

Introduction
KIT juxtamembrane oncogenic mutations (encoded by KIT exon 11) are found in 67% of GI stromal tumors (GISTs) and are inhibited potently by imatinib. Virtually all patients with these mutations therefore achieve clinical benefit when treated with imatinib. Imatinib resistance in KIT exon 11–mutant GISTs typically occurs after 18 to 24 months of response or disease stabilization, most often resulting from expansion of multiple tumor clones harboring secondary KIT kinase domain mutations.1 However, approximately 10% of patients with GISTs have primary imatinib resistance, defined by clinical progression within 3 to 6 months after initiating therapy. Such GISTs typically lack KIT and platelet-derived growth factor receptor alpha (PDGFRA) mutations, or contain particular mutations, such as PDGFRA D842V, that are intrinsically imatinib resistant.2 To our knowledge, this is the first report of polyclonal heterogeneity—including KRAS mutation— as a mechanism of primary imatinib resistance in a patient with GIST.
Case Report
A 61-year-old man presented to an outpatient clinic in October 2003 with an 8-week history of progressive left shoulder pain, nausea, and fatigue. A left upper quadrant mass was palpable on physical examination. Laboratory data were normal, other than a hematocrit level of 28.3% and a platelet count of 536,000/uL. An abdominal computed tomography (CT) scan revealed a 19.7 × 13.1-cm mass arising from the anterior wall of the stomach, accompanied by five liver metastases, all less than 1 cm in maximal diameter. Endoscopic biopsy demonstrated a spindle cell GIST (Fig 1A) with 20 mitoses per 50 high-power fields (hpf), diffuse cytoplasmic KIT expression (Fig 1B), CD34 expression, and no expression of smooth muscle actin and cytokeratins.
Fig 1.

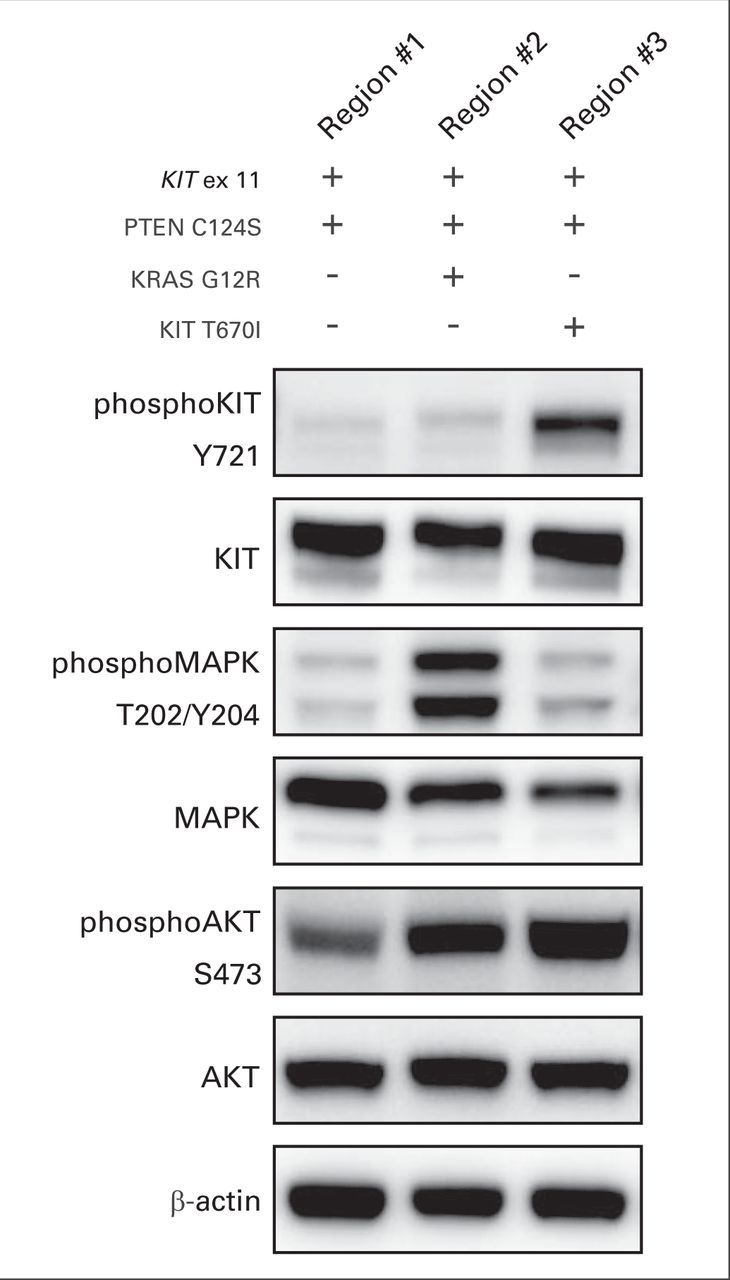
The patient received imatinib 400 mg per day and experienced symptomatic improvement within 1 month, including resolution of shoulder pain, softening of the palpable mass, and normalization of the blood counts. A follow-up CT scan after 6 weeks of treatment with imatinib showed that the gastric mass (22.7 × 13.1 cm) had typical post-therapy changes, including hypodensity and a decrease in wall thickness (Fig 2A). The liver metastases were unchanged. A CT scan at week 16 of imatinib treatment showed reduction of the hypodense overall gastric residual mass to 15.4 × 11.6 cm; however, a new, hyperdense 2.7 × 2.0-cm nodule was present at the caudal aspect of the mass (Fig 2B). The patient