

Mutations in PLA2G6, which encodes ‘calcium-independent phospholipase A2 beta’, have been implicated in parkinsonian disorders. Kinghorn et al. show, in a Drosophila model and in human fibroblasts, that reduced PLA2G6 activity is associated with elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Treatment with deuterated polyunsaturated fatty acids reverses the deficits.
Keywords: PLA2G6, infantile neuroaxonal dystrophy, neurodegeneration with brain iron accumulation, Drosophila
Mutations in PLA2G6, which encodes ‘calcium-independent phospholipase A2 beta’, have been implicated in parkinsonian disorders. Kinghorn et al. show, in a Drosophila model and in human fibroblasts, that reduced PLA2G6 activity is associated with elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Treatment with deuterated polyunsaturated fatty acids reverses the deficits.
Abstract
The PLA2G6 gene encodes a group VIA calcium-independent phospholipase A2 beta enzyme that selectively hydrolyses glycerophospholipids to release free fatty acids. Mutations in PLA2G6 have been associated with disorders such as infantile neuroaxonal dystrophy, neurodegeneration with brain iron accumulation type II and Karak syndrome. More recently, PLA2G6 was identified as the causative gene in a subgroup of patients with autosomal recessive early-onset dystonia-parkinsonism. Neuropathological examination revealed widespread Lewy body pathology and the accumulation of hyperphosphorylated tau, supporting a link between PLA2G6 mutations and parkinsonian disorders. Here we show that knockout of the Drosophila homologue of the PLA2G6 gene, iPLA2-VIA, results in reduced survival, locomotor deficits and organismal hypersensitivity to oxidative stress. Furthermore, we demonstrate that loss of iPLA2-VIA function leads to a number of mitochondrial abnormalities, including mitochondrial respiratory chain dysfunction, reduced ATP synthesis and abnormal mitochondrial morphology. Moreover, we show that loss of iPLA2-VIA is strongly associated with increased lipid peroxidation levels. We confirmed our findings using cultured fibroblasts taken from two patients with mutations in the PLA2G6 gene. Similar abnormalities were seen including elevated mitochondrial lipid peroxidation and mitochondrial membrane defects, as well as raised levels of cytoplasmic and mitochondrial reactive oxygen species. Finally, we demonstrated that deuterated polyunsaturated fatty acids, which inhibit lipid peroxidation, were able to partially rescue the locomotor abnormalities seen in aged flies lacking iPLA2-VIA gene function, and restore mitochondrial membrane potential in fibroblasts from patients with PLA2G6 mutations. Taken together, our findings demonstrate that loss of normal PLA2G6 gene activity leads to lipid peroxidation, mitochondrial dysfunction and subsequent mitochondrial membrane abnormalities. Furthermore we show that the iPLA2-VIA knockout fly model provides a useful platform for the further study of PLA2G6-associated neurodegeneration.
Introduction
The PLA2G6 gene encodes an 85-kDa group VI calcium-independent phospholipase A2 beta (PLA2G6). This enzyme hydrolyses the sn-2 acyl chain of glycerophospholipids to release free fatty acids and lysophospholipids (Wolf and Gross, 1996; Ma and Turk, 2001). PLA2G6 localizes to the mitochondria, and has proposed roles in the remodelling of membrane phospholipids, signal transduction and calcium signalling, cell proliferation and apoptosis (Gadd et al., 2006; Seleznev et al., 2006; Strokin et al., 2012).
Humans with PLA2G6 mutations can show progressive cognitive and motor skill regression, as displayed in disorders such as infantile neuroaxonal dystrophy, neurodegeneration with brain iron accumulation type II and Karak syndrome (Khateeb et al., 2006; Morgan et al., 2006). Infantile neuroaxonal dystrophy is a neurodegenerative disease with onset in infancy and fatality in the teenage years or in early adulthood. It is characterized neuropathologically by axonal swelling and the presence of spheroid bodies in the central and peripheral nervous systems in addition to hallmark cerebellar atrophy. Neurodegeneration with brain iron accumulation comprises a clinically and genetically heterogeneous group of disorders with a progressive extrapyramidal syndrome and high basal ganglia iron, and includes pantothenate kinase-associated neurodegeneration caused by mutations in PANK2 (neurodegeneration with brain iron accumulation type I). Post-mortem examination of the brain of a patient with neurodegeneration with brain iron accumulation associated with homozygous PLA2G6 mutations demonstrated both Parkinson’s and Alzheimer’s disease pathology with widespread Lewy bodies, dystrophic neurites and cortical neuronal neurofibrillary tangles (Gregory et al., 2008).
Furthermore, PLA2G6 has also been implicated in a number of other brain diseases including Alzheimer’s disease (Schaeffer and Gattaz, 2008) and bipolar disorder (Xu et al., 2013). More recently, PLA2G6, at the PARK14 locus, has also been characterized as the causative gene in a subgroup of patients with autosomal recessive early-onset dystonia-parkinsonism. Interestingly these patients did not display cerebellar atrophy or basal ganglia iron on MRI (Paisan-Ruiz et al., 2009), and neuropathological examination revealed widespread Lewy body pathology and the accumulation of hyperphosphorylated tau (Paisan-Ruiz et al., 2012). These clinical and neuropathological features further support a link between PLA2G6 mutations and parkinsonian disorders, and demonstrate the clinical heterogeneity in PLA2G6-associated neurodegeneration.
