

Abstract
The term early age-of-onset Alzheimer’s disease (EOAD) identifies patients who meet criteria for AD, but show onset of symptoms before the age of 65. We map progression of gray matter (GM) atrophy in EOAD patients compared to late onset AD (LOAD). T1-weighted MRI scans were obtained at diagnosis and one-year follow-up from 15 EOAD, 10 LOAD, and 38 age-matched controls. Voxel-based and tensor-based morphometry were used, respectively, to assess the baseline and progression of atrophy. At baseline, EOAD patients already showed a widespread atrophy in temporal, parietal, occipital and frontal cortices. After one year, EOAD had atrophy progression in medial temporal and medial parietal cortices. At baseline, LOAD patients showed atrophy in the medial temporal regions only, and, after one year, an extensive pattern of atrophy progression in the same neocortical cortices of EOAD. Although atrophy mainly involved different lateral neocortical or medial temporal hubs at baseline, it eventually progressed along the same brain default-network regions in both groups. The cortical region showing a significant progression in both groups was the medial precuneus/posterior cingulate.
Keywords: Voxel Based Morphometry, Tensor Based Morphometry, atrophy progression, Alzheimer’s disease, age of onset, default mode network
1. INTRODUCTION
Alzheimer’s disease (AD) is not a unitary syndrome. Patients with early onset (<65 years, EOAD) and late onset (>65 years, LOAD) have different cognitive [1–5], and neuroimaging profiles [6–10]. More recently, in vivo salient differences in anatomo-functional network involved in EO and LOAD have been detected [11, 12].
Clinically, episodic memory impairment is the central symptom in LOAD, even in the early, prodromal “mild cognitive impairment” (MCI) phase. As the disease progresses, and with the appearance of other cognitive deficits, memory impairment remains the central core of LOAD dementia. Conversely, EOAD patients present early with a multidomain cognitive impairment, performing poorly in visuo-spatial, language, executive, and attention tasks [13–15]. Focal, atypical presentations of EOAD comprise logopenic primary progressive aphasia and posterior cortical atrophy [16, 17]. Episodic memory deficits are frequently present in EOAD, but they are not predominant nor isolated, and can be difficult to be evaluated because of concomitant language and visuo-spatial difficulties [18].
Accordingly with its clinical profile, EOAD has been associated with a posterior pattern of cortical atrophy, centered on the parietal and lateral temporal lobes, with relatively less involvement of the medial temporal structures [8, 17], while the amnestic syndrome of typical LOAD has been associated with hippocampal atrophy.
In general, cognitive longitudinal studies have demonstrated more rapid decline in EOAD than LOAD [2, 19]. One of the first studies assessing the cognitive differences (using the Mini Mental State Examination, MMSE [20]) between EO- and LOAD showed greater decline in executive domains (attentional items of MMSE) in EOAD patients over a 2-year follow-up period [21].
Greater efforts have been devoted to the study of the anatomical progression of typical LOAD. In the early nineties, Braak and Braak conducted the first pathological studies to identify the trajectory of neurodegeneration in AD [22, 23]. In the last decades, imaging studies have investigated in vivo the progression of atrophy in LOAD, showing a spreading of the pathology through a widespread pattern including medial and lateral temporal, frontal, and parietal lobes bilaterally, with maximal differences found in the left medial temporal regions [24] (see for a review [25]). To our knowledge, only one study has assessed atrophy progression in EO relative to LOAD so far showing more rapid decline and more rapid cortical thinning in widespread association cortices in EOAD group [26]. Some studies, conducted between 80s and 90s, already demonstrated that heterogeneous nonmemory language and visuospatial impairments in early AD were related to and predicted by the earlier-appearing distribution of metabolic reductions in the association neocortex [27, 28].
Within this framework, we used tensor based morphometry (TBM) to map in vivo progression of regional gray matter (GM) atrophy in EOAD. We investigated the GM loss at the time of diagnosis and whether GM contraction during the follow-up period of one year. The pattern of atrophy progression of EOAD patients was compared with that of a group of LOAD cases.
2. METHODS
2.1 Subjects
We searched the University of California, San Francisco Memory and Aging Center (UCSF MAC) database for all patients with a diagnosis of probable AD [29]..
Clinical diagnosis was based on a multi-disciplinary evaluation including a history and neurological examination by a neurologist, caregiver interview by a nurse, and a neuropsychological test battery. From this group, we selected patients with two structural MRIs one-year apart (at presentation and one-year follow-up) and clinical and cognitive evaluation at the same time of scan acquisitions. Fifteen EOAD patients (<65 years at disease onset) were included in the study. Ten LOAD (>65 years at onset) with similar symptom duration and global cognitive performance (MMSE) were included. No patients had family history suggestive of autosomal dominant disease or psychiatric illness. Table 1 shows main demographic and clinical features of the two groups of patients studied.
Table 1.
Mean demographic and clinical characteristics of EO and LOAD patients and of groups of normal controls.
| EOAD | LOAD | Young controls | Old controls | |
|---|---|---|---|---|
| Number of subjects | 15 | 10 | 17 | 21 |
| Men/Women | 11/4 | 5/5 | 6/11* | 8/13 |
| Mean age (SD) [years] | 56 (5) | 76 (4) | 60 (4) | 74 (2.5) |
| Education (SD) [years] | 15. 9 (2.9) | 15.8 (2.9) | 17.2 (2.4) | 16.7 (2.8) |
| Symptom duration (SD) [years between symptom onset and time 1st scan] | 2.1 (0.7) | 1.7 (0.5) | - | - |
| Months between MRI scans (SD) [months] | 14 (3) | 15 (4) | 13 (4) | 13 (3.5) |
| MMSE at baseline | 22.3 (4.0) | 22.9 (6.9) | 29.8 (0.4) | 29.7 (0.4) |
| MMSE at follow up | 17.8 (5.6)** | 19.4 (6.6) | 29.4 (0.8) | 29.3 (0.8) |
EOAD vs. young controls; p=0.03
baseline vs. follow up MMSE in EOAD; p< .01
Abbreviations: EOAD=early onset Alzheimer Disease, LOAD=late onset Alzheimer Disease, MMSE= mini-mental state examination; SD= standard deviation.
Age at onset and symptom duration were determined based on an interview with caregivers who lived with the patient or regularly saw the patient. We obtained the initial symptoms of cognitive decline, and the disease onset was defined as the first cognitive symptom reported. All the patients had memory deficits as first complaint. Also, we excluded patients with atypical variant of AD (posterior cortical atrophy, logopenic variant of primary progressive aphasia, or frontal-variant AD).
Thirty-eight healthy subjects matched separately to each group for age and sex, with no history of neurological or psychiatric disorders, were used as controls (Table 1). The study was approved by the UCSF committee on human research. All subjects or their caregivers provided written informed consent before participating.