continued to receive imatinib, and a follow-up CT scan 2 months later showed progression of the hyperdense nodule to 4.9 × 5.6 cm, now accompanied by additional progressing nodules in the bed of the gastric primary (Fig 2C). An upper GI bleed prompted resection of the gastric mass, which was performed 24 hours after the last imatinib dose. During this surgery, subcentimeter peritoneal implants were observed but not removed. Histologically, the gastric mass was spindle cell–type GIST. Genomic analyses by Sanger sequencing, Ion Torrent (Life Technologies, Carlsbad, CA), and Sequenom MassArray System (Sequenom, San Diego, CA) were performed in clinically responding (region No. 1) versus clinically progressing (regions No. 2 and No. 3) aspects of the mass. Region No. 1 was hypocellular, nonmitotic, and therefore consistent with stable/responding disease, whereas regions No. 2 and No. 3 had 60 and 55 mitoses per 50 hpf, respectively, and were therefore consistent with progressing, imatinib-resistant disease. Each of these three regions expressed KIT strongly and had a homozygous KIT exon 11 E554_V559del mutation (Fig 3A) and a homozygous PTEN missense mutation, C124S (Fig 3B), which is known to abrogate PTEN lipid- and protein-phosphatase activity and PTEN-mediated phospholipase regulation.3 The imatinib-responsive region No. 1 had no additional mutations, whereas imatinib-resistant region No. 2 had a KRAS G12R mutation by Sequenom analysis, which was corroborated by KRAS genomic sequencing and transcript allelic subcloning and sequencing (Fig 3C). Imatinib-resistant region No. 3 had a KIT gatekeeper T670I mutation (Fig 3D), which is known to confer imatinib resistance.4 Immunoblotting evaluations confirmed strong KIT expression in both imatinib-responsive and -resistant regions (Fig 4); however, KIT was activated, as assessed by phosphoKIT Y721 expression, only in region No. 3 with the KIT T670I mutation, mitogen-activated protein kinase was hyperactivated only in region No. 2 with KRAS G12R, whereas AKT was hyperactivated in both of these regions (Fig 4).
Fig 2.

Fig 3.

Fig 4.
Imatinib was resumed, but the patient manifested further progression of intra-abdominal disease 4 weeks postoperatively and died 5 months later while receiving high-dose imatinib (800 mg per day). He was unable to receive second-line therapy because progression occurred during the window between completion of a phase III trial and US Food and Drug Administration approval of sunitinib for imatinib-resistant GIST.