It is not known how mutations in PLA2G6 cause neuropathology. However, it has been suggested that the loss of normal PLA2G6 activity in neurodegenerative disease involves structural abnormalities of cell or mitochondrial membranes and disturbances in lipid homeostasis and lipid metabolism. Indeed, a study in a mouse Pla2g6 knockout model demonstrated ultrastructural abnormalities in mitochondrial and synaptic membranes (Beck et al., 2011). As well as possible mitochondrial defects, it is thought that changes in the composition of the plasma membrane and other intracellular membranes may in turn affect the normal processes responsible for the movement of membranes within axons and dendrites, subsequently leading to accumulation of membranes as so-called spheroids.
Support for a loss-of-function of PLA2G6 enzyme activity in causing disease comes from a recent study on recombinant wild-type and mutant human PLA2G6 proteins. This demonstrated that mutations in PLA2G6 associated with infantile neuroaxonal dystrophy or neurodegeneration with brain iron accumulation resulted in encoded proteins that exhibited <20% of control levels of phospholipase and lysophospholipase activities. Conversely, mutations associated with dystonia-parkinsonism did not impair catalytic activity. The differential enzymatic activities associated with PLA2G6 mutations in infantile neuroaxonal dystrophy/neurodegeneration with brain iron accumulation and dystonia-parkinsonism may explain the relatively later onset and milder phenotype seen in the latter. Furthermore, the preserved enzymatic function of dystonia-parkinsonism causing mutations must be interpreted with caution as there may be differences in PLA2G6 activity in vivo that are not detected by these in vitro assays. For example, there may be alternative mechanisms that alter the regulation of PLA2G6 activity, such as changes in the binding to calmodulin or other proteins (Engel et al., 2010). Further support for the loss of enzymatic function hypothesis comes from a recent study of a Chinese population with Parkinson’s disease, which identified novel PLA2G6 mutations occurring in the heterozygous state, associated with an inhibition in the phospholipid-hydrolysing functions of PLA2G6 (Gui et al., 2013). Moreover Gregory et al. (2008) found a genotype–phenotype correlation in patients with infantile neuroaxonal dystrophy and neurodegeneration with brain iron accumulation: mutations that are predicted to lead to an absence of protein were associated with more severe infantile neuroaxonal dystrophy-type clinical phenotypes, while those with compound heterozygous missense mutations correlated with the less severe phenotype of neurodegeneration with brain iron accumulation and are predictedto result in protein with some residual enzyme function.
Genetic ablation of Pla2g6 in mice is associated with an in vivo disturbance of brain phospholipid metabolism (Cheon et al., 2012), mitochondrial membrane degeneration (Beck et al., 2011) and marked cerebellar atrophy (Zhao et al., 2011). Furthermore, in vitro experiments demonstrated a disturbance in calcium signalling in astrocytes from PLA2G6-deficient mice (Strokin et al., 2012). However, despite these studies, the precise molecular mechanisms linking loss of PLA2G6 activity to mitochondrial morphological abnormalities and neurodegeneration remain unclear and further studies are required.
The fruit fly Drosophila melanogaster has proven to be an excellent model system for studying neurodegenerative diseases, especially the role of mitochondrial activity in Parkinson’s disease (Clark et al., 2006; Park et al., 2006). Once a faithful model is established, the fly has the advantage over mouse models in that it can be used to perform high-throughput screening to identify genetic modifiers and novel therapeutic targets. Here we show that iPLA2-VIA, the Drosophila homologue of PLA2G6, plays an essential role in maintaining normal mitochondrial function. We demonstrate that knockout of the iPLA2-VIA gene activity in the fly results in reduced survival, locomotor deficits and organismal hypersensitivity to oxidative stress. Furthermore, loss of iPLA2-VIA function leads to a number of mitochondrial abnormalities, including reduced mitochondrial membrane potential, mitochondrial respiratory chain dysfunction and reduced ATP synthesis. We also show that levels of lipid peroxidation are significantly elevated in the brains of flies lacking iPLA2-VIA. In addition, we examined cultured fibroblasts taken from two patients with known PLA2G6 mutations and found similar abnormalities to those seen in the fly, including mitochondrial membrane abnormalities and raised levels of cytoplasmic and mitochondrial reactive oxygen species. Moreover, lipid peroxidation levels, especially in the mitochondria, were elevated in the PLA2G6 mutant human fibroblasts compared to controls. Taken together, our findings suggest that loss of normal PLA2G6 gene activity leads to mitochondrial lipid peroxidation, mitochondrial dysfunction and mitochondrial membrane abnormalities. Finally we demonstrate that reduction of lipid peroxidation, by treatment with deuterated polyunsaturated fatty acids (D-PUFAs), is able to recover the mitochondrial membrane potential of PLA2G6 mutant human fibroblasts and partially rescue the locomotor deficits in iPLA2-VIA knockout flies. We have therefore shown that the iPLA2-VIA knockout fly appears to be a useful model organism for studying PLA2G6-associated neurodegeneration.