2.2 Cognitive testing
EO-AD patients underwent a comparable neuropsychological screening battery testing at study entry and follow-up visit. The neuropsychological measures included in our bedside screening protocol have been described previously [30]. Briefly, general intellectual functioning was assessed using the MMSE [20]. The California Verbal Learning Test – Short Form (CVLT-SF) [31, 32] was used to evaluate verbal episodic memory; visual-nonverbal episodic memory was measured using the 10–minute free recall of the Benson Figure [33]. Language assessment included an abbreviated (15-item) Boston Naming Test (BNT) [34], and semantic fluency (animals in 1 minute). Tests of executive functioning included phonemic fluency (D-words in 1 minute), and backwards digit span. Copy of the Benson Figure was used to assess visuospatial functioning.
The same detailed cognitive evaluation was obtained in LOAD patients at study entry (supplementary table 1). The MMSE was used to assess cognitive status in LOAD, longitudinally.
2.3 Image acquisition
Baseline and follow-up MRI scans were obtained on a 1.5 Tesla Magnetom VISION system (Siemens, Iselin, NJ). Structural MRI sequences included the following: (1) double spin echo sequence (repetition time [TR]=5000 ms, echo time [TE]=20/80 ms, 51 contiguous axial slices, thickness=3 mm, 1.0 × 1.25 mm2 in-plane resolution); and (2) volumetric magnetization prepared rapid gradient echo (MP-RAGE) sequence (TR=10 ms, TE=4 ms, inversion time=300 ms, flip angle=15°, coronal orientation perpendicular to the double echo sequence, matrix size=256 × 192, voxel resolution=1.0 x 1.0 x 1.0 mm, slab thickness=1.5 mm).
2.4 Voxel-based morphometry analysis
VBM analysis [35, 36] includes two steps: spatial preprocessing (normalization, segmentation, Jacobian modulation, and smoothing) and statistical analysis. Both steps were implemented using the Statistical Parametric Mapping (SPM2) software package (Wellcome Department of Imaging Neuroscience, London; http://www.fil.ion.ucl.ac.uk/spm) running on Matlab 7.13 (Math-Works, Natick, MA). MRI images were preprocessed using an optimized method for the spatial normalization of GM [36]. Ad hoc template and a priori images were created by averaging both age- and sex-matched normal controls and patients’ scans. This procedure involved spatial normalization of the original T1-weighted images to the Montreal Neurological Institute (MNI) stereotaxic space, segmentation into GM, WM, and cerebrospinal fluid (CSF), averaging of the images and smoothing with an 12-mm full-width half-maximum (FWHM) isotropic Gaussian kernel. An analysis of variance [ANOVA] model was performed, and age, gender, and total intracranial volume were entered into the design matrix as nuisance variables. Regionally specific differences in GM volumes were assessed using the general linear model and the significance of each effect was determined by using the theory of Gaussian fields. We created a study-specific template from all the scans of all the subjects, and then specific contrasts were performed comparing GM volumes in each AD group vs. the subset of age matched controls (EO or LOAD groups vs. younger or older age-matched controls). Moreover, an interaction analysis was applied in order to evaluate the differences in the pattern of atrophy at baseline between EO and LOAD patients (e.g., set contrast EOAD>LOAD: EOAD, LOAD, young controls, old controls, −1 1 1 −1). A conjunction analysis was performed to assess the common areas of atrophy across the AD groups, at baseline. A significance threshold of p<0.05 corrected for multiple comparisons (Family Wise Error [FWE]) was accepted. Results were also tested at p<0.001 uncorrected for multiple comparisons.
2.5 Tensor-based morphometry analysis
TBM, as implemented in SPM2, was used to map progression of regional GM atrophy over time in EO and LOAD patients. Details regarding TBM image pre-processing are described in previous studies [37–40]. We applied a bias correction to the follow-up T1-weighted scan, previously coregistered with the baseline image, to make it comparable to the early one. A high-dimensional deformation field was then used to warp the corrected late image to match the early one within subject. The amount of volume change was quantified by taking the determinant of the gradient of deformation at a single-voxel level (Jacobian determinants). The following formula was applied to the segmented GM image obtained from the first scan and the Jacobian determinant map: (Jacobian value – 1) × GM. The resulting product image represented a measure of the GM specific volume change between the first and the second scan. Study-specific template and a priori images were created by averaging each subject’s early and late normalized images. The normalization parameters were estimated by matching the customized GM template with the segmented GM image from the first scan, and then applied to the product image. Normalized images were smoothed using a 12 mm isotropic Gaussian kernel. Smoothed images were multiplied by an inclusive binary mask identifying only GM tissue. Normalized, smoothed maps of GM loss over time for each subject were entered into the statistical analysis. Using SPM, an ANOVA model was performed, and gender, age, follow-up duration, and total intracranial volume were entered into the statistical model as confounding variables. Specific contrasts were performed comparing GM contraction in all AD patients vs. controls, and in EO and LOAD groups vs. age-matched controls. Interaction and conjunction analyses were also performed in order to evaluate differences and commonalities of GM atrophy progression across the groups (for details see VBM section). Finally, maps of the average percentage GM tissue loss in EOAD and LOAD patients vs. controls were created. A significance threshold of p<0.05 corrected for multiple comparisons (FWE) was accepted. Results were also tested at p<0.001 uncorrected for multiple comparisons.
3. RESULTS
3.1 Cognitive results
MMSE mean scores at baseline were 22.3 (SD=4.0) in the EOAD group and 22.9 (SD =6.9) in the LOAD group (table 1). Symptom duration was also similar in EO (mean = 2.1 years) and LOAD (1.7 years) (p =.13). At both baseline and follow up visits, EOAD patients showed abnormal neuropsychological test scores for all cognitive domains compared with healthy controls (table 2). After one year of follow up, EOAD patients showed significant decline on the MMSE with an average loss of 4.5 points (table 2). EOAD also showed significant decline on animal fluency, and calculation compared with baseline (p<.01 after Bonferroni correction) (table 2). The MMSE was administered to 7 of the 10 LOAD patients after 1 year and it was not significantly different compared with baseline score (p = .32; average loss of 3.5 points).
Table 2.
EOAD and controls cognitive progression.
| Group | Controls | EOAD | ||
|---|---|---|---|---|
| T1 | T2 | T1 | T2 | |
| Mini-Mental State Examination score (0–30) | 29.8 (0.4) | 29.4 (0.8) | 22.3 (4.0) | 17.8 (5.6)* |
| Memory | ||||
| CVLT-SF 4 trials | 5.2 (1.4) | 3.9 (1.7) | ||
| CVLT-SF LDFR | 1.8 (1.8) | 1.1 (1.4) | ||
| Modified Rey 10-minute recall (0–17) | 12.6 (2.7) | 13.1 (2.5) | 2.2 (2.3) | 1.6 (2.5) |
| Visuospatial function | ||||
| Modified Rey copy (0–17) | 16.1 (1.1) | 15.9 (1.0) | 9.5 (4.8) | 6.1 (4.5) |
| Language | ||||
| Boston Naming Test (0–15) | 14.7 (0.6) | 14.6 (0.6) | 12.4 (3.1) | 11.3 (3.6) |
| Animal Fluency (1 min) | 24.1 (5.4) | 24.6 (5.9) | 10.4 (4.0) | 7.1 (2.6)* |
| Executive Function | ||||
| Backwards Digit Span | 5.9 (1.2) | 5.7 (1.2) | 3.3 (0.6) | 2.8 (1.1) |
| D-word generation (1 min) | 17.0 (5.2) | 17.4 (4.7) | 11.1 (5.4) | 7.2 (4.7) |
| Calculations | ||||
| Calculation Screen (0–5) | 4.7 (0.5) | 4.8 (0.5) | 3.0 (1.2) | 1.9 (1.2)* |
For each test mean and standard deviation are reported.
