

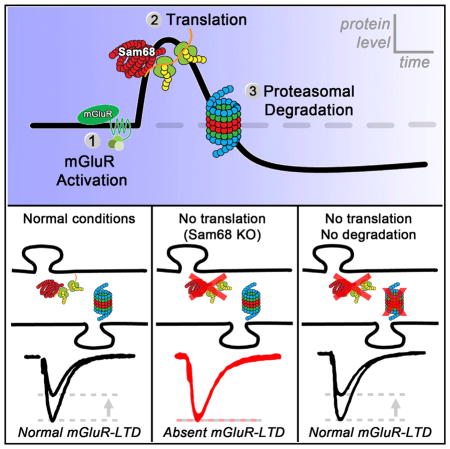
SUMMARY
Dendritic protein homeostasis is crucial for most forms of long-term synaptic plasticity, and its dysregulation is linked to a wide range of brain disorders. Current models of metabotropic glutamate receptor mediated long-term depression (mGluR-LTD) suggest that rapid, local synthesis of key proteins is necessary for the induction and expression of LTD. Here, we find that mGluR-LTD can be induced in the absence of translation if the proteasome is concurrently inhibited. We report that enhanced pro-teasomal degradation during the expression of mGluR-LTD depletes dendritic proteins and inhibits subsequent inductions of LTD. Moreover, proteasome inhibition can rescue mGluR-LTD in mice null for the RNA binding protein Sam68, which we show here lack mGluR-dependent translation and LTD. Our study provides mechanistic insights for how changes in dendritic protein abundance regulate mGluR-LTD induction. We propose that Sam68-mediated translation helps to counterbalance degradation, ensuring that protein levels briefly remain above a permissive threshold during LTD induction.
Graphical Abstract
INTRODUCTION
Coordination between the translational machinery, RNA binding proteins (RBPs), and the proteasome regulates dendritic proteostasis in response to neuronal activity (Hanus and Schuman, 2013). Mutations in components of these systems are associated with altered long-term synaptic depression induced by metabotropic glutamate receptor activation (mGluR-LTD) and may underlie the pathogenesis of certain autism spectrum disorders. Current models of mGluR-LTD suggest that elevated levels of key synaptic proteins are required for LTD induction and expression (Lüscher and Huber, 2010). Several plasticity-related proteins (PRPs), including ARC (Park et al., 2008; Waung et al., 2008), OPHN1 (Nadif Kasri et al., 2011), FMRP (Todd et al., 2003), APP (Westmark and Malter, 2007), and PSD95 (Muddashetty et al., 2007), are rapidly synthesized following mGluR activation. Knockdown experiments have demonstrated a necessity for ARC and OPHN1 in LTD (Nadif Kasri et al., 2011; Waung et al., 2008). However, whether an acute increase in PRP levels is sufficient to induce mGluR-LTD is unclear (Di Prisco et al., 2014; Niere et al., 2012; Park et al., 2008).
Along with protein synthesis, proteasomal degradation regulates synaptic protein abundance (Ehlers, 2003). Proteasomal subunits and E3 ligases present in dendrites can be transported into active spines to alter synaptic PRP levels (Bingol and Schuman, 2006). Degradation of ARC, FMRP, and PSD95 by the proteasome is important for regulating AMPA receptor endocytosis and spine morphology (Greer et al., 2010; Mabb et al., 2014; Nalavadi et al., 2012; Tsai et al., 2012). However, previous reports on the role of proteasome in mGluR-LTD are conflicting (Citri et al., 2009; Hou et al., 2006).
Functional impairment of the RBP Src-associated in Mitosis 68kD (Sam68) has been observed in patients diagnosed with the neurodegenerative disorder fragile X tremor ataxia syndrome (FXTAS), which is characterized by adult-onset ataxia and cognitive decline (Lukong and Richard, 2008; Sellier et al., 2010). We previously showed that Sam68 acts as a positive regulator of local translation by promoting the association of β actin mRNA with synaptic ribosomes (Klein et al., 2013). Sam68 binds to the mRNAs of several PRPs, including ARC (Grange et al., 2009), and likely coordinates mRNA metabolism in response to neuronal activity (Ben Fredj et al., 2004; Iijima et al., 2011).
In this study, we elaborate on a current model of mGluR-LTD, which states that rapid increases in translation of key proteins are necessary for the induction and expression of LTD (Costa-Mattioli et al., 2009; Lüscher and Huber, 2010). We demonstrate that activation of mGluRs rapidly depletes dendritic protein levels by proteasomal degradation. This effect occurs despite the well-established increase in protein synthesis during mGluR-LTD induction. The concurrent increase in degradation and translation during mGluR-LTD mediates metaplasticity by elevating the threshold for subsequent inductions of LTD. Our findings suggest that mGluR-LTD does not require an acute increase in dendritic PRP levels per se. Rather, protein translation is necessary to counterbalance degradation and ensure that PRP levels briefly remain above a permissive threshold during LTD induction.