Materials and methods
Fly stocks and husbandry
The fly stocks y[1] w[67c23]; P{w[+mC] y[+mDint2]=EPgy2}iPLA2-VIA[EY05103], actin-5C-GAL4, elav-GAL4c155 were obtained from the Bloomington Drosophila Stock Centre. The iPLA2-VIA RNAi (HMS01544) line was provided by the TRiP at Harvard Medical School. All fly strains used were backcrossed at least six generations into the w1118 background to obtain isogenic lines. All fly stocks were maintained at 25°C on a 12:12 hour light: dark cycle at constant humidity on a standard sugar-yeast (SY) medium (15 g/l agar, 50 g/l sugar, 100 g/l autolysed yeast, 100 g/l nipagin and 3 ml l/ propionic acid). For all experiments, flies were raised at a standard density on standard SY medium in 200 ml bottles unless otherwise stated. Tissue-specific expression of iPLA2-VIA RNAi constructs was achieved by using the GAL4-UAS system [GAL4-dependant upstream activator sequence (Brand and Perrimon, 1993)].
Lifespan analyses
The survival assays were performed using newly eclosed flies that were allowed to mature and mate for 24 h before the females and males were separated and collected. One hundred and fifty flies were housed in groups of 15 and the flies were transferred every 2 to 3 days onto fresh food and the number of dead flies recorded. Data are presented as cumulative survival curves, and survival rates were compared using log-rank tests.
Climbing assays
The climbing assay was performed using a negative geotaxis assay according to previously published methods (Rival et al., 2004). Briefly, 15 adult flies were placed in a vertical column (25 ml tissue culture pipette), and allowed to climb for 45 s before the number of flies at the top and bottom was determined. A performance index (PI) defined as 0.5(n(total) + n(top) − n(bottom)/ n(total)) was calculated. The experiment was repeated three times for each time point.
Fertility tests
For female fecundity tests, female flies were housed with males for 48 h post-eclosion and then separated into vials at a density of 10 females per vial. Eggs were collected over a 24-h period at different time points. The number of eggs laid per vial at each time point was counted.
Quantitative RT-PCR
Total RNA was extracted from 15 flies per sample using TRIzol® (GIBCO) according to the manufacturer’s instructions. The concentration of total RNA purified for each sample was measured using an Eppendorf biophotometer. One microgram of total RNA was then subjected to DNA digestion using DNase I (Ambion), immediately followed by reverse transcription using the SuperScript® II system (Invitrogen) with oligo(dT) primers. Quantitative PCR was performed using the PRISM 7000 sequence-detection system (Applied Biosystems), SYBR® Green (Molecular Probes), ROX Reference Dye (Invitrogen), and HotStarTaq (Qiagen) by following the manufacturer’s instructions. Each sample was analysed in triplicate with both target gene (iPLA2-VIA) and reference gene (RP49) primers in parallel. The end products of the RT-PCR reactions were also visualized by staining with ethidium bromide following separation with agarose-gel electrophoresis. The primers for the Drosophila iPLA2-VIA used in the RT-PCR of the EY05103 fly line were as follows: forward 5’-TACTGGAATTGTGCGATAAGG-‘3 and reverse 5’-GATGTGGTATTGGAATCCGAG-‘3. The primers used for the RT-PCR performed on the iPLA2-VIA RNAi (HMS01544) fly line were as follows: forward 5’- AACtriacylglycerolTAGTGCCGATCGTCCAA-‘3 and reverse 5’-GAACCAGTATCCTTGCAGCG-‘3. The reference gene primers were as follows: forward 5’-ATGACCATCCGCCCAGCATCAGG-‘3 and reverse 5’-ATCTCGCCGCAGTAAACG-‘3.
Stress experiments
For all stress assays, flies were reared and housed as for lifespan experiments. For oxidative stress assays, 7-day or 15-day old flies were transferred onto 5% sucrose/agar containing 5% hydrogen peroxide (Sigma) or 20 mM paraquat (Sigma). For starvation and osmotic stress experiments, 7-day or 15-day old flies were transferred to 1% agar or 500 mM NaCl respectively. For hypoxia experiments 15-day old flies were transferred to a hypoxia chamber with oxygen levels of 7%. After 15 h the flies were removed from the chamber and the number of dead flies was counted.
Triacylglyceride and ATP assays
Levels of triacylglyceride in adult females were measured using the Triglyceride Infinity Reagent (ThermoScientific) and normalized to total body protein using a BCA assay (Sigma). The ATP concentration of whole female flies was determined using the Roche ATP Bioluminescence Assay Kit HS II (Roche). Briefly, two live whole flies or eight fly heads were homogenized in 100 μl ice-cold lysis buffer (provided in the kit) for 1 min using a Kontes pellet pestle. The lysate was then boiled for 5 min and centrifuged at 20 000g for 1 min. 2.5 μl of cleared lysate was added to 187.5 μl dilution buffer and 10 μl luciferase and the luminescence was immediately measured using a Tecan Infinite M2000 microplate reader and Magellan V6.5 software. Each reading was converted to the amount of ATP per fly based on the standard curve generated with ATP standards. At least three measurements were made for each genotype and the experiment was repeated three times.