Significant EOAD patients vs. healthy controls differences were found for all tests both at T1 and T2 (after Bonferroni correction).
p<.01: T1 vs. T2 in EOAD patients (after Bonferroni correction).
Abbreviations: CVLT-SF LDFR= California verbal learning test-short form long delay free recall; EO-AD= early age of onset Alzheimer’s disease.
3.2 Neuroimaging results
Tables 3 and 4 and figures 1–4 summarize cross-sectional and longitudinal MRI results.
Table 3.
VBM results: regions showing gray matter (GM) loss in all AD patients vs. controls, and in EOAD and LOAD vs. age-matched controls.
| Anatomical region | SPM coordinates | All vs. controls | EOAD vs. age-matched controls | LOAD vs. age-matched controls | ||
|---|---|---|---|---|---|---|
| X | Y | Z | T value | T value | T value | |
| L anterior hippocampus | −29 | −14 | −16 | 5.92* | 4.07° (−32,−16,−18) | 4.15 ° (−30,−14,−15) |
| R anterior hippocampus | 27 | −15 | −17 | 5.25* | 4.49° (28,−17,−19) | - |
| L hippocampus tail | −32 | −36 | −4 | 5.00° | 3.94° (−32,−36,−4) | - |
| R hippocampus tail | 26 | −31 | −8 | 3.73° | 3.82° (26,−31,−8) | - |
| L angular gyrus | −41 | −54 | 36 | 5.65* | 6.71* (−40,−52,37) | - |
| R angular gyrus | 46 | −55 | 42 | 5.82* | 7.20* (44,−53,40) | - |
| R precuneus | 20 | −58 | 20 | 3.50° | 3.5° (16,−60,17) | - |
| L precuneus | −14 | −68 | 18 | 3.73° | 4.28° (−15,−69,18) | - |
| R PCC | 6 | −37 | 29 | 3.48° (6,−37,29) | - | |
| L PCC | −1 | −44 | 27 | 3.42° (−1,−44,27) | - | |
| L posterior middle temporal gyrus | −47 | −56 | 5 | 5.63* | 5.72* (−49,−53,7) | - |
| R posterior middle temporal gyrus | 57 | −47 | 14 | 5.47* | 6.28* (48,−63,1) | - |
| R middle temporal gyrus | 54 | −29 | −4 | 5.29* | 6.25* (55,−35,5) | - |
| L middle occipital | −32 | −86 | 5 | 5.04* | 5.49* (−36,−86,4) | - |
| R middle occipital | 35 | −84 | 7 | 5.32* | 6.14* (47,−80,14) | - |
| L precentral gyrus | −27 | −11 | 47 | 4.03° | 4.78° (−30,−12,48) | - |
| R precentral gyrus | 42 31 |
−2 −13 |
30 49 |
4.99° 4.14° |
5.82* (45,−1,32) 4.48° |
- |
| L IFG | −37 | 9 | 26 | 4.52° | 5.13* (−38,11,25) | - |
| R IFG | 45 | 14 | 30 | 4.28° | 4.97* (43,18,29) | - |
| L anterior insula | −34 | −3 | 13 | 3.49° | 3.28° (−36,12,16) | - |
| R anterior insula | 39 | 6 | 11 | 3.42° | 3.68° (39,18,11) | - |
p <0.05 (FWE),
p <0.001 uncorrected.
Abbreviations: EO-AD= early onset Alzheimer Disease, IFG= inferior frontal gyrus, LO-AD= late onset Alzheimer Disease, SPM= Statistical Parametric Mapping, PCC= posterior cingulate cortex.
Table 4.
TBM results: regions showing gray matter (GM) loss progression in all AD patients vs. controls, and in EOAD and LOAD vs. age-matched controls.
| Anatomical region | SPM coordinates | All vs. controls | EOAD vs. age-matched controls | LOAD vs. age-matched controls | ||
|---|---|---|---|---|---|---|
| X | Y | Z | T value | T value | T value | |
| L anterior hippocampus/parahippocam pus | −14 | 2 | −20 | 4.86° | 4.21° (−14,−16,−20) | - |
| R parahippocampus | 20 | −14 | −28 | 5.39* | - | 4.84° |
| R hippocampus tail | 16 | −36 | 6 | 3.59° | - | 4.26°(20,−32,8) |
| L amygdala | −34 | 2 | −22 | 4.49° | 3.57°(−32,2,−20) | - |
| L precuneus | −2 | −60 | 18 | 6.29* | 4.74° (0,−62,18) | 4.49° (−6,−60,20) |
| L PCC | −2 | −40 | 34 | 5.61* | 3.89° (2,−42,30) | 4.35° (−4,−42,34) |
| L inferior parietal lobule | −54 | −48 | 46 | 3.95° | - | 3.51°(−54,−34,42) |
| L posterior middle temporal gyrus | −64 | −30 | −10 | 4.47° | 3.69°(−68,−40,0) | - |
| R posterior middle temporal gyrus | 48 | −58 | 20 | - | - | 3.49° |
| L anterior middle temporal gyrus | −58 | −10 | −20 | 5.46* | - | 5.29* (−56,−12,−20) |
| R inferior temporal gyrus | −56 | −26 | −22 | 4.60° | 3.86° (68,−34,−22) | 4.05° (50,−24,−30) |
| L fusiform gyrus | −38 | −12 | −32 | 3.97° | - | 3.99°(−36,−14,−32) |
| L middle occipital | −38 | −76 | 20 | 3.25° | - | 3.90°(−38,−70,36) |
| R rectus gyrus | 8 | 24 | −22 | - | - | 3.44° |
| L superior medial frontal gyrus | 2 | 36 | 40 | 4.64° | 3.69° | - |
| R SMA | 2 | 4 | 58 | - | 3.34° | - |
| L medial orbito-frontal gyrus | −2 | 52 | −2 | 4.67° | - | 3.73°(0,50,−6) |
| L IFG | −36 | 16 | 26 | - | - | 3.60° |
p <0.05 (FWE),
p <0.001 uncorrected.
Abbreviations: EOAD= early onset Alzheimer Disease, IFG= inferior frontal gyrus, LOAD= late onset Alzheimer Disease, PCC= posterior cingulate cortex, SMA= supplementary motor area, SPM= Statistical Parametric Mapping.
Figure 1.