RESULTS
Lack of ARC Translation and mGluR-LTD in Sam68 KO Mice Reveals Proteasomal Degradation of ARC
To investigate how PRP levels change during mGluR-LTD induction, we examined mice null for Sam68, an RBP that was previously shown to promote translation of its mRNA cargos (Klein et al., 2013; Paronetto et al., 2009). Sam68 binds to the mRNA of ARC (Grange et al., 2009), a PRP necessary for mGluR-LTD. We found no difference in the basal levels of ARC protein in acute hippocampal slices prepared from Sam68 KO mice and WT littermates. However, a brief application of the mGluR group I (mGluR-I) agonist DHPG reduced levels of ARC in slices from knockout (KO) animals, in contrast to the expected increase in ARC protein (Park et al., 2008; Waung et al., 2008) in slices from WT littermates (Figure 1A). This result not only indicated that Sam68 promotes mGluR-dependent translation of ARC, but also revealed that ARC may be degraded in an activity-dependent manner. Indeed, pretreating slices with the proteasome inhibitor MG132 blocked this decrease (Figures 1A and S2B), suggesting that mGluR-I stimulation led to proteasomal degradation of ARC, evident in the KO mice presumably due to their lack of mGluR-I-stimulated ARC protein synthesis. This degradation was not unique to Sam68 KO mice as DHPG application in WT slices induced a rapid and transient increase in ARC levels followed by a prolonged decrease, which could similarly be blocked by MG132. In contrast, levels of the cytoskeletal protein β actin did not change following DHPG application (Figure 1B). Therefore, we found that ARC is rapidly depleted by proteasomal degradation following mGluR activation in both Sam68 KO and WT mice.
Figure 1. Proteasome Inhibition Rescues mGluR-Triggered Translation of ARC and mGluR-LTD.

(A) Hippocampal slices from Sam68 KO or WT mice were treated with vehicle (VEH) or 50 μM (R,S)-3,5-DHPG for 5 min. (S)-MG132 (5 μM) was added for 1 hr before and during DHPG treatment. (Right) Densitometric quantification of western blots normalized to GapDH. Each dot represents a separate experiment consisting of pooled lysate from three slices. The average for each condition was generated from eight separate experiments using slices from four KO and WT mice. An asterisk denotes a significant difference from WT (VEH). ([VEH] WT, 100.0 ± 4.2; KO, 99.0 ± 6.8; [DHPG] WT, 161.6 ± 12.7; KO, 60.5 ± 13.1; KO [MG132], 99.2 ± 14.9).
(B) Western blots (left) and scatter plot (right) showing time courses of protein levels after three different treatments. (Left) Rat hippocampal slices were treated with 100 μM DHPG for 6 min (top, ARC [red circles] and actb [not plotted]); vehicle (middle, ARC [open circles]) or 5 μM MG132 for 1 hr before and during DHPG treatment (bottom, ARC [black circles]). Each time point represents pooled lysate from three slices. (Right) Time course plot showing DHPG application (shaded red area) results in sustained depletion in ARC protein levels that is blocked by proteasome inhibition. Each dot represents average ARC levels at indicated time points (n = 4 rats for DHPG, n = 2 rats for VEH and MG132/DHPG).
(C) Extracellular field recordings (fEPSP) in acute hippocampal slices show Sam68 KO mice (red filled circles) lack mGluR-LTD at Schaffer collateral synapses induced by 50 μM DHPG for 5 min. MG132 (5 μM) for 1 hr before and during DHPG treatment rescued LTD (red open circles). Dashed box indicates time of DHPG application. Scale bars indicate 10 ms and 0.25 mV. (Right) Average fEPSP slope during the last 5 min of recording (WT, 59.4 ± 3.9; KO, 91.9 ± 8.1; KO MG132, 59.8 ± 3.9).
(D) Sam68 KO mice lack mGluR-LTD induced by PP-LFS for 15 min (2 pulses at 50 ms ISI, 1Hz). MG132 (5 μM) for 1 hr before and during PP-LFS rescued LTD (WT, 67.7 ± 7.3; KO, 99.8 ± 6.5; KO MG132, 70.4 ± 8.1).
(E) Whole-cell recordings in voltage clamp (−60 mV) in acute hippocampal slices (mouse) with an antisense oligonucleotide against ARC (AS, 150 μM, filled red circles) or a scrambled oligo (Scr, 150 μM, filled black circles) loaded into the patch pipette. Proteasomal inhibition rescues mGluR-LTD blocked by acute knockdown of ARC synthesis by antisense oligonucleotides (AS+MG132, red open circles) (Scr, 72.2 ± 8.2; AS, 120.3 ± 9.5; AS+MG132, 70.0 ± 9.1).
Summary data consist of mean ± SEM.
As ARC is necessary for mGluR-LTD, we measured plasticity in the Sam68 KO mice and found that both chemically induced (DHPG; Figure 1C) and synaptically induced (PP-LFS; Figures 1D and S1A) mGluR-LTD were reduced in KO animals compared with WT littermates. The absence of Sam68 did not affect basal synaptic transmission (Figures S1C–S1E) or translation-independent NMDA-LTD (Figures S1F and S1G). While mammalian target of rapamycin (mTOR) activity is reportedly decreased in adipose tissue of Sam68 KO mice (Huot et al., 2012), we observed normal phosphorylation of mTOR and S6 Kinase in response to DHPG application, suggesting that mGluR signaling and mTOR activity are intact in the hippocampus of Sam68 KO mice (Figure S2A).