Isolation of Drosophila mitochondria
Fly mitochondria were isolated by differential centrifugation, essentially as described in Miwa et al. (2003). Briefly, adult flies (n = 500 for each time point) were chilled on ice and gently pressed using a pre-chilled pestle and mortar. Importantly, the pestle was moved in a vertical motion (with no horizontal motion) until the shape of the flies was no longer visible. The flies were then washed in STE + BSA buffer [250 mM sucrose, 5 mM Tris, 2 mM EGTA, pH 7.4 (4°C), 1% bovine serum albumin]. The flies were then pressed further in 5 ml of STE + BSA buffer. The squashed flies were then passed through double-layered muslin cloth. The collected pulp was then spun for 3 min at 4°C at 1500 rpm to remove debris. The supernatant was then passed through a single layer of muslin into a clean centrifuge tube and spun for 10 min at 4°C and 10 000 rpm to collect the mitochondria. The mitochondrial pellet was then suspended in 250 μl STE + BSA buffer and stored on ice. A BCA assay (Sigma) was used to determine the protein concentration of the mitochondrial preparations. The isolated mitochondria were kept on ice and used immediately for experiments.
Drosophila mitochondrial respiration measurement
Oxygen consumption was measured using a Clark-type oxygen electrode thermostatically maintained at 25°C. Glutamate (5 mM) and malate (5 mM) or pyruvate (5 mM) were added to measure Complex I-linked respiration, and succinate (5 mM) with rotenone (5 µM) were added to measure Complex II-linked respiration. All data were obtained using an Oxygraph Plus system with Chart recording software.
Light and electron microscopy of fly brains
Flies were decapitated and the proboscis was removed to allow penetration of the fixative. The heads were perfused with 3% glutaraldehyde in phosphate buffer overnight. Brains were then treated with 1% osmium tetroxide for 3 h at 4°C and embedded in Araldite® CY212 resin. Thin sections were stained with toluidine blue and the brain visualized with the light microscope. Ultrathin sections (70 µm) were stained with lead citrate and uranyl acetate and digital images taken on a Phillips CM10 electron microscope with a Megaview III digital camera (Olympus).
Human fibroblast collection and culture
The p.R747W PLA2G6 fibroblasts were generated from a 4 mm skin punch biopsy taken under local anaesthetic. Biopsies were dissected into ∼1 mm pieces and cultured in 5 cm petri dishes in Dulbecco's modified Eagle's medium (DMEM), 10% foetal bovine serum, 1% L-glutamine until fibroblasts were seen to grow out from the explants. When fibroblasts reached confluency, they were detached from culture dishes using TrypleE™ (Invitrogen) and transferred to larger culture vessels for further expansion and cryopreservation. This sample was collected with the written consent of the participant and formal ethical approval from the National Hospital for Neurology and Neurosurgery–Institute of Neurology Joint Research Ethics Committee (London, UK). The GM11529 mutant PLA2G6 human fibroblast cell cultures were obtained from the Coriell Cell Repositories. This line was sampled from a patient with infantile neuroaxonal dystrophy.
Fibroblasts were seeded at a density of 4 × 104 cells/cm2, grown to 75–80% confluence and maintained at 37°C and 5% CO2 in DMEM supplemented with 10% (v/v) foetal bovine serum, 2 mM L-glutamine and 1% (v/v) penicillin/streptomycin.
Measurement of mitochondrial membrane potential and cytosolic and mitochondrial reactive oxygen species
For measurements of mitochondrial membrane potential (ΔΨm), dissected fly brains or cells were loaded with 25 nM tetramethylrhodamine methylester (TMRM) for 30 min at room temperature and the dye was present for the duration of the experiment. TMRM is used in the redistribution mode to assess ΔΨm, and therefore a reduction in TMRM fluorescence represents depolarization.
Confocal microscopy of mitochondrial membranes in fly brains and human fibroblasts
Confocal images were obtained using a Zeiss 710 VIS CLSM equipped with a META detection system and a ×40 oil immersion objective. Illumination intensity was kept to a minimum (at 0.1–0.2% of laser output) to avoid phototoxicity and the pinhole set to give an optical slice of ∼2 µm. TMRM was excited using the 560 nm laser line and fluorescence measured above 580 nm. All data presented were obtained from at least five coverslips and two to three different cell preparations for cell experiments, and six coverslips and at least six counts for fly brain experiments. The same anatomical area of the fly brain was used each time.
Measurement of reactive oxygen species levels in human fibroblasts
For measurement of mitochondrial reactive oxygen species production, cells were preincubated with 5 µM MitoSOX™ (Molecular Probes) for 10 min at room temperature. Cytosolic reactive oxygen species generation was measured with 2 µM dihydroethidium with the dye present in all solutions throughout the duration of the experiment.
Fluorescence images were obtained on an epifluoresence inverted microscope equipped with a ×20 fluorite objective (Cairn Research). Ratiometric dihydroethidium and MitoSOX fluorescence was recorded with an excitation light at 380 and 530 nm and emission at 440 and 630 nm, respectively.