The figure shows grey matter (GM) atrophy at baseline (on the top; p< 0.05 FWE) and atrophy progression after one year (at the bottom) in early onset (EOAD) Alzheimer’s disease patients compared with matched healthy controls. Results are displayed on the Montreal Neurological Institute (MNI) template available on MRIcron software (http://www.mccauslandcenter.sc.edu/mricro/mricron/). Color bars denote T values (on the top) and percentage of GM reduction during follow up (at the bottom).
Figure 4.
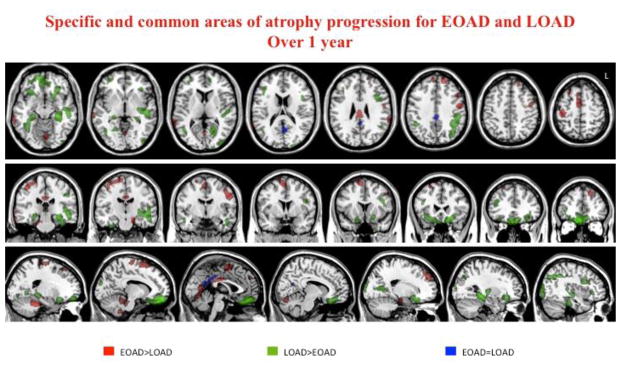
Common and specific patterns of atrophy progression in EO and LOAD patients. Common area (blue) is centered on precuneus and posterior cingulum (conjunction analysis). More severe atrophy progression in EOAD (red) vs. LOAD were found in lateral and medial frontal areas, cingulate gyrus, lateral temporal regions as well as in peri-central regions, such the post-central gyrus, and the supplementary motor areas. LOAD patients (green) showed a greater atrophy progression in ventral frontal and temporal regions, included parahippocampal and fusiform gyri, as well as in lateral frontal and temporal, and occipital regions.
3.2.1 Cross-sectional neuroimaging analysis at baseline: voxel-based morphometry
EOAD vs. younger controls
In EOAD patients at presentation, extensive GM volume loss was observed within bilateral temporal and parietal lobes (p <0.05, FWE) (figure 1 and table 3). GM atrophy was mainly found in the angular gyrus and middle temporal gyrus. GM loss extended posteriorly to occipital lobes, and anteriorly to bilateral frontal dorsal regions, including precentral and inferior frontal gyri (figure 1). Using a less stringent statistical threshold (p <0.001 uncorrected), EOAD patients also showed GM atrophy in bilateral anterior and posterior hippocampus, precuneus, posterior cingulate gyrus and anterior insula (table 3).
LOAD vs. older controls
In LOAD patients at presentation, GM volume loss was observed in left hippocampus compared to older controls (p<0.001 uncorrected) (figure 2 and table 3).
Figure 2.
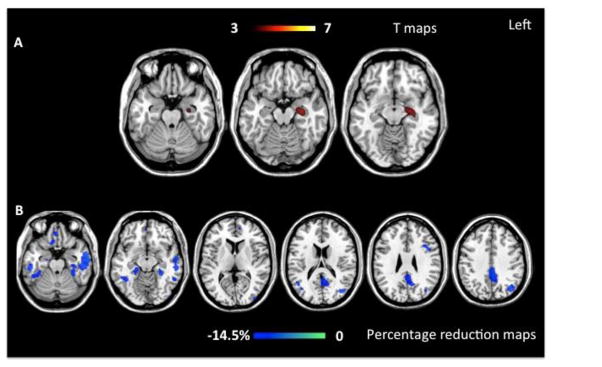
The figure shows grey matter (WM) atrophy at baseline (on the top; p< 0.001 uncorrected) and atrophy progression after one year (at the bottom) in late onset (LOAD) Alzheimer’s disease patients compared with matched healthy controls. Results are displayed on the Montreal Neurological Institute (MNI) template available on MRIcron software (http://www.mccauslandcenter.sc.edu/mricro/mricron/). Color bars denote T values (on the top) and percentage of GM reduction during follow up (at the bottom).
Specific patterns of atrophy
At baseline, GM loss specific of EOAD included the lateral inferior and superior parietal regions, and occipital lobes bilaterally, as well as left posterior temporal and left fusiform areas. No specific areas of atrophy were found for LOAD (p<0.001 uncorrected) (figure 3).
Figure 3.
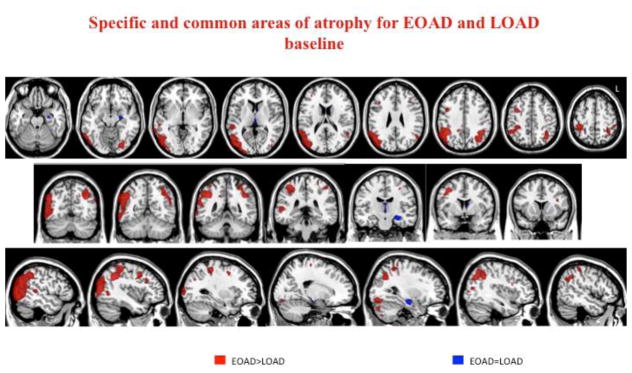
Common and specific patterns of atrophy at baseline in EO and LOAD patients. Common area (blue) is centered on left hippocampus (conjunction analysis). More severe atrophy in EOAD (red) vs. LOAD were found in the lateral inferior and superior parietal regions, and occipital lobes bilaterally, as well as left posterior temporal and left fusiform areas. No specific areas of atrophy were found for LOAD.
Common pattern of atrophy in EOAD and LOAD
The left hippocampus was the region of common GM loss in EO and LOAD at baseline (p<0.001 uncorrected) (figure 3).
3.2.2 Longitudinal neuroimaging analysis: tensor-based morphometry
EOAD vs. younger controls
Over one year, EOAD patients showed GM contraction compared to younger controls in a large medial area including left precuneus and posterior cingulate (p<0.001 uncorrected). GM contraction was also found within the temporal lobes in left hippocampus, amygdala, and posterior middle temporal and right inferior temporal gyri, and within the frontal lobes, in superior medial frontal gyrus and supplementary motor area (p<0.001 uncorrected) (figure 1 and table 4). The greatest GM loss occurred in the medial parietal areas (5%).
LOAD vs. older controls
Over one year, LOAD patients showed GM contraction compared to older controls mainly in temporal and parietal lobes. Within the temporal lobes, GM contraction was observed in right parahippocampus, hippocampus tail, posterior middle and inferior temporal gyrus, and fusiform gyri (p<0.001 uncorrected), as well as in left anterior middle temporal (p <0.05 FWE). Within the parietal lobes LOAD showed GM atrophy progression in the left precuneus and posterior cingulate, and inferior parietal lobule. Smaller regions of GM contraction were also found in left medial orbito-frontal, inferior frontal, and middle occipital gyri, and right rectus gyrus (p<0.001 uncorrected) (figure 2 and table 4). The percentage of tissue loss was of 14.5%, higher than EOAD group.