Inhibition of the Proteasome Rescues mGluR-LTD from Loss of ARC Translation in Both Sam68 KO and WT Mice
As treatment with MG132 prevented mGluR-mediated degradation of ARC in both Sam68 KO and WT mice, we next investigated the consequences of proteasome inhibition on mGluR-LTD. Blocking the degradation of ARC with MG132 fully rescued both chemically and synaptically induced mGluR-LTD in the Sam68 KO mice (Figures 1C and 1D). This rescue occurred without a change in the paired-pulse ratio (PPR; Figure S1B) and was associated with changes in AMPAR surface expression, consistent with classical, postsynaptic mGluR-LTD. Virally mediated knockdown of Sam68 blocked GluR1 internalization in primary neuronal cultures following DHPG application and pre-treatment with MG132 could rescue this effect (Figure S2D). To determine whether proteasome inhibition could rescue acute deficits in ARC translation, we introduced ARC antisense (ARC AS) oligonucleotides into CA1 pyramidal neurons in acute slices from WT mice by whole-cell patch pipette. Infusion of ARC AS oligos, but not scrambled oligos (Scr), blocked mGluR-LTD (Waung et al., 2008) (Figure 1E). Consistent with our experiments in the Sam68 KO mice, inhibition of the proteasome was able to rescue mGluR-LTD from the effects of the ARC AS oligos in WT neurons (Figure 1E). Together, these results suggest that proteasome inhibition allows for LTD to be induced in the absence of mGluR-dependent ARC translation in either Sam68 KO or WT animals.
Proteasome Inhibition Increases Dendritic PRP Levels with No Change in mGluR-LTD Magnitude
We next investigated the effects of proteasome inhibition on dendritic protein abundance in WT animals. Inhibition of the proteasome for 1 hr significantly raised levels of ARC and other proteins implicated in mGluR-LTD such as OPHN1 and PSD95, above baseline in either microdissected S. Radiatum (Figure 2A) or synaptosomal fractions prepared from acute hippocampal slices (Figure S3A). This acute increase in local PRP levels was not accompanied by a change in baseline transmission or PPR (Figure S3B), indicating that increased abundance of PRPs alone, in the context of proteasomal blockade, is not sufficient to induce a depression in baseline transmission. Additionally, inhibition of the proteasome for 1 hr had no effect on the magnitude of mGluR-LTD recorded in slices prepared from either mice or rats (Figures 2B and 2C), suggesting that elevated PRP levels may not be correlated with enhanced magnitude of mGluR-LTD.
Figure 2. Proteasomal Inhibition Increases Dendritic PRP Levels with No Change in mGluR-LTD Magnitude.

(A) Western blots of PRPs from rat microdissected S. Radiatum incubated in 5 μM MG132 for 1 hr or vehicle (APP, amyloid precursor protein; OPHN1, oligophrenin 1; PSD95, post-synaptic density 95; FMRP, fragile X mental retardation protein; ARC, activity-regulated cytoskeletal-associated protein; actb, β-actin). (Right) Densitometric quantification shows brief proteasome inhibition significantly increases dendritic levels of PSD95, ARC, and OPHN1 compared with vehicle condition (APP, 3.1 ± 6.5; PSD95, 27.4 ± 7.0; ARC, 61.5 ± 5.9; FMRP, 7.0 ± 4.7; OPHN1, 49.7 ± 10.4). The asterisk denotes significantly different than vehicle.
(B and C) MG132 does not affect mGluR-LTD at Schaffer collateral synapses in acute hippocampal slices. Dashed boxes show time of DHPG application (mouse, 50 μM, 5 min; rat, 100 μM, 6 min). MG132 (5 μM) for 1 hr before and during DHPG treatment does not change mGluR-LTD magnitude in mice (B) or rats (C). Scale bars indicate 10 ms and 0.25 mV. (Right) Average fEPSP slope during the last 5 min of the recording ([mouse] VEH, 61.3 ± 2.6; MG132, 57.2 ± 2.8; [rat] VEH, 71.6 ± 4.7; MG132, 70.2 ± 8.1).
Summary data consist of mean ± SEM.