Measurement of lipid peroxidation in fly brains and human fibroblasts
C11-BODIPY581/591 (Molecular Probes) is a sensitive fluorescent probe for indexing lipid peroxidation in model membrane systems and living cells undergoing a shift from red to green fluorescence emission upon oxidation, even under physiological oxidation (Vaarmann et al., 2010). Fly brains were dissected in phosphate-buffered saline and placed into Hank’s balanced salt solution (HBSS). C11-BODIPY581/591 (5 μM) was loaded for 15 min at 20°C. Coverslips were washed once and resuspended in 1 ml of HBSS. Ratiometric measurements of probe oxidation were taken over a 12 min time course. Fluorescence excitation was at 488 nm, green fluorescence was detected at 530 nm and red fluorescence at 670 nm.
Cardiolipin analysis by mass spectrometry
Cardiolipin and oxidized cardiolipin were measured by mass spectrometry essentially as described previously (Pope et al., 2008; Heather et al., 2010; Rodriguez-Cuenca et al., 2010) with some minor modifications to make the analysis specific for Drosophila cardiolipin species. In brief, 500 flies of each genotype were homogenized in NaPi buffer (50 mM NaPi, 1 mM EDTA, 100 µM DTPA, pH 7.5) using a Kontes pellet pestle. The homogenates were then spun for 2 min at 13 000 rpm to remove fly debris. Lipids were extracted using a modified Folch extraction and the combined organic layers were evaporated to dryness under nitrogen. Before analysis, samples were resuspended in 1 ml of a 1:1 mix of buffers A and B (buffer A, 15% water, 85% methanol containing 1 ml of 250 ml/l aqueous ammonia per litre; buffer B, 97% chloroform, 3% methanol containing 0.1 ml of 250 ml/l aqueous ammonia per litre). 1 nmol of (C14:0)4-cardiolipin (Avanti Polar Lipids) was used as an internal standard and was added to Drosophila head homogenates.
The LC/MS/MS system (Waters 2795 separation unit and a Quattro Micro API triple quadrupole mass spectrometer) and parameters were as described previously (Heather et al., 2010; Rodriguez-Cuenca et al., 2010) except that the multiple reaction monitoring (MRM) transitions were specific for Drosophila cardiolipin. Mass spectra from retention times 5.5–6.0 min were combined to generate cardiolipin spectra. Cardiolipin species were identified in MRM mode with the collision gas at 0.25 Pa and collision energy at 35 eV. Cardiolipin species were analysed as their dianions [CL–2H]2−. In these samples, the dominant form of cardiolipin was (C16:1)4-cardiolipin (m/z 671), which was therefore selected for analysis based on its fragmentation to a C16:1 fatty acid (m/z 253) by monitoring the 671 > 253 transition. Similarly, other cardiolpin species (C18:2)1(C16:1)3-cardiolipin (m/z 684) and (C18:2)2(C16:1)2-cardiolipin (m/z 697) were analysed by assessing transitions of 684 > 253 and 697 > 253, respectively. Oxidized cardiolipin was analysed by the transitions 678 > 253 and 691 > 253 transition. The internal standard (C14:0)4-cardiolipin (m/z 619) was analysed by its fragmentation to a C14:0 fatty acid (m/z 227) using the transition 619 > 227. Data were analysed with QuanLynx software (Waters). Total cardiolipin content was expressed as peak area relative to that of the internal standard. The amounts of each cardiolipin and oxidized cardiolipin were expressed relative to the total amount of cardiolipin in the sample, giving dimensionless ratios.
Deuterated polyunsaturated fatty acids
Deuterated polyunsaturated fatty acids (D-PUFAs) 11,11-D2-linoleic acid (D2-linoleic acid) and 11,11,14,14-D4-α-linolenic acid (D4-linoleic acid) were kindly provided by Dr Mikhail S. Shchepinov (Retrotope, Inc), and their synthesis has been previously described (Hill et al., 2012). Hydrogenated polyunsaturated fatty acids (H-PUFAs) were obtained from Sigma–Aldrich (99%). Cells were preincubated with 10 µM H- or D-PUFA (D4-linoleic acid) in the culture media for 24 h and then washed with HBSS before experiments. For the fly experiments, D2-linoleic acid was diluted in water and added to the standard fly medium to give a final concentration of 10 or 100 μM.
Statistics
Data were expressed as means ± standard error of the mean (SEM). Significant differences were evaluated by unpaired two-tailed Student’s t-test and significance levels are described in individual figure legends. Climbing assays were analysed using a two-way ANOVA or a one-way ANOVA with Bonferroni correction.
Results
Drosophila lacking iPLA2-VIA activity display age-dependent locomotor deficits and reduced lifespan
A null mutant fly stock completely lacking the Drosophila orthologue of the PLA2G6 gene, iPLA2-VIA, was obtained from the Bloomington Drosophila Stock Centre. We will refer to null mutant flies lacking both iPLA2-VIA genes as iPLA2-VIA−/− henceforth. This fly strain carries a P-element insertion (EY05103) in the iPLA2-VIA gene (Fig. 1A). As previously shown, RT-PCR analysis confirmed that there was complete knockout of iPLA2-VIA gene expression in this strain with no detectable SYBR green signal produced. The end products of the RT-PCR reactions were also visualized on agarose gel electrophoresis, which confirmed the complete absence of iPLA2-VIA gene expression in the iPLA2-VIA−/− flies (Fig. 1B) (Malhotra et al., 2009). Knockout of iPLA2-VIA activity in the fly did not produce any gross changes in body size or obvious differences in morphology (data not shown). iPLA2-VIA is expressed throughout the adult fly body, including the brain and eye. The iPLA2-VIA gene displays good homology with the human PLA2G6 gene, with a 51% identity and 67% positivity (FlyBase).