Specific patterns of atrophy progression
Comparing the pattern of atrophy progression between EO and LOAD groups each other, we found that EOAD patients accumulated greater atrophy in lateral and medial frontal areas, cingulate gyrus, lateral temporal regions as well as in peri-central regions, such the post-central gyrus, and the supplementary motor areas relative to LOAD patients (figure 4). LOAD patients showed greater atrophy in ventral frontal and temporal regions, included parahippocampal and fusiform gyri, but also in lateral frontal and temporal, and occipital regions (p<0.001 uncorrected) (figure 3).
Common pattern of atrophy progression in EOAD and LOAD
The conjunction analyses showed a common area of atrophy progression in the two AD groups relative to controls in precuneus (MNI coordinates −4,−60,18; T value 4.34) and posterior cingulate (−2,−40,32; T value 3.81) (p<0.001 uncorrected) (figure 4).
4. DISCUSSION
In this study we aimed at tracking the progression of structural MRI changes in the first year of disease in EOAD patients compared with LOAD. We found that EOAD were much more atrophic than LOAD at first anatomical evaluation, and that the atrophy progression over one year was less severe in EOAD (maximum 5.9%) compared with LOAD. LOAD patients showed widespread atrophy progression with a maximum percentage of GM loss of 14.5%. We also found that atrophy in both groups, although mainly involving different lateral neocortical or medial temporal hubs at baseline, eventually progressed along the same brain default-network regions. The cortical region that showed a significant progression in both groups was the medial precuneus/posterior cingulate. We will discuss our results taking into account the current knowledge on lesion burden and atrophy progression, as well as the theory of network vulnerability, in AD according to age at onset.
To date, only one study has explored atrophy progression in EOAD relative to LOAD patients [26]. Cho and collaborators analyzed cognitive and anatomical progression, after three years of follow up, in the two AD populations, and concluded that EOAD group showed more rapid decline in attention, language, and frontal-executive, along with greater cortical thickness reduction in widespread association cortices, compared with LOAD cases. Their findings are in agreement with the cognitive reserve theory suggesting that patients who develop AD at a younger age have greater cognitive/brain reserve than older patients and thus require more extensive atrophy relative to the severity of cognitive impairment [41–43]. Our results provide further evidence in favor of this differential cognitive reserve.
In line with this consideration, pathological studies have shown that EOAD patients had greater pathologic burden [44], higher neurofibrillary tangles density [45] and greater synapses loss [46] than LOAD, at autopsy. A recent study, comparing metabolic dysfunction and amyloid burden in EO and LOAD patients, has shown a greater hypometabolism and pathological burden in the parietal lobe of very young AD patients (median: 56 years) [47].
In the present study, EO and LOAD patients initially show differential patterns of atrophy, but, as disease progresses, gray matter volume changes occur in regions that are, in both cases, part of the so-called default mode network. This network has been identified using resting state functional MRI (fMRI) [48, 49]. Resting state functional connectivity changes within the default mode network have been observed all across the spectrum from healthy aging [50], to mild cognitive impairment [51, 52] to AD [53–55]. This network encompasses several cortical regions, including the posterior cingulate cortex/precuneus, lateral temporal and parietal cortex, as well as the hippocampus and medial frontal cortex. Recently, Damoiseaux and collaborators [56] conducted a longitudinal study to assess connectivity changes in the default mode network over time in a group of AD patients with a mean of age of 64.2 (SD= 8.7). They have shown that, earlier in the disease, regions of the posterior default mode network started to disengage, whereas regions within the anterior and ventral networks enhanced their connectivity [56]. However, as the disease progresses, the authors found a global deterioration of connectivity, including all default mode network regions [56]. The functional changes seemed to follow a posterior-to-anterior axis. The patterns of structural damage we observed in EOAD at study entry and after one year closely involve these regions, quite mirroring the functional changes. In agreement with functional data [56], we found atrophy progression mainly in temporal and frontal regions included in default mode network.
The precuneus and posterior cingulate cortex were the areas that showed clear progression in both EO and LOAD. Neuroimaging cross-sectional studies have shown that the precuneus and posterior cingulate cortex are among the most severely involved regions in AD patients, especially EOAD [17, 57, 58]. Resting state fMRI studies have shown that medial parietal signal changes occur in the early stages of disease in AD [55]. In addition, Rabinovici et al. [58] have found the precuneus as one of the most metabolically vulnerable regions in EOAD patients. The precuneus/posterior cingulate is a key node in the default mode network and shows increased activity during the ‘rest’ condition in many fMRI activation studies [59]. In AD, the posterior cingulate has an abnormal metabolism in turn associated with amyloid deposition and brain atrophy [60]. One possible explanation is that a high metabolic activity in brain network hubs, such as the posterior cingulate, predisposes to the neuropathological cascade that leads to AD [61]. The involvement of medial parietal areas is considered an element of peculiarity of EOAD, distinguishing from other early non-AD neurodegenerative diseases, for example semantic dementia [62], or cortico-basal degeneration [63]. The results of the present study emphasize the role of the medial parietal regions in particular during the progression of the disease in both EO and LOAD.
Recent studies suggested that the damage of the white matter pathways is greater in EO than in LOAD patients. We demonstrated that white matter atrophy in EOAD (and its focal variants) is mainly centered on the posterior and medial parietal areas, including the connections of posterior cingulate region [64]. Using a less stringent statistical threshold, we found a similar but less severe result in the LOAD [64]. A recent diffusion tensor MRI study showed that, compared with LOAD, EOAD patients had a more severe and distributed pattern of white matter tract damage, in particular in the posterior fibers of cingulum and corpus callosum [65]. These data provide the anatomical substrate for the spread of misfolded protein aggregates over time in AD. In typical LOAD patients, the clinical progression will follow the cortical connectivity structure, in a Braak and Braak-like sequence [22] such as “1-temporal medial, 2-parietal, 3-frontal medial, 4-occipital secondary, 5-occipital primary” (for details see [66]). Most likely, EOAD patients develop stages 1 and 2 very rapidly or even at the same time, as shown by the greater white matter atrophy observed [64, 67]. This could explain why EOAD patients are already very impaired at first clinical and cognitive evaluation. An alternative explanation is that younger patients have a completely different distribution of neurofibrillary tangles. Recent anatomopathological studies suggest that in 11% of AD patients the hippocampi were spared from neurofibrillary pathology, and the mean age of those patients at disease onset was 63 (+/−10) years [68].
The main limitations of the present study are the relatively small number of patients included and the lack of an extensive neuropsychological evaluation for LOAD patients at follow up. In particular, the low number of LOAD studied is due to the recruitment of “Memory and Aging center”, which is mostly dedicated to younger and rare dementia patients. However, since many previous studies have investigated clinical and cognitive progression in LOAD, we do not consider this as a major shortcoming. Present findings on medial temporal lobe atrophy in LOAD are in agreement with previous MRI studies [5, 7, 8, 10, 26, 57, 69, 70] and recent updated pathologic evidence [71], which have highlighted the anatomical differences between EOAD and LOAD. Previous and present MRI findings support the view that medial temporal damage is a key structural substrate in LOAD.