Proteasome Inhibition Rescues mGluR-LTD Blocked by Inhibitors of Diverse Translational Pathways
Inhibition of the proteasome maintains basal ARC levels and rescues mGluR-LTD in Sam68 KO animals (Figure 1). We next tested whether proteasome inhibition could generally rescue mGluR-LTD from deficits in other translational pathways. Application of the translational blockers cycloheximide (CHX; Figure 3A) or ansiomycin (ANISO; Figure S3C) inhibited mGluR-LTD in WT rats and mice as expected (Huber et al., 2000) and resulted in the rapid depletion of several PRPs from synaptosomal fractions following DHPG application (Figure S4A). In contrast, coincubation with MG132 and CHX clamped synaptic protein levels at baseline (Figure S2C) and fully rescued the ability of mGluR-LTD to be induced with DHPG. Therefore, LTD could be reliably induced if synaptic protein levels were clamped at baseline by concurrently inhibiting translation and the proteasome (Figures 3A and S3C). This mGluR-LTD under protein-clamped conditions was similar to translation-dependent, postsynaptically expressed mGluR-LTD in that it did not manifest with a change in PPR and could be transiently reversed by mGluR antagonists (Figure S4B) (Lodge et al., 2013). To determine whether additional translational pathways were similarly affected by proteasome inhibition, we also measured mTOR-sensitive mGluR-LTD (Hou and Klann, 2004). Consistent with our previous results, inhibition of the proteasome rescues mGluR-LTD blocked by rapamycin (Figure S3D). Thus, rescue of translation-dependent mGluR-LTD by proteasome inhibition is observed not only in Sam68 KO mice but also under various conditions where PRP synthesis is inhibited.
Figure 3. mGluR-LTD Persists in the Absence of Translation if the Proteasome Is Inhibited.

(A) Field recordings from acute hippocampal slices (mouse and rat) treated with either 60 μM CHX (blue) or 60 μM CHX and 5 μM MG132 (CHX/MG, orange) for 1 hr. Dashed boxes show time of DHPG application (mouse, 50 μM, 5 min; rat, 100 μM, 6 min). Scale bars indicate 10 ms and 0.25 mV. (Right) Average fEPSP slope during the last 5 min of recording indicates inhibiting the proteasome rescues mGluR-LTD blocked by translational inhibition ([mouse] VEH, 61.3 ± 2.6; CHX, 94.7 ± 4.7; CHX+MG132, 66.9 ± 3.9; [rat] VEH, 71.6 ± 5.2; CHX, 91.2 ± 3.3; CHX+MG132, 67.7 ± 6.0).
(B) Concurrent field (black) and whole-cell recordings (red) of mGluR-LTD (50 μM DHPG, 5 min) from mouse slices. (Left) CHX (60 μM) included in the perfusate blocked LTD (WC, 109 ± 12; field, 110 ± 6). (Middle) Lactacystin (1 μM) included in the recording pipette, with vehicle in the perfusate resulted in no significant difference in the amount of LTD between the field and whole-cell recordings (WC, 68.5 ± 7.7; field 67.7 ± 6.9). (Right) Lactacystin (1 μM) in the recording pipette and 60 μM CHX in bath blocked LTD in the field recordings, but not in the whole-cell recordings (WC, 68.7 ± 11.5; field, 108.8 ± 10.3). Scale bars indicate 0.25 mV/50 pA and 10 ms.
Summary data consist of mean ± SEM.
To confirm that mGluR-LTD can be induced in the absence of translation, we devised a set of internally controlled experiments using concurrent field and whole-cell recordings in the same slice. For these experiments, lactacystin, a proteasome inhibitor mechanistically distinct from MG132, was loaded into the patch pipette, and CHX or a vehicle was bath applied to the slice during the recordings. Similar to MG132, application of lactacystin resulted in increased PRP levels in synaptosomal fractions (Figure S3A). With CHX in the perfusate and no lactacystin in the pipette, mGluR-LTD was blocked in both the field and whole-cell recordings (Figure 3B, left). With a vehicle in the perfusate the magnitude of LTD measured from a single neuron filled with lactacystin was indistinguishable from the magnitude of LTD in the field recording (Figure 3B, middle). Finally, adding CHX to the perfusate while simultaneously recording from a single neuron loaded with lactacystin blocked extracellularly measured LTD, but intracellularly measured LTD was normal (Figure 3B, right). These results provide strong evidence that mGluR-LTD can persist in the absence of an acute increase in translation as long as PRP abundance is clamped at basal levels by inhibiting the proteasome.
Proteasomal Degradation following mGluR Activation Rapidly Depletes PRPs and Inhibits Subsequent Inductions of LTD
Our results thus far indicated an important role for the proteasome in regulating dendritic ARC abundance during mGluR-LTD induction. In addition to ARC we found decreased levels of several other PRPs (APP, OPHN1, PSD95, and FMRP) in S. Radiatum following DHPG application (e.g., 30 min post-DHPG washout) (Figures 4A and 4C). This decrease in PRP levels was likely due to proteasomal degradation as inhibition of the proteasome during LTD induction (DHPG+MG wash 30) resulted in PRP levels remaining elevated 30 min after DHPG washout (Figures 4B and 4C). Moreover, the decrease in dendritic PRP levels was correlated with an increase in the ubiquitination of ARC, APP, and PSD95 (Figure 4D), suggesting that enhanced proteasomal degradation is a general mechanism for regulating local PRP abundance during the induction of mGluR-LTD.
Figure 4. Proteasomal Degradation following mGluR Activation Rapidly Depletes PRPs and Inhibits Subsequent Inductions of LTD.

(A) Western blots of PRPs from rat microdissected S. Radiatum prepared immediately after DHPG treatment (100 μM, 6 min) or 30 min after DHPG washout (DHPG wash 30).
(B) Western blots of PRPs from rat microdissected S. Radiatum prepared 30 min after DHPG+MG132 washout (DHPG+MG wash 30). Slices in vehicle condition were treated with only MG132 and no DHPG.