Figure 1.

Lack of iPLA2-VIA gene activity leads to reduced survival and locomotor deficits. (A) Schematic of the Drosophila iPLA2-VIA gene, showing the position of the P-element insertion (arrowhead). The iPLA2-VIA gene has four main transcripts: iPLA2-VIA-RB, iPLA2-VIA-RC, iPLA2-VIA-RD and iPLA2-VIA-RA (FlyBase). The position of the primers has been previously shown (Malhotra et al., 2009). (B) RT-PCR analysis of iPLA2-VIA−/− flies shows that there is no mRNA expression of the iPLA2-VIA gene compared with w1118 control flies. The end products of the RT-PCR reaction were run on an agarose gel. RP49 is an internal control gene and is expressed in both the iPLA2-VIA−/− and control w1118 flies. (C) iPLA2-VIA−/− flies display an age-dependent reduction in climbing ability compared with w1118 controls (n = 45 flies per genotype, three repeats per genotype, P < 0.05). Climbing ability is expressed as a perfomance index (PI) over time in days (d). (D) Female flies lacking both iPLA2-VIA genes (iPLA2-VIA−/−) have a significantly reduced lifespan compared with w1118 control flies (P < 0.0001). Furthermore female flies lacking only one iPLA2-VIA gene (iPLA2-VIA+/−; n = 150 flies per genotype) do not display a reduction in lifespan. (E) Ubiquitous downregulation of iPLA2-VIA activity using sRNAi (UAS-iPLA2-VIA-RNAi) driven by Act5c-Gal4 (Act5c-Gal4 > UAS-iPLA2-VIA-RNAi) results in an age-dependent reduction in climbing ability compared with control UAS-iPLA2-VIA-RNAi flies (P < 0.05; n = 45 flies per genotype, three repeats each). (F) Knockdown of iPLA2-VIA activity using iPLA2-VIA sRNAi (UAS-iPLA2-VIA-RNAi) driven ubiquitously by Act5c-Gal4 (Act5c-Gal4 > UAS-iPLA2-VIA-RNAi) results in a decrease in lifespan compared with control non-expressing UAS-iPLA2-VIA-RNAi flies (P < 0.0001) or driver Act5c-Gal4 flies (P < 0.0001), similar to that seen with the null mutant flies (iPLA2-VIA−/−; n = 150 flies per genotype). (G) Knockdown of iPLA2-VIA activity using sRNAi (UAS-iPLA2-VIA-RNAi) driven in the fly nervous system using the elav-Gal4 driver results in a decrease in lifespan compared with control non-expressing UAS-iPLA2-VIA-RNAi flies (P < 0.0001), or driver elav-Gal4 (P < 0.0001; n = 150 flies per genotype), similar to that seen with the null mutant flies (iPLA2-VIA−/−).
The iPLA2-VIA−/− flies displayed progressive locomotor deficits, with the climbing ability of iPLA2-VIA−/− flies in response to light tapping progressively falling below that of w1118 controls from ∼20 days of age (Fig. 1C). Accompanying this climbing defect the iPLA2-VIA−/−flies also showed an ∼50% reduction in lifespan, in both females (Fig. 1D) and males (Supplementary Fig. 1A). Furthermore, flies heterozygous for the EY05103 P-element insertion (iPLA2-VIA), with presumed 50% gene activity only, did not display reduced survival (Fig. 1D and Supplementary Fig. 1A). To ensure that the reduced lifespan was, indeed, due to the EY05103 P-element insertion, we generated flies with precise excisions of the P-element, by introducing a transposase in an appropriate cross, and found that this restored a normal lifespan (Supplementary Fig. 1B).
To confirm the effect of iPLA2-VIA deficiency in the fly, we also inhibited iPLA2-VIA gene expression by transgenic RNA interference (RNAi) technology, using the GAL4/UAS system (Brand and Perrimon, 1993). Ubiquitous expression of iPLA2-VIA dsRNAi (UAS-iPLA2-VIA-RNAi) with the actin-GAL4 driver resulted in a ∼40% reduction in whole body iPLA2-VIA gene expression using RT-PCR analysis (Supplementary Fig. 1C). This reduction was associated with an age-dependent decrease in climbing ability (Fig. 1E) and a shortened lifespan (Fig. 1F). It can therefore be robustly concluded that genetic inactivation of iPLA2-VIA activity results in progressive locomotor deficits and a reduction in lifespan in Drosophila. To determine whether these phenotypes were specific to loss of iPLA2-VIA activity in the brain, we knocked down iPLA2-VIA in fly neural tissue using UAS-iPLA2-VIA-RNAi driven by the elavc155-Gal4 driver. We observed a significant reduction in lifespan in these flies (Fig. 1G), confirming the brain-specific effects of iPLA2-VIA deficiency.