Further longitudinal studies, coupling structural and functional neuroimaging explorations in a larger population of EO and LOAD will provide additional knowledge on the way AD pathology not only begins but also progresses.
In conclusion, the present study demonstrated that despite the baseline differences, EO and LOAD patients seemed to converge to a similar pattern of atrophy matching the brain areas that are included in the so-called default mode network.
Supplementary Material
Acknowledgments
Raffaella Migliaccio is supported by “France Alzheimer” Foundation. The authors also acknowledge the National Institutes of Health (NINDS R01 NS050915, NIA P50 AG03006, NINDS K23 AG037566-01A1, NIA P01 AG019724, NIA K23-AG031861, K23 K23AG037566), the state of California (DHS 04-35516), the Alzheimer’s Disease Research Centre of California (03-75271 DHS/ADP/ARCC), the Larry L. Hillblom Foundation, the John Douglas French Alzheimer’s Foundation, the Koret Family Foundation, and the McBean Family Foundation.
References
- 1.Seltzer B, Sherwin I. A comparison of clinical features in early- and late-onset primary degenerative dementia. One entity or two? Arch Neurol. 1983;40:143–146. doi: 10.1001/archneur.1983.04050030037006. [DOI] [PubMed] [Google Scholar]
- 2.Koss E, Edland S, Fillenbaum G, Mohs R, Clark C, Galasko D, Morris JC. Clinical and neuropsychological differences between patients with earlier and later onset of Alzheimer’s disease: A CERAD analysis, Part XII. Neurology. 1996;46:136–141. doi: 10.1212/wnl.46.1.136. [DOI] [PubMed] [Google Scholar]
- 3.Grady CL, Haxby JV, Horwitz B, Berg G, Rapoport SI. Neuropsychological and cerebral metabolic function in early vs late onset dementia of the Alzheimer type. Neuropsychologia. 1987;25:807–816. doi: 10.1016/0028-3932(87)90118-7. [DOI] [PubMed] [Google Scholar]
- 4.Smits LL, Pijnenburg YA, Koedam EL, van der Vlies AE, Reuling IE, Koene T, Teunissen CE, Scheltens P, van der Flier WM. Early onset Alzheimer’s disease is associated with a distinct neuropsychological profile. J Alzheimers Dis. 2012;30:101–108. doi: 10.3233/JAD-2012-111934. [DOI] [PubMed] [Google Scholar]
- 5.van der Vlies AE, Staekenborg SS, Admiraal-Behloul F, Prins ND, Barkhof F, Vrenken H, Reiber JH, Scheltens P, van der Flier WM. Associations between magnetic resonance imaging measures and neuropsychological impairment in early and late onset alzheimer’s disease. J Alzheimers Dis. 2013;35:169–178. doi: 10.3233/JAD-121291. [DOI] [PubMed] [Google Scholar]
- 6.Rogaeva E. The solved and unsolved mysteries of the genetics of early-onset Alzheimer’s disease. Neuromolecular Med. 2002;2:1–10. doi: 10.1385/NMM:2:1:01. [DOI] [PubMed] [Google Scholar]
- 7.Ishii K, Kawachi T, Sasaki H, Kono AK, Fukuda T, Kojima Y, Mori E. Voxel-based morphometric comparison between early- and late-onset mild Alzheimer’s disease and assessment of diagnostic performance of z score images. AJNR Am J Neuroradiol. 2005;26:333–340. [PMC free article] [PubMed] [Google Scholar]
- 8.Frisoni GB, Pievani M, Testa C, Sabattoli F, Bresciani L, Bonetti M, Beltramello A, Hayashi KM, Toga AW, Thompson PM. The topography of grey matter involvement in early and late onset Alzheimer’s disease. Brain. 2007;130:720–730. doi: 10.1093/brain/awl377. [DOI] [PubMed] [Google Scholar]
- 9.Rossor MN, Fox NC, Mummery CJ, Schott JM, Warren JD. The diagnosis of young-onset dementia. Lancet Neurol. 2010;9:793–806. doi: 10.1016/S1474-4422(10)70159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moller C, Vrenken H, Jiskoot L, Versteeg A, Barkhof F, Scheltens P, van der Flier WM. Different patterns of gray matter atrophy in early- and late-onset Alzheimer’s disease. Neurobiol Aging. 2013;34:2014–2022. doi: 10.1016/j.neurobiolaging.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Lehmann M, Madison CM, Ghosh PM, Seeley WW, Mormino E, Greicius MD, Gorno-Tempini ML, Kramer JH, Miller BL, Jagust WJ, Rabinovici GD. Intrinsic connectivity networks in healthy subjects explain clinical variability in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2013;110:11606–11611. doi: 10.1073/pnas.1221536110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann M, Ghosh PM, Madison C, Laforce R, Jr, Corbetta-Rastelli C, Weiner MW, Greicius MD, Seeley WW, Gorno-Tempini ML, Rosen HJ, Miller BL, Jagust WJ, Rabinovici GD. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain. 2013;136:844–858. doi: 10.1093/brain/aws327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujimori M, Imamura T, Yamashita H, Hirono N, Ikejiri Y, Shimomura T, Mori E. Age at onset and visuocognitive disturbances in Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12:163–166. doi: 10.1097/00002093-199809000-00007. [DOI] [PubMed] [Google Scholar]
- 14.Imamura T, Takatsuki Y, Fujimori M, Hirono N, Ikejiri Y, Shimomura T, Hashimoto M, Yamashita H, Mori E. Age at onset and language disturbances in Alzheimer’s disease. Neuropsychologia. 1998;36:945–949. doi: 10.1016/s0028-3932(98)00010-4. [DOI] [PubMed] [Google Scholar]
- 15.Mendez MF, Lee AS, Joshi A, Shapira JS. Nonamnestic presentations of early-onset Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2012;27:413–420. doi: 10.1177/1533317512454711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grady CL, Haxby JV, Schapiro MB, Gonzalez-Aviles A, Kumar A, Ball MJ, Heston L, Rapoport SI. Subgroups in dementia of the Alzheimer type identified using positron emission tomography. J Neuropsychiatry Clin Neurosci. 1990;2:373–384. doi: 10.1176/jnp.2.4.373. [DOI] [PubMed] [Google Scholar]
- 17.Migliaccio R, Agosta F, Rascovsky K, Karydas A, Bonasera S, Rabinovici GD, Miller BL, Gorno-Tempini ML. Clinical syndromes associated with posterior atrophy: early age at onset AD spectrum. Neurology. 2009;73:1571–1578. doi: 10.1212/WNL.0b013e3181c0d427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Licht EA, McMurtray AM, Saul RE, Mendez MF. Cognitive differences between early- and late-onset Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2007;22:218–222. doi: 10.1177/1533317506299156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heyman A, Wilkinson WE, Hurwitz BJ, Helms MJ, Haynes CS, Utley CM, Gwyther LP. Early-onset Alzheimer’s disease: clinical predictors of institutionalization and death. Neurology. 1987;37:980–984. doi: 10.1212/wnl.37.6.980. [DOI] [PubMed] [Google Scholar]