(C) Densitometric analyses reveal DHPG application transiently increased dendritic PRP levels (DHPG) followed by a rapid depletion from peak levels 30 min after washout (DHPG wash30). PRP levels remain elevated if the proteasome was inhibited with MG132 (DHPG+MG wash30) during mGluR-LTD induction. Black bars indicate PRP levels after DHPG treatment relative to baseline conditions. Red bars indicate PRP levels 30 min after DHPG washout relative to levels immediately following DHPG treatment. Blue bars indicate PRP levels 30 min after DHPG+MG132 washout relative to control (MG132 alone).
(D) Western blots show increased ubiquitination of ARC, APP, and PSD95 after mGluR activation. Acute hippocampal slices were treated with vehicle, or DHPG (100 μM, 6 min), and lysed after 30 min of DHPG washout. Immunoprecipitations were performed as indicated, and ubiquitination was assessed by an anti-Ub Ab.
(E) (Left) Field recordings from acute hippocampal slices (rat) treated with a vehicle (VEH, black) or 5 μM MG132 (MG132, red) for 1 hr. An initial round of LTD was induced (100 μM DHPG, 6 min), followed by a subthreshold stimulus (100 μM DHPG, 3 min). Dashed boxes show time of DHPG application. (Right) For each experiment, the magnitude of the initial LTD (1) is plotted on the x axis with the magnitude of the subsequent round of LTD (2), relative to the first, plotted on the y axis. Under vehicle conditions a subthreshold stimulus did not, on average, produce a second round of LTD. However, inhibition of the proteasome allows for a subsequent round of LTD to be induced.
Summary data consist of mean ± SEM.
We hypothesized that proteasomal degradation of PRPs might regulate the inducibility of mGluR-LTD by rapidly depleting PRP levels below a permissive threshold after an initial round of LTD, thereby limiting subsequent inductions. Application of a sub-threshold DHPG stimulus (3 min, 100 μM) produced a transient depression, but failed to induce LTD in either vehicle or MG132-treated hippocampal slices (Figure S4D). Furthermore, if the proteasome was intact this subthreshold stimulus did not result in any further depression if applied following a standard stimulation protocol that was able to reliably induce LTD (DHPG 6 min, 100 μM; Figures 4E and S4C). However, if the proteasome was inhibited during the initial induction of LTD, then a subsequent subthreshold stimulus elicited a subsequent round of LTD (Figure 4E). This metaplastic effect was sustained throughout multiple rounds of subthreshold inductions (Figure S4E). Therefore, while inhibition of the proteasome does not lower the threshold for induction of an initial round of LTD, it does so for subsequent inductions of LTD. These results suggest that enhanced proteasomal degradation ensures PRP levels remain below a permissive threshold following an initial LTD induction to limit subsequent inductions of LTD.
DISCUSSION
We find that the increase in PRPs following mGluR activation is transient with dendritic levels of certain PRPs depleted to below baseline within 30 min due to proteasomal degradation. In addition, we report that Sam68 KO mice lack mGluR-dependent ARC translation and LTD. In these mice, blocking the proteasome rescues mGluR-LTD, indicating that preserving basal ARC is sufficient for LTD induction. In WT animals, mGluR-LTD can be induced if levels of PRPs are clamped during mGluR activation by concurrently blocking translation and proteasomal degradation. Similarly, concurrent inhibition of the proteasome also relieves the block of mGluR-LTD due to ARC AS oligos or rapamycin. Our findings show that increases in dendritic PRPs are not necessary for translation-dependent mGluR-LTD. We propose that mGluR-triggered translation serves to counterbalance enhanced degradation ensuring PRP levels remain above a permissive threshold during LTD induction. Increased ubiquitination following mGluR activation allows for rapid depletion of local PRP levels by the proteasome, which engages metaplasticity by inhibiting subsequent inductions of LTD. Therefore, coordination between translation and degradation by the ubiquitin-proteasome-system regulates local protein abundance following mGluR activation and the inducibility of synaptic plasticity.
The need to deplete PRP levels after an initial round of LTD may arise from the persistence of mGluR signaling after agonist washout. mGluR-LTD can be de-depressed (Figure S4B) by transient application of mGluR antagonists, indicating sustained mGluR signaling throughout the expression of LTD (Lodge et al., 2013). In our model, mGluR-LTD induction is achieved by the coincidence of PRP levels above a threshold and signaling from mGluRs. If PRPs remained above threshold following an initial round of LTD, persistent mGluR signaling could lead to subsequent unintended inductions of LTD. How mGluR signaling persists in the absence of ligand is unknown. One interesting possibility is that a brief application of agonist may alter the binding of mGluRs to intracellular partners such as Homer leading to agonist-independent activity (Ango et al., 2001). Our results indicate that enhanced proteasomal degradation following an initial bout of mGluR-LTD ensures PRPs remain below threshold until an appropriate activation is received for a subsequent round of LTD. Increased ubiquitination of ARC, FMRP, and PSD95 has been observed following synaptic stimulation, raising the possibility that the enhanced degradation of PRPs we observe during LTD induction is due to an mGluR-dependent increase in E3 ligase activity (Figures 4C and 4D) (Colledge et al., 2003; Hou et al., 2006; Mabb et al., 2014; Nalavadi et al., 2012).
