. Accessory function of human thymic dendritic cells in Con A–induced proliferation of autologous thymocyte subsets. J Immunol. 1990;144:836–843. [PubMed] [Google Scholar]
- Lee AW, Truong T, Bickham K, Fonteneau JF, Larsson M, Da Silva I, Somersan S, Thomas EK, Bhardwaj N. A clinical grade cocktail of cytokines and PGE2 results in uniform maturation of human monocyte-derived dendritic cells: implications for immunotherapy. Vaccine. 2002;20(Suppl 4):A8–A22. doi: 10.1016/s0264-410x(02)00382-1. [DOI] [PubMed] [Google Scholar]
- Lenz A, Heine M, Schuler G, Romani N. Human and murine dermis contain dendritic cells: Isolation by means of a novel method and phenotypical and functional characterization. J Clin Invest. 1993;92:2587–2596. doi: 10.1172/JCI116873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macagno A, Napolitani G, Lanzavecchia A, Sallusto F. Duration, combination and timing: the signal integration model of dendritic cell activation. Trends Immunol. 2007;28:227–233. doi: 10.1016/j.it.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Markowicz S, Engleman EG. Granulocyte-macrophage colony-stimulating factor promotes differentiation and survival of human peripheral blood dendritic cells in vitro. J Clin Invest. 1990;85:955–961. doi: 10.1172/JCI114525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels A, Tuyaerts S, Bonehill A, Corthals J, Breckpot K, Heirman C, Van Meirvenne S, Dullaers M, Allard S, Brasseur F, van der Bruggen P, Thielemans K. Electroporation of immature and mature dendritic cells: implications for dendritic cell-based vaccines. Gene Ther. 2005;12:772–782. doi: 10.1038/sj.gt.3302471. [DOI] [PubMed] [Google Scholar]
- Najar HM, Bru-Capdeville AC, Gieseler RKH, Peters JH. Differentiation of human monocytes into accessory cells at serum-free conditions. Eur J Immunol. 1990;51:339–346. [PubMed] [Google Scholar]
- Nestle FO, Zheng XG, Thompson CB, Turka LA, Nickoloff BJ. Characterization of dermal dendritic cells obtained from normal human skin reveals phenotypic and functionally distinctive subsets. J Immunol. 1993;151:6535–6545. [PubMed] [Google Scholar]
- Nicod LP, Lipscomb MF, Weissler JC, Lyons CR, Alberton J, Toews GB. Mononuclear cells in human lung parenchyma: Characterization of a potent accessory cell not obtained by bronchoalveolar lavage. Am Rev Respir Dis. 1987;136:818–823. doi: 10.1164/ajrccm/136.4.818. [DOI] [PubMed] [Google Scholar]
- O’Doherty U, Steinman RM, Peng M, Cameron PU, Gezelter S, Kopeloff I, Swiggard WJ, Pope M, Bhardwaj N. Dendritic cells freshly isolated from human blood express CD4 and mature into typical immunostimulatory dendritic cells after culture in monocyte- conditioned medium. J Exp Med. 1993;178:1067–1078. doi: 10.1084/jem.178.3.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, Ruppert J, Gieseler RK, Najar HM, Xu H. Differentiation of human monocytes into CD14 negative accessory cells: Do dendritic cells derive from the monocyte lineage? Pathobiology. 1991;59:122–126. doi: 10.1159/000163628. [DOI] [PubMed] [Google Scholar]
- Prickett TCR, McKenzie JL, Hart DNJ. Adhesion molecules on human tonsil dendritic cells. Transplantation. 1992;53:483–490. doi: 10.1097/00007890-199202010-00041. [DOI] [PubMed] [Google Scholar]
- Reid CD, Fryer PR, Clifford C, Kirk A, Tikerpae J, Knight SC. Identification of hematopoietic progenitors of macrophages and dendritic Langerhans cells (DL-CFU) in human bone marrow and peripheral blood. Blood. 1990;76:1139–1149. [PubMed] [Google Scholar]
- Reid CDL, Stackpoole A, Meager A, Tikerpae J. Interactions of tumor necrosis factor with granulocyte-macrophage colony-stimulating factor and other cytokines in the regulation of dendritic cell growth in vitro from early bipotent CD34+ progenitors in human bone marrow. J Immunol. 1992;149:2681–2688. [PubMed] [Google Scholar]
- Romani N, Lenz A, Glassel H, Stossel H, Stanzl U, Majdic O, Fritsch P, Schuler G. Cultured human Langerhans cells resemble lymphoid dendritic cells in phenotype and function. J Invest Dermatol. 1989;93:600–609. doi: 10.1111/1523-1747.ep12319727. [DOI] [PubMed] [Google Scholar]
- Romani N, Gruner S, Brang D, Kampgen E, Lenz A, Trockenbacher B, Konwalinka G, Fritsch PO, Steinman RM, Schuler G. Proliferating dendritic cell progenitors in human blood. J Exp Med. 1994;180:83–93. doi: 10.1084/jem.180.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani N, Reider D, Heuer M, Ebner S, Kampgen E, Eibl B, Niederwieser D, Schuler G. Generation of mature dendritic cells from human blood. An improved method with special regard to clinical applicability. J Immunol Methods. 1996;196:137–151. doi: 10.1016/0022-1759(96)00078-6. [DOI] [PubMed] [Google Scholar]
- Ruppert J, Friedrichs D, Xu H, Peters JH. IL-4 decreases the expression of the monocyte differentiation marker CD14, paralleled by an increasing accessory potency. Immunobiology. 1991;182:449–464. doi: 10.1016/S0171-2985(11)80209-3. [DOI] [PubMed] [Google Scholar]
- Santiago-Schwarz F, Belilos E, Diamond B, Carsons SE. TNF in combination with GM-CSF enhances the differentiation of neonatal cord blood stem cells into dendritic cells and macrophages. J Leukocyte Biol. 1992;52:274–281. [PubMed] [Google Scholar]