- 20.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 21.Jacobs D, Sano M, Marder K, Bell K, Bylsma F, Lafleche G, Albert M, Brandt J, Stern Y. Age at onset of Alzheimer’s disease: relation to pattern of cognitive dysfunction and rate of decline. Neurology. 1994;44:1215–1220. doi: 10.1212/wnl.44.7.1215. [DOI] [PubMed] [Google Scholar]
- 22.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 23.Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–278. doi: 10.1016/0197-4580(95)00021-6. discussion 278–284. [DOI] [PubMed] [Google Scholar]
- 24.Risacher SL, Shen L, West JD, Kim S, McDonald BC, Beckett LA, Harvey DJ, Jack CR, Jr, Weiner MW, Saykin AJ. Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiol Aging. 2010;31:1401–1418. doi: 10.1016/j.neurobiolaging.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitwell JL. Progression of atrophy in Alzheimer’s disease and related disorders. Neurotox Res. 2010;18:339–346. doi: 10.1007/s12640-010-9175-1. [DOI] [PubMed] [Google Scholar]
- 26.Cho H, Jeon S, Kang SJ, Lee JM, Lee JH, Kim GH, Shin JS, Kim CH, Noh Y, Im K, Kim ST, Chin J, Seo SW, Na DL. Longitudinal changes of cortical thickness in early- versus late-onset Alzheimer’s disease. Neurobiol Aging. 2013;34:1921 e1929–1921 e1915. doi: 10.1016/j.neurobiolaging.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Haxby JV, Grady CL, Friedland RP, Rapoport SI. Neocortical metabolic abnormalities precede nonmemory cognitive impairments in early dementia of the Alzheimer type: longitudinal confirmation. J Neural Transm Suppl. 1987;24:49–53. [PubMed] [Google Scholar]
- 28.Haxby JV, Grady CL, Koss E, Horwitz B, Heston L, Schapiro M, Friedland RP, Rapoport SI. Longitudinal study of cerebral metabolic asymmetries and associated neuropsychological patterns in early dementia of the Alzheimer type. Arch Neurol. 1990;47:753–760. doi: 10.1001/archneur.1990.00530070043010. [DOI] [PubMed] [Google Scholar]
- 29.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 30.Kramer JH, Jurik J, Sha SJ, Rankin KP, Rosen HJ, Johnson JK, Miller BL. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol. 2003;16:211–218. doi: 10.1097/00146965-200312000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Delis DC, Freeland J, Kramer JH, Kaplan E. Integrating clinical assessment with cognitive neuroscience: construct validation of the California Verbal Learning Test. J Consult Clin Psychol. 1988;56:123–130. doi: 10.1037//0022-006x.56.1.123. [DOI] [PubMed] [Google Scholar]
- 32.Delis DC, Kramer JH, Kaplan E, Ober BA. The California Verbal Learning Test. 2. San Antonio: The Psychological Corporation; 2000. [Google Scholar]
- 33.Possin KL, Laluz VR, Alcantar OZ, Miller BL, Kramer JH. Distinct neuroanatomical substrates and cognitive mechanisms of figure copy performance in Alzheimer’s disease and behavioral variant frontotemporal dementia. Neuropsychologia. 2011;49:43–48. doi: 10.1016/j.neuropsychologia.2010.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaplan E, Goodglass H, Weintraub S. Boston Naming Test. Philadelphia: Lea & Febiger; 1983. [Google Scholar]
- 35.Ashburner J, Friston KJ. Voxel-based morphometry--the methods. Neuroimage. 2000;11:805–821. doi: 10.1006/nimg.2000.0582. [DOI] [PubMed] [Google Scholar]
- 36.Good CD, Scahill RI, Fox NC, Ashburner J, Friston KJ, Chan D, Crum WR, Rossor MN, Frackowiak RS. Automatic differentiation of anatomical patterns in the human brain: validation with studies of degenerative dementias. Neuroimage. 2002;17:29–46. doi: 10.1006/nimg.2002.1202. [DOI] [PubMed] [Google Scholar]
- 37.Brambati SM, Renda NC, Rankin KP, Rosen HJ, Seeley WW, Ashburner J, Weiner MW, Miller BL, Gorno-Tempini ML. A tensor based morphometry study of longitudinal gray matter contraction in FTD. Neuroimage. 2007;35:998–1003. doi: 10.1016/j.neuroimage.2007.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brambati SM, Rankin KP, Narvid J, Seeley WW, Dean D, Rosen HJ, Miller BL, Ashburner J, Gorno-Tempini ML. Atrophy progression in semantic dementia with asymmetric temporal involvement: a tensor-based morphometry study. Neurobiol Aging. 2009;30:103–111. doi: 10.1016/j.neurobiolaging.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leow AD, Yanovsky I, Parikshak N, Hua X, Lee S, Toga AW, Jack CR, Jr, Bernstein MA, Britson PJ, Gunter JL, Ward CP, Borowski B, Shaw LM, Trojanowski JQ, Fleisher AS, Harvey D, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM. Alzheimer’s disease neuroimaging initiative: a one-year follow up study using tensor-based morphometry correlating degenerative rates, biomarkers and cognition. Neuroimage. 2009;45:645–655. doi: 10.1016/j.neuroimage.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hua X, Lee S, Yanovsky I, Leow AD, Chou YY, Ho AJ, Gutman B, Toga AW, Jack CR, Jr, Bernstein MA, Reiman EM, Harvey DJ, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM. Optimizing power to track brain degeneration in Alzheimer’s disease and mild cognitive impairment with tensor-based morphometry: an ADNI study of 515 subjects. Neuroimage. 2009;48:668–681. doi: 10.1016/j.neuroimage.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scarmeas N, Stern Y. Cognitive reserve: implications for diagnosis and prevention of Alzheimer’s disease. Curr Neurol Neurosci Rep. 2004;4:374–380. doi: 10.1007/s11910-004-0084-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robertson IH. A right hemisphere role in cognitive reserve. Neurobiol Aging. 2013 doi: 10.1016/j.neurobiolaging.2013.11.028. [DOI] [PubMed] [Google Scholar]
- 43.Lo RY, Jagust WJ. Effect of cognitive reserve markers on Alzheimer pathologic progression. Alzheimer Dis Assoc Disord. 2013;27:343–350. doi: 10.1097/WAD.0b013e3182900b2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marshall GA, Fairbanks LA, Tekin S, Vinters HV, Cummings JL. Early-onset Alzheimer’s disease is associated with greater pathologic burden. J Geriatr Psychiatry Neurol. 2007;20:29–33. doi: 10.1177/0891988706297086. [DOI] [PubMed] [Google Scholar]
- 45.Mizuno Y, Ikeda K, Tsuchiya K, Ishihara R, Shibayama H. Two distinct subgroups of senile dementia of Alzheimer type: quantitative study of neurofibrillary tangles. Neuropathology. 2003;23:282–289. doi: 10.1046/j.1440-1789.2003.00514.x. [DOI] [PubMed] [Google Scholar]
- 46.Bigio EH, Hynan LS, Sontag E, Satumtira S, White CL. Synapse loss is greater in presenile than senile onset Alzheimer disease: implications for the cognitive reserve hypothesis. Neuropathol Appl Neurobiol. 2002;28:218–227. doi: 10.1046/j.1365-2990.2002.00385.x. [DOI] [PubMed] [Google Scholar]