Inhibition of the protein synthesis-dependent, late-phase long-term potentiation (L-LTP) by translational blockers is alleviated by co-application of proteasome inhibitors (Fonseca et al., 2006), suggesting common roles for translation and the proteasome in regulating PRP abundance during mGluR-LTD and L-LTP (Dong et al., 2014). In our model, synthesis and degradation are precisely coordinated during LTD induction to briefly maintain dendritic PRP levels at a permissive threshold for mGluR-LTD induction. Conversely, PRP levels below threshold inhibit the induction of LTD even if the mGluR signal is generated. In this way, dendritic levels of ARC and other PRPs can act as molecular switches for plasticity by determining if mGluR signaling will induce LTD. If translation is inhibited either pharmacologically or genetically, as in Sam68 KO mice, degradation is uncompensated, and DHPG application results in the rapid depletion of PRPs from synapses (Figures 1A and S4A). Our results suggest that the block of LTD by translational inhibitors may be due to uncompensated depletion of synaptic PRPs below a permissive threshold, rather than a lack of acute PRP synthesis.
Basal levels of ARC protein can be influenced by previous activation, varying substantially between individual neurons (Jakkamsetti et al., 2013). Several mechanisms exist to suppress ARC expression, including activity-dependent as well as nonsense-mediated mRNA decay (Farris et al., 2014; Giorgi et al., 2007), poor initiation of ARC mRNA (Park et al., 2008), and rapid turnover of ARC protein by the proteasome (Soulé et al., 2012). The relative scarcity of ARC during basal conditions allows for stimulus-induced increases in ARC to act as a plasticity switch. For example, the dendritic seeding of ARC mRNA in response to a brief, non-LTD inducing, novel experience increases the likelihood that the activated neuron will subsequently undergo mGluR-LTD ex vivo (Jakkamsetti et al., 2013). In the context of our findings, this dendritic seeding may ensure enough substrate for mGluR-triggered translation to keep ARC protein above the permissive threshold. Interestingly, repeated exposures to a novel environment that may induce mGluR-LTD in vivo, inhibits subsequent induction of mGluR-LTD ex vivo (Jakkamsetti et al., 2013). This observation is consistent with our model that enhanced proteasomal degradation following an initial round of LTD inhibits subsequent inductions.
We found that acute inhibition of the proteasome did not decrease basal transmission or PPR even though certain dendritic PRPs increased to comparable levels to those observed immediately following DHPG application (Figures 2A and 4A). Thus, elevated PRPs alone are not sufficient to induce LTD, which extends previous findings from dissociated cultures (Niere et al., 2012; Park et al., 2008). Furthermore, increasing dendritic PRP levels by inhibiting the proteasome prior to DHPG application did not alter the magnitude of LTD, which is consistent with a previous report showing no effect of MG132 on LTD in mice (Citri et al., 2009). In another study, however, inhibiting the proteasome reduced the magnitude of mGluR-LTD (Hou et al., 2006). The reason for this discrepancy is unclear, but it could reflect differences in the experimental designs. Because of the widespread actions of MG132 or lactacystin on the neuronal proteome, we cannot solely attribute the effects, or lack thereof, specifically to the subset of PRPs we examined. However, we find that blocking proteasome degradation during mGluR-LTD unmasks a novel form of metaplasticity that facilitates subsequent rounds of LTD. As mutations in several components of the proteasomal machinery results in neurological disorders, such as Angelman syndrome, and X-linked mental retardation, we predict that altered metaplasticity could be involved in the pathogenesis of these disorders.
Although previous studies have shown that mGluR-LTD can occur independently of protein synthesis under some circumstances, for example, in the fragile X syndrome disease mouse model (Hou et al., 2006; Nosyreva and Huber, 2006), in 3- to 4-month-old rodents (Moult et al., 2008), or in early postnatal life (Nosyreva and Huber, 2005), the mGluR-LTD that we studied in young-adult rodents is dependent on protein translation. Our conclusions that mGluR-LTD does not require an acute increase in dendritic protein levels and that the magnitude of LTD does not correlate with PRP abundance may seem at odds with current hypotheses that elevated levels of PRPs contribute to enhanced mGluR-LTD in FMRP KO mice. FMRP-null mice display a general increase in basal protein levels, but an absence of mGluR-triggered translation (Darnell and Klann, 2013). One theory is that elevated basal PRP levels in the FMRP-null mice bypass the need for mGluR-triggered translation and result in exaggerated LTD, which may contribute to the neurological symptoms of FXS (Bear et al., 2004). An important difference between our study and previous results is that FMRP KO mice display generally increased basal protein levels while inhibition of the proteasome will elevate the levels of proteins actively undergoing degradation. A robust finding in FMRP KO mice is the insensitivity of mGluR-LTD to translational inhibition (Nosyreva and Huber, 2006). In this case, we draw a parallel to our clamped mGluR-LTD paradigm. Several reports have suggested that loss of FMRP results in disruption of the proteasomal machinery (Tsai et al., 2012; Zhao et al., 2011). Perhaps decreased proteasomal activity in the FMRP KO mouse allows for normal induction of mGluR-LTD in the presence of translational inhibitors, similar to our demonstration of clamped mGluR-LTD. In addition to current efforts to modulate translation and mGluR signaling, pharmacological manipulation of the proteasomal machinery might be an additional avenue for ameliorating the neurological symptoms of fragile X syndrome.