- Santiago-Schwarz F, Divaris N, Kay C, Carsons SE. Mechanisms of tumor necrosis factor–granulocyte-macrophage colony-stimulating factor–induced dendritic cell development. Blood. 1993;82:3019–3028. [PubMed] [Google Scholar]
- Schreibelt G, Tel J, Sliepen KH, Benitez-Ribas D, Figdor CG, Adema GJ, de Vries IJ. Toll-like receptor expression and function in human dendritic cell subsets: implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol Immunother. 2010;59:1573–1582. doi: 10.1007/s00262-010-0833-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalova K, Mollova K, Michalek J. Human myeloid dendritic cells for cancer therapy: does maturation matter? Vaccine. 2010;28:5153–5160. doi: 10.1016/j.vaccine.2010.05.042. [DOI] [PubMed] [Google Scholar]
- Spisek R, Bretaudeau L, Barbieux I, Meflah K, Gregoire M. Standardized generation of fully mature p70 IL-12 secreting monocyte-derived dendritic cells for clinical use. Cancer Immunol Immunother. 2001;50:417–427. doi: 10.1007/s002620100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–1162. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- Szabolcs P, Moore MAS, Young JW. Expansion of immunostimulatory dendritic cells among the myeloid progeny of human CD34+ bone marrow precursors cultured with c-kit ligand, granulocyte-macrophage colony-stimulating factor and TNF-α. J Immunol. 1995;154:5851–5861. [PubMed] [Google Scholar]
- Thomas R, Lipsky PE. Human peripheral blood dendritic cell subsets. Isolation and characterization of precursor and mature antigen-presenting cells. J Immunol. 1994;153:4016–4028. [PubMed] [Google Scholar]
- Thomas R, Davis LS, Lipsky PE. Isolation and characterization of human peripheral blood dendritic cells. J Immunol. 1993;150:821–834. [PubMed] [Google Scholar]
- Thomas R, Davis LS, Lipsky PE. Rheumatoid synovium is enriched in mature antigen-presenting dendritic cells. J Immunol. 1994;152:2613–2623. [PubMed] [Google Scholar]
- Ueno H, Schmitt N, Klechevsky E, Pedroza-Gonzalez A, Matsui T, Zurawski G, Oh S, Fay J, Pascual V, Banchereau J, Palucka K. Harnessing human dendritic cell subsets for medicine. Immunol Rev. 2010;234:199–212. doi: 10.1111/j.0105-2896.2009.00884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Voorhis WC, Hair SL, Steinman RM, Kaplan G. Human dendritic cells: Enrichment and characterization from peripheral blood. J Exp Med. 1982;155:1172–1187. doi: 10.1084/jem.155.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman D, Li Y, Ananworanich J, Zhou LJ, Adelsberger J, Tedder TF, Baseler M, Fauci AS. Three populations of cells with dendritic morphology exist in peripheral blood, only one of which is infectable with human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1995;92:826–830. doi: 10.1073/pnas.92.3.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J, Cullup H, Laurie R, Sheng Y, Palkova A, Radford KJ, Dickinson AM, Rice AM, Hart DNJ, Munster DJ. Antibody to the dendritic cell surface activation antigen CD83 prevents acute graft-versus-host disease. J Exp Med. 2009;206:387–398. doi: 10.1084/jem.20070723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood GS, Freudenthal RS. CD5 monoclonal antibody reacts with human peripheral blood dendritic cells. Am J Path. 1992;141:789–795. [PMC free article] [PubMed] [Google Scholar]
- Xu H, Friendrichs U, Gieseler RKH, Ruppert J, Ocklind G, Peters JH. Human blood dendritic cells exhibit a distinct T-cell-stimulatory mechanism and differentiation pattern. Scand J Immunol. 1992;36:689–696. doi: 10.1111/j.1365-3083.1992.tb03129.x. [DOI] [PubMed] [Google Scholar]
- Young JW, Steinman RM. Accessory cell requirements for the mixed-leukocyte reaction and polyclonal mitogens, as studied with a new technique for enriching blood dendritic cells. Cell Immunol. 1988;111:167–182. doi: 10.1016/0008-8749(88)90061-5. [DOI] [PubMed] [Google Scholar]
- Zhou LJ, Tedder TF. Human blood dendritic cells selectively express CD83, a member of the immunoglobulin superfamily. J Immunol. 1995a;154:3821–3835. [PubMed] [Google Scholar]
- Zhou LJ, Tedder TF. A distinct pattern of cytokine gene expression by human CD83+ blood dendritic cells. Blood. 1995b;86:3295–3301. [PubMed] [Google Scholar]
- Zhou LJ, Tedder TF. CD14+ blood monocytes can differentiate into functionally mature CD83+ dendritic cells. Proc Natl Acad Sci USA. 1996;93:2588–2592. doi: 10.1073/pnas.93.6.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LJ, Schwarting R, Smith HM, Tedder TF. A novel cell-surface molecule expressed by human interdigitating reticulum cells, Langerhans cells and activated lymphocytes is a new member of the immunoglobulin superfamily. J Immunol. 1992;149:735–742. [PubMed] [Google Scholar]
- Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJM, Liu YJ, MacPherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–e80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- Zvaifler NJ, Steinman RM, Kaplan G, Lau LL, Rivelis M. Identification of immunostimulatory dendritic cells in the synovial effusions of patients with rheumatoid arthritis. J Clin Invest. 1985;76:789–800. doi: 10.1172/JCI112036. [DOI] [PMC free article] [PubMed] [Google Scholar]