We also assessed the fecundity of iPLA2-VIA−/− female flies. The number of eggs laid per female significantly declined with age compared with age-matched w1118 control flies, and by Day 25 the null mutant flies were almost completely infertile (Supplementary Fig. 1D).
Brains lacking iPLA2-VIA display severe, widespread neurodegeneration and mitochondrial degeneration
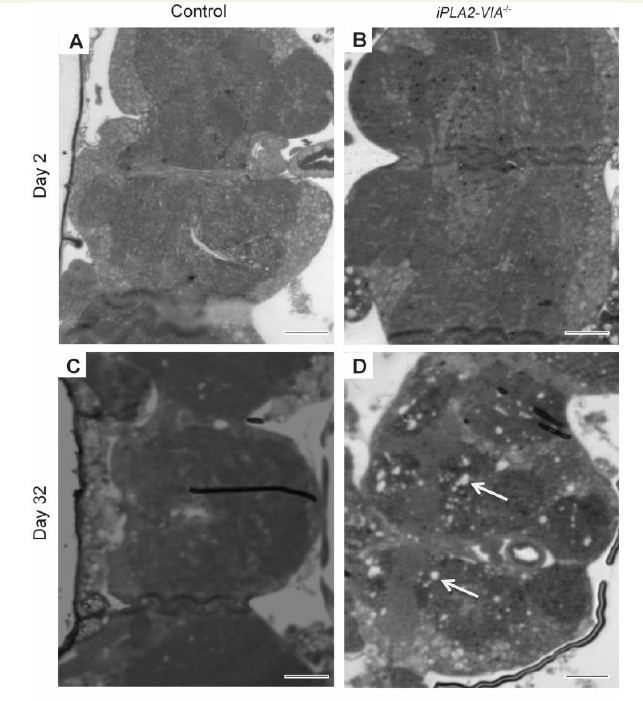
Light microscopic examination of thin paraffin sections of iPLA2-VIA−/− fly brain revealed normal brain architecture in young Day 2 flies that was indistinguishable from w1118 controls (Fig. 2A and B). However, by Day 32 there was significant widespread vacuolation in the iPLA2-VIA−/− fly brains that was not present in w1118 control flies (Fig. 2C and D). Ultrastructural examination using electron microscopy revealed that the mitochondria were grossly abnormal and swollen with fragmented cristae in aged Day 32 iPLA2-VIA−/− fly brains as compared to the mitochondria in age-matched w1118 control brains (Fig. 2E–H) or younger Day 2 iPLA2-VIA−/− flies (data not shown), which were both filled with densely packed cristae. Furthermore, examination of the fly eye using electron microscopy revealed abnormal retinal structure in the aged Day 32 iPLA2-VIA−/− flies compared with age-matched control flies, with grossly abnormal ommatidial structure (Supplementary Fig. 2A and B).
Figure 2.

Fly brains lacking iPLA2-VIA display severe mitochondrial degeneration at the ultrastructural level. (A) Light microsopic examination of Day 2 w1118 control (scale bar = 20 μm) and (B) iPLA2-VIA−/− fly brains (scale bar = 30 μm). (C) At Day 32 w1118 control fly brains display healthy brain tissue compared with (D) iPLA2-VIA−/− flies brains, which display widespread vacuolation (arrows) (×60 magnification; scale bars 20 = μm). (E and F) Ultrastructural examination of aged Day 32 w1118 control fly brains (×2500; scale bar = 0.6 μm and × 5300; scale bar = 0.2 μm) revealed normal mitochondrial morphology, which is in contrast to (G and H) Day 32 iPLA2-VIA−/− fly brains, which display significant degeneration of the mitochondrial cristae (×3100; scale bar = 0.5 μm and × 5300; scale bar = 0.2 μm). Images representative of six fly brains examined for each genotype. No abnormal mitochondria were seen in the w1118 control fly brains.
Loss of iPLA2-VIA resulted in reduced mitochondrial membrane potential and abnormal mitochondrial respiratory chain activity
Given the abnormal mitochondrial membrane morphology in older iPLA2-VIA−/− flies, we investigated whether loss of iPLA2-VIA affected mitochondrial membrane potential (ΔΨm), an indicator of mitochondrial health and function. Mitochondrial membrane potential was assessed in the brains of 25-day-old flies using TMRM fluorescence and imaging with confocal microscopy. This revealed a significant reduction in mitochondrial membrane potential in iPLA2-VIA−/− fly brains compared to controls (Fig. 3A). We next isolated mitochondria from iPLA2-VIA−/− flies and assessed respiratory chain activity by measuring oxygen consumption using a Clark-type oxygen electrode. The steps in respiration were compared in mitochondria isolated from iPLA2-VIA−/− flies and control w1118 flies with substrates and inhibitors specific to individual respiratory complexes. We found that knockout of iPLA2-VIA activity conferred a decrease in complex I and II-dependent respiration (Fig. 3B), manifested as significantly decreased oxygen consumption in state 3 respiration, whether using substrates for complex I (5 mM glutamate and 5 mM malate) or for complex II (5 mM succinate in the presence of rotenone) (Fig. 3B). The respiratory control ratio is the ratio of state 3 respiration (ADP stimulated) to state 4 respiration (no ADP present), and is considered an indication of the degree of coupling of mitochondrial respiratory chain activity to oxidative phosphorylation (Chance and Williams, 1955). The respiratory control ratio was significantly lower in iPLA2-VIA−/− mitochondria compared with age-matched control w1118 flies (Fig. 3C), because of the inhibition of state 3 respiration. Furthermore, the respiratory control ratio of iPLA2-VIA−/− flies in the presence of glutamate/malate was dependent on the age of the flies: older flies having lower respiratory control ratio values (Fig. 3C). Thus, loss of iPLA2-VIA activity reduces mitochondrial membrane potential as a result of mitochondrial uncoupling, which increases with age in the iPLA2-VIA−/− flies.