EXPERIMENTAL PROCEDURES
Electrophysiology
Rats and mice (P20–P25) were anesthetized and killed in compliance with the guidelines of the Institutional Animal Care and Use Committee. Acute hippocampal slices were prepared (400 μm) in chilled sucrose solution and recovered in 50/50 sucrose/ACSF at 30°C for 20 min before switching to 30°C ACSF for an additional 1 hr recovery at room temperature. All recordings were conducted at 30°C with 100 μM picrotoxin (Sigma). For whole-cell recordings, a K-gluconate-based internal solution was used. See the Supplemental Experimental Procedures for extended recording conditions.
Western Blotting
Acute hippocampal slices were prepared as above and treated with drugs as indicated. Following treatment, slices were transferred to ice-cold ACSF, and S. Radiatum was excised. Lysates were prepared in radioimmunoprecipitation assay buffer (RIPA) buffer and loaded onto 10% Bis-tris acrylamide gels. For all main figure blots two separate experiments (lanes) are shown for each experimental condition. For Figures 2A and 4C, each dot represents a separate experiment consisting of pooled lysate from eight microdissected slices normalized to β actin (actb). The average for each condition was generated from four separate experiments using slices from four rats. See the Supplemental Experimental Procedures for antibodies and synaptosomal preparation.
Statistical Analysis
N values are displayed as (slices/cells; animals). Statistical analyses were performed using OriginPro 9.0 (OriginLab). ANOVAs were performed on experiments with multiple groups followed by post hoc pairwise analyses with Bonferroni correction. An asterisk denotes a p value of < 0.05, and n.s. indicates a p value > 0.05. All values are expressed as mean ± SEM (percentage of baseline).
Supplementary Material
Highlights.
mGluR-LTD can occur in the absence of an acute increase in dendritic protein levels
mGluR-LTD leads to both synthesis and degradation of plasticity related proteins
Proteasome inhibition rescues deficits in mGluR-LTD caused by blocking translation
Sam68 is required for mGluR-induced ARC synthesis and LTD
Acknowledgments
We thank all members of the Jordan and Castillo laboratories for help with experiments and valuable discussions. We are especially grateful to Adina R. Buxbaum for her critical reading of the manuscript. This work supported by funding from the NIH (R01-MH081935 and R01-DA017392 to P.E.C., R01-AG039521 to B.A.J., and F31-NS073200 and T32-GM007288 to M.E.K.). We thank Dr. Stéphane Richard (McGill University) for the Sam68 mice.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.02.020.
AUTHOR CONTRIBUTIONS
M.E.K. and B.A.J. carried out research and analyzed data. All authors designed research and wrote and edited the manuscript.
References
- Ango F, Prézeau L, Muller T, Tu JC, Xiao B, Worley PF, Pin JP, Bockaert J, Fagni L. Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein Homer. Nature. 2001;411:962–965. doi: 10.1038/35082096. [DOI] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Ben Fredj N, Grange J, Sadoul R, Richard S, Goldberg Y, Boyer V. Depolarization-induced translocation of the RNA-binding protein Sam68 to the dendrites of hippocampal neurons. J Cell Sci. 2004;117:1079–1090. doi: 10.1242/jcs.00927. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:1144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- Citri A, Soler-Llavina G, Bhattacharyya S, Malenka RC. N-methyl-D-aspartate receptor- and metabotropic glutamate receptor-dependent long-term depression are differentially regulated by the ubiquitin-proteasome system. Eur J Neurosci. 2009;30:1443–1450. doi: 10.1111/j.1460-9568.2009.06950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci. 2013;16:1530–1536. doi: 10.1038/nn.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Prisco GV, Huang W, Buffington SA, Hsu CC, Bonnen PE, Placzek AN, Sidrauski C, Krnjević K, Kaufman RJ, Walter P, Costa-Mattioli M. Translational control of mGluR-dependent long-term depression and object-place learning by eIF2α. Nat Neurosci. 2014;17:1073–1082. doi: 10.1038/nn.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Bach SV, Haynes KA, Hegde AN. Proteasome modulates positive and negative translational regulators in long-term synaptic plasticity. J Neurosci. 2014;34:3171–3182. doi: 10.1523/JNEUROSCI.3291-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Farris S, Lewandowski G, Cox CD, Steward O. Selective localization of arc mRNA in dendrites involves activity- and translation-dependent mRNA degradation. J Neurosci. 2014;34:4481–4493. doi: 10.1523/JNEUROSCI.4944-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nägerl UV. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron. 2006;52:239–245. doi: 10.1016/j.neuron.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Giorgi C, Yeo GW, Stone ME, Katz DB, Burge C, Turrigiano G, Moore MJ. The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell. 2007;130:179–191. doi: 10.1016/j.cell.