- 47.Ossenkoppele R, Zwan MD, Tolboom N, van Assema DM, Adriaanse SF, Kloet RW, Boellaard R, Windhorst AD, Barkhof F, Lammertsma AA, Scheltens P, van der Flier WM, van Berckel BN. Amyloid burden and metabolic function in early-onset Alzheimer’s disease: parietal lobe involvement. Brain. 2012;135:2115–2125. doi: 10.1093/brain/aws113. [DOI] [PubMed] [Google Scholar]
- 48.Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci U S A. 2001;98:676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Damoiseaux JS, Greicius MD. Greater than the sum of its parts: a review of studies combining structural connectivity and resting-state functional connectivity. Brain Struct Funct. 2009;213:525–533. doi: 10.1007/s00429-009-0208-6. [DOI] [PubMed] [Google Scholar]
- 50.Andrews-Hanna JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, Buckner RL. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56:924–935. doi: 10.1016/j.neuron.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bai F, Watson DR, Yu H, Shi Y, Yuan Y, Zhang Z. Abnormal resting-state functional connectivity of posterior cingulate cortex in amnestic type mild cognitive impairment. Brain Res. 2009;1302:167–174. doi: 10.1016/j.brainres.2009.09.028. [DOI] [PubMed] [Google Scholar]
- 52.Sorg C, Riedl V, Muhlau M, Calhoun VD, Eichele T, Laer L, Drzezga A, Forstl H, Kurz A, Zimmer C, Wohlschlager AM. Selective changes of resting-state networks in individuals at risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104:18760–18765. doi: 10.1073/pnas.0708803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101:4637–4642. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gili T, Cercignani M, Serra L, Perri R, Giove F, Maraviglia B, Caltagirone C, Bozzali M. Regional brain atrophy and functional disconnection across Alzheimer’s disease evolution. J Neurol Neurosurg Psychiatry. 2011;82:58–66. doi: 10.1136/jnnp.2009.199935. [DOI] [PubMed] [Google Scholar]
- 55.Agosta F, Pievani M, Geroldi C, Copetti M, Frisoni GB, Filippi M. Resting state fMRI in Alzheimer’s disease: beyond the default mode network. Neurobiol Aging. 2012;33:1564–1578. doi: 10.1016/j.neurobiolaging.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 56.Damoiseaux JS, Prater KE, Miller BL, Greicius MD. Functional connectivity tracks clinical deterioration in Alzheimer’s disease. Neurobiol Aging. 2012;33:828, e819–830. doi: 10.1016/j.neurobiolaging.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shiino A, Watanabe T, Maeda K, Kotani E, Akiguchi I, Matsuda M. Four subgroups of Alzheimer’s disease based on patterns of atrophy using VBM and a unique pattern for early onset disease. Neuroimage. 2006;33:17–26. doi: 10.1016/j.neuroimage.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 58.Rabinovici GD, Furst AJ, Alkalay A, Racine CA, O’Neil JP, Janabi M, Baker SL, Agarwal N, Bonasera SJ, Mormino EC, Weiner MW, Gorno-Tempini ML, Rosen HJ, Miller BL, Jagust WJ. Increased metabolic vulnerability in early-onset Alzheimer’s disease is not related to amyloid burden. Brain. 2010;133:512–528. doi: 10.1093/brain/awp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leech R, Sharp DJ. The role of the posterior cingulate cortex in cognition and disease. Brain. 2013 doi: 10.1093/brain/awt162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T, Andrews-Hanna JR, Sperling RA, Johnson KA. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. J Neurosci. 2009;29:1860–1873. doi: 10.1523/JNEUROSCI.5062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boxer AL, Rankin KP, Miller BL, Schuff N, Weiner M, Gorno-Tempini ML, Rosen HJ. Cinguloparietal atrophy distinguishes Alzheimer disease from semantic dementia. Arch Neurol. 2003;60:949–956. doi: 10.1001/archneur.60.7.949. [DOI] [PubMed] [Google Scholar]
- 63.Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, Huang EJ, Trojanowski JQ, Growdon ME, Jang JY, Sidhu M, See TM, Karydas AM, Gorno-Tempini ML, Boxer AL, Weiner MW, Geschwind MD, Rankin KP, Miller BL. Clinicopathological correlations in corticobasal degeneration. Ann Neurol. 2011;70:327–340. doi: 10.1002/ana.22424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Migliaccio R, Agosta F, Possin KL, Rabinovici GD, Miller BL, Gorno-Tempini ML. White matter atrophy in Alzheimer’s disease variants. Alzheimers Dement. 2012;8:S78–87. e71–72. doi: 10.1016/j.jalz.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Canu E, Agosta F, Spinelli EG, Magnani G, Marcone A, Scola E, Falautano M, Comi G, Falini A, Filippi M. White matter microstructural damage in Alzheimer’s disease at different ages of onset. Neurobiol Aging. 2013;34:2331–2340. doi: 10.1016/j.neurobiolaging.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 66.Reid AT, Evans AC. Structural networks in Alzheimer’s disease. Eur Neuropsychopharmacol. 2013;23:63–77. doi: 10.1016/j.euroneuro.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 67.Canu E, Frisoni GB, Agosta F, Pievani M, Bonetti M, Filippi M. Early and late onset Alzheimer’s disease patients have distinct patterns of white matter damage. Neurobiol Aging. 2012;33:1023–1033. doi: 10.1016/j.neurobiolaging.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 68.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10:785–796. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karas G, Scheltens P, Rombouts S, van Schijndel R, Klein M, Jones B, van der Flier W, Vrenken H, Barkhof F. Precuneus atrophy in early-onset Alzheimer’s disease: a morphometric structural MRI study. Neuroradiology. 2007;49:967–976. doi: 10.1007/s00234-007-0269-2. [DOI] [PubMed] [Google Scholar]
- 70.van de Pol LA, Hensel A, van der Flier WM, Visser PJ, Pijnenburg YA, Barkhof F, Gertz HJ, Scheltens P. Hippocampal atrophy on MRI in frontotemporal lobar degeneration and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2006;77:439–442. doi: 10.1136/jnnp.2005.075341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Whitwell JL, Dickson DW, Murray ME, Weigand SD, Tosakulwong N, Senjem ML, Knopman DS, Boeve BF, Parisi JE, Petersen RC, Jack CR, Jr, Josephs KA. Neuroimaging correlates of pathologically defined subtypes of Alzheimer’s disease: a case-control study. Lancet Neurol. 2012;11:868–877. doi: 10.1016/S1474-4422(12)70200-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.