Wild-type GISTs lacking KIT and PDGFRA mutations frequently show primary imatinib resistance, and although some of these are succinate dehydrogenase-deficient because of SDHA, SDHB, or SDHC mutations,5,6 others have no known genetic mutations. To test the hypothesis that such GISTs might contain RAS mutations or other KIT downstream mutations, we used a Sequenom panel to screen for RAS, BRAF, and PI3KCA mutations in KIT/PDGFRA wild-type GISTs from 27 patients. Only one of these 27 GISTs contained demonstrable mutation(s): this was a high-risk GIST (8-cm gastric primary with 62 mitoses per 50 hpf) that contained both HRAS G12V and PIK3CA H1047R mutations. PIK3CA H1047R is a gain-of-function mutation that accounts for approximately 20% of PIK3CA mutations in advanced human cancers7 and is associated with response to phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin pathway inhibitors.8
Discussion
Although pretreatment genomic intratumoral heterogeneity has been implicated as a cause of treatment failure in several solid tumors,2 this report provides the first evidence, to our knowledge, of primary imatinib resistance resulting from intratumoral genomic heterogeneity. This resistance, manifested already at 16 weeks of treatment, was related (in separate lesions) to KRAS mutation and the KIT gatekeeper mutation T670I. Recently, Miranda et al9 reported in vitro evidence that KRAS mutations might bestow imatinib resistance in GIST, and our case report corroborates that the KRAS gain-of-function mutation is a contributor to clinical imatinib resistance, even in the face of therapeutic KIT oncoprotein inhibition. Notably, Diaz et al10 predicted by mathematical modeling that KRAS-mutant subclones are present before initiation of anti–epidermal growth factor receptor treatment in some colorectal cancers. Similarly, it is conceivable that KRAS mutations are present as minor subclones in more untreated GISTs than previously appreciated, and are then enriched for by KIT/PDGFRA-inhibitor therapies.
Most GISTs with RAS pathway dysregulation by BRAF or NF1 alterations have been low-grade and low-risk tumors,11–15 whereas the two RAS-mutant cases reported here—both containing concurrent PI3K-PTEN mutations— are decidedly high grade. Notably, our case report relates to a KIT exon 11–mutant GIST in which the KRAS mutation was restricted to a subregion of the tumor and was therefore acquired after the KIT exon 11 mutation. However, our mutation screens in 27 KIT/PDGFRA wild-type GISTs demonstrated a case with concomitant PIK3CA and HRAS mutations, suggesting that PI3K and RAS pathway genetic coactivation provides a transforming equivalent to KIT activation in some GISTs lacking KIT mutations. In this sense, we propose that KIT/PDGFRA oncogenesis in high-grade GISTs is most effectively supplanted when the PI3K/AKT and RAS/RAF/MEK pathways are both constitutively activated by independent mutations. This hypothesis is appealing in that GIST KIT/PDGFRA mutations are known to coactivate both PI3K and RAS downstream pathways.16 The hypothesis warrants evaluation in a larger group of patients with GISTs, but if true, adds a level of complexity to KIT/PDGFRA downstream resistance mechanisms and accounts for why such mechanisms, potentially requiring mutational hits to genes in two pathways, infrequently cause imatinib resistance. In keeping with this hypothesis, we note that the PTEN C124S mutation reported here was demonstrated along with KIT exon 11 mutation in both imatinib-sensitive and imatinib-resistant aspects of the GIST. Therefore, the PTEN mutation was not directly responsible for imatinib resistance, but likely created a biologic state that was permissive for KRAS G12R–transforming activity, with KRAS G12R being a known imatinib-resistance mechanism.9
In summary, our findings demonstrate KRAS mutation and polyclonal heterogeneity as mechanisms of primary imatinib resistance in GIST, show that both KRAS and HRAS isoforms can contribute to GIST oncogenesis, and highlight the conjoined nature of the PI3K/AKT and RAS/RAF signaling pathways in GIST tumorigenesis. These findings validate the PI3K/AKT/ mammalian target of rapamycin pathway and RAS/RAF/MEK pathways as concurrently relevant in GIST oncogenic signaling.