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Grange J, Belly A, Dupas S, Trembleau A, Sadoul R, Goldberg Y. Specific interaction between Sam68 and neuronal mRNAs: implication for the activity-dependent biosynthesis of elongation factor eEF1A. J Neurosci Res. 2009;87:12–25. doi: 10.1002/jnr.21824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z, Maehr R, et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140:704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus C, Schuman EM. Proteostasis in complex dendrites. Nat Rev Neurosci. 2013;14:638–648. doi: 10.1038/nrn3546. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huot ME, Vogel G, Zabarauskas A, Ngo CT, Coulombe-Huntington J, Majewski J, Richard S. The Sam68 STAR RNA-binding protein regulates mTOR alternative splicing during adipogenesis. Mol Cell. 2012;46:187–199. doi: 10.1016/j.molcel.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Iijima T, Wu K, Witte H, Hanno-Iijima Y, Glatter T, Richard S, Scheiffele P. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell. 2011;147:1601–1614. doi: 10.1016/j.cell.2011.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakkamsetti V, Tsai NP, Gross C, Molinaro G, Collins KA, Nicoletti F, Wang KH, Osten P, Bassell GJ, Gibson JR, Huber KM. Experience-induced Arc/Arg3.1 primes CA1 pyramidal neurons for metabotropic glutamate receptor-dependent long-term synaptic depression. Neuron. 2013;80:72–79. doi: 10.1016/j.neuron.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein ME, Younts TJ, Castillo PE, Jordan BA. RNA-binding protein Sam68 controls synapse number and local β-actin mRNA metabolism in dendrites. Proc Natl Acad Sci USA. 2013;110:3125–3130. doi: 10.1073/pnas.1209811110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge D, Tidball P, Mercier MS, Lucas SJ, Hanna L, Ceolin L, Kritikos M, Fitzjohn SM, Sherwood JL, Bannister N, et al. Antagonists reversibly reverse chemical LTD induced by group I, group II and group III metabotropic glutamate receptors. Neuropharmacology. 2013;74:135–146. doi: 10.1016/j.neuropharm.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Lukong KE, Richard S. Motor coordination defects in mice deficient for the Sam68 RNA-binding protein. Behav Brain Res. 2008;189:357–363. doi: 10.1016/j.bbr.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Je HS, Wall MJ, Robinson CG, Larsen RS, Qiang Y, Corrêa SA, Ehlers MD. Triad3A regulates synaptic strength by ubiquitination of Arc. Neuron. 2014;82:1299–1316. doi: 10.1016/j.neuron.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Corrêa SA, Collingridge GL, Fitzjohn SM, Bashir ZI. Co-activation of p38 mitogen-activated protein kinase and protein tyrosine phosphatase underlies metabotropic glutamate receptor-dependent long-term depression. J Physiol. 2008;586:2499–2510. doi: 10.1113/jphysiol.2008.153122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dys- regulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadif Kasri N, Nakano-Kobayashi A, Van Aelst L. Rapid synthesis of the X-linked mental retardation protein OPHN1 mediates mGluR-dependent LTD through interaction with the endocytic machinery. Neuron. 2011;72:300–315. doi: 10.1016/j.neuron.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalavadi VC, Muddashetty RS, Gross C, Bassell GJ. Dephosphorylation-induced ubiquitination and degradation of FMRP in dendrites: a role in immediate early mGluR-stimulated translation. J Neurosci. 2012;32:2582–2587. doi: 10.1523/JNEUROSCI.5057-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niere F, Wilkerson JR, Huber KM. Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. J Neurosci. 2012;32:5924–5936. doi: 10.1523/JNEUROSCI.4650-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2005;25:2992–3001. doi: 10.1523/JNEUROSCI.3652-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronetto MP, Messina V, Bianchi E, Barchi M, Vogel G, Moretti C, Palombi F, Stefanini M, Geremia R, Richard S, Sette C. Sam68 regulates translation of target mRNAs in male germ cells, necessary for mouse spermatogenesis. J Cell Biol. 2009;185:235–249. doi: 10.1083/jcb.200811138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29:1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulé J, Alme M, Myrum C, Schubert M, Kanhema T, Bramham CR. Balancing Arc synthesis, mRNA decay, and proteasomal degradation: maximal protein expression triggered by rapid eye movement sleep-like bursts of muscarinic cholinergic receptor stimulation. J Biol Chem. 2012;287:22354–22366. doi: 10.1074/jbc.M112.376491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Mack KJ, Malter JS. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci USA. 2003;100:14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai NP, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN, Cowan CW, Huber KM. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 2012;151:1581–1594. doi: 10.1016/j.cell.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007;5:e52. doi: 10.1371/journal.pbio.0050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Chuang SC, Bianchi R, Wong RK. Dual regulation of fragile X mental retardation protein by group I metabotropic glutamate receptors controls translation-dependent epileptogenesis in the hippocampus. J Neurosci. 2011;31:725–734. doi: 10.1523/JNEUROSCI.2915-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.