Acknowledgment
This work was supported by GI Specialized Program of Research Excellence Grant No. 1P50CA12703-05 (J.A.F., G.D.D.), grants from the Virginia and Daniel K. Ludwig Trust for Cancer Research (J.A.F., G.D.D.), the GI Stromal Tumor (GIST) Cancer Research Fund (C.L.C., M.C.H., J.A.F.), the Life Raft Group (C.L.C., M.C.H., J.A.F.), the B.P. Lester Foundation (C.L.C., M.C.H.), a Veterans Affairs Merit Review Grant from the Department of Veterans' Affairs (M.C.H.), the Cesarini Pan Mass Challenge for GIST (J.A.F., G.D.D.), the Italian Society of Pharmacology and Fondazione del Monte di Bologna e Ravenna (G.R.), the Sarcoma Alliance for Research through Collaboration (A.M.E.), and the Spanish Society of Medical Oncology (C.S.). Both C.S. and Y.W. contributed equally to this work.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: George D. Demetri, Novartis (C), Pfizer (C), sanofi-aventis (C), GlaxoSmithKline (C), Johnson & Johnson (C), Merrimack Pharma (C), Foundation Medicine (C), Merck (C), ZIOPHARM Oncology (C), N-of-One (U), Champions Oncology (C), G1 Therapeutics (C), Kolltan Pharmaceuticals (C), Blueprint Medicines (C); Christopher L. Corless, Novartis (C), Pfizer (C); Michael C. Heinrich, Molecular Insight (C), Novartis (C), Ariad Pharmaceuticals (C), Pfizer (C); Jonathan A. Fletcher, Novartis (C) Stock Ownership: George D. Demetri, N-of-One, Champions Oncology, G1 Therapeutics, Kolltan Pharmaceuticals, Blueprint Medicines; Michael C. Heinrich, Molecular Insight Honoraria: Christopher L. Corless, Novartis, Pfizer; Michael C. Heinrich, Novartis, Pfizer; Jonathan A. Fletcher, Novartis Research Funding: George D. Demetri, Novartis, Pfizer, sanofi-aventis, GlaxoSmithKline, Johnson & Johnson, Merck; Christopher L. Corless, Novartis, Pfizer; Michael C. Heinrich, Ariad Pharmaceuticals, Arog Pharmaceuticals, ImClone Systems, Novartis; Jonathan A. Fletcher, Deciphera Pharmaceuticals Expert Testimony: None Patents: None Other Remuneration: Christopher L. Corless, Novartis
References
- 1.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:7510–7518. doi: 10.1158/1078-0432.CCR-09-0190. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez-Breckenridge CA, Waite KA, Eng C. PTEN regulates phospholipase D and phospholipase C. Hum Mol Genet. 2007;16:1157–1163. doi: 10.1093/hmg/ddm063. [DOI] [PubMed] [Google Scholar]
- 4.Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–4774. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 5.Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miettinen M, Wang ZF, Sarlomo-Rikala M, et al. Succinate dehydrogenase-deficient GISTs: A clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol. 2011;35:1712–1721. doi: 10.1097/PAS.0b013e3182260752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janku F, Wheler JJ, Naing A, et al. PIK3CA mutations in advanced cancers: Characteristics and outcomes. Oncotarget. 2012;3:1566–1575. doi: 10.18632/oncotarget.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janku F, Wheler JJ, Naing A, et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013;73:276–284. doi: 10.1158/0008-5472.CAN-12-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miranda C, Nucifora M, Molinari F, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:1769–1776. doi: 10.1158/1078-0432.CCR-11-2230. [DOI] [PubMed] [Google Scholar]
- 10.Diaz LA, Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agaram NP, Wong GC, Guo T, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008;47:853–859. doi: 10.1002/gcc.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hostein I, Faur N, Primois C, et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol. 2010;133:141–148. doi: 10.1309/AJCPPCKGA2QGBJ1R. [DOI] [PubMed] [Google Scholar]
- 13.Agaimy A, Terracciano LM, Dirnhofer S, et al. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin Pathol. 2009;62:613–616. doi: 10.1136/jcp.2009.064550. [DOI] [PubMed] [Google Scholar]
- 14.Daniels M, Lurkin I, Pauli R, et al. Spectrum of KIT/PDGFRA/BRAF mutations and phosphatidylinositol-3-kinase pathway gene alterations in gastrointestinal stromal tumors (GIST) Cancer Lett. 2011;312:43–54. doi: 10.1016/j.canlet.2011.07.029. [DOI] [PubMed] [Google Scholar]
- 15.Miettinen M, Fetsch JF, Sobin LH, et al. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: A clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30:90–96. doi: 10.1097/01.pas.0000176433.81079.bd. [DOI] [PubMed] [Google Scholar]
- 16.Bauer S, Duensing A, Demetri GD, et al. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007;26:7560–7568. doi: 10.1038/sj.onc.1210558. [DOI] [PubMed] [Google Scholar]