Figure 3.

Flies lacking iPLA2-VIA have reduced mitochondrial membrane potential and decreased mitochondrial respiratory chain activity. (A) The mitochondrial membrane potential measured using TMRM fluorescence in the brains of 25-day-old flies was significantly decreased in iPLA2-VIA−/− brains compared to w1118 controls (***P < 0.001; n = 6 brains for each genotype). (B) The respiratory chain activity was measured by assessing oxygen consumption and demonstrated that mitochondria from young Day 2 and old Day 32 iPLA2-VIA−/− flies have reduced complex I and II–dependent respiration compared with w1118 control flies, manifested as significantly decreased oxygen consumption in state 3 respiration (*P < 0.05, **P < 0.001). (C) The respiratory control ratio was significantly lower in iPLA2-VIA−/− mitochondria compared with age-matched control w1118 mitochondria. Furthermore, the respiratory control ratio of iPLA2-VIA−/− flies in the presence of glutamate/malate was dependent on the age of the flies: older flies having lower respiratory control ratio values (*P < 0.05, **P < 0.001).
Loss of iPLA2-VIA activity results in reduced ATP levels and increased sensitivity to oxidative stress
In keeping with the mitochondrial dysfunction caused by loss of iPLA2-VIA activity, we also demonstrated that ATP levels in aged, Day 25, iPLA2-VIA−/− fly heads were reduced by 40% compared with control flies (Fig. 4A). Levels were also reduced in whole iPLA2-VIA−/− flies compared with controls (Supplementary Fig. 3A).
Figure 4.

iPLA2-VIA−/− flies have reduced ATP levels and are more sensitive to oxidative stress. (A) ATP levels are reduced in the heads of Day 25 iPLA2-VIA−/− flies compared with age-matched w1118 control flies (*P < 0.05). (B) Aged Day 15 iPLA2-VIA−/− flies show reduced survival when treated with 5% hydrogen peroxide (H2O2) (P = 5.6 × 10−7) and (C) 20 mM paraquat (P = 4.6 × 10−7) compared with w1118 control flies (n = 150 flies per genotype).
Mitochondrial dysfunction can lead to decreased resistance to reactive oxygen species, which has been implicated in many neurodegenerative diseases. To explore the role of iPLA2-VIA in resistance to oxidative stress, we examined the survival of iPLA2-VIA−/− flies after exposure to the potent oxidizer hydrogen peroxide and paraquat, a free radical generator. Consistent with the structural mitochondrial abnormalities in aged flies, iPLA2-VIA−/− flies showed an age-dependent, mild sensitivity at Day 15 to hydrogen peroxide and, more so, to paraquat (Fig. 4B and C) that was not consistently present in younger 7-day-old flies (data not shown). Both young Day 7 and older Day 15 iPLA2-VIA−/− flies were also sensitive to osmotic stress and showed reduced survival compared to w1118 control flies when fed 500 mM NaCl (Supplementary Fig. 3B). Day 15 iPLA2-VIA−/− flies also displayed increased sensitivity to hypoxic and xenobiotic (dichlorodiphenyltrichloroethane, DDT) stresses (Supplementary Fig. 3C and D).
Loss of iPLA2-VIA was not associated with changes in cardiolipin composition
Given that mitochondrial membrane morphology appeared to be abnormal in the iPLA2-VIA−/− flies, we performed mass spectroscopic analysis of cardiolipin, one of the major phospholipids in mitochondrial membranes. It has been hypothesized that PLA2G6 is critical for the normal repair and remodelling of damage to cardiolipin caused by mitochondrial reactive oxygen species, and that loss of PLA2G6 activity may lead to cardiolipin oxidation and pathogenic downstream events such as induction of apoptosis. To test this hypothesis, we performed mass spectroscopic analysis on iPLA2-VIA−/− fly heads to compare the cardiolipin composition with that in w1118 control flies. We found no significant differences in the cardiolipin content, monolysocardiolipin content or fatty acid composition between iPLA2-VIA−/− and control w1118 fly brains (Supplementary Fig. 4). Similar results have previously been reported in whole flies lacking iPLA2-VIA (Malhotra et al., 2009). Furthermore, we found that oxidized cardiolipin was not detected in significant amounts in either the iPLA2-VIA−/− or control w1118 fly head homogenates (Supplementary Fig. 4). Oxidized cardiolipin species would be expected to be present as additions of +7/8 m/z on the scan spectra. Our results therefore do not support a role for oxidized cardiolipin in causing mitochondrial dysfunction and neurodegeneration in flies lacking iPLA2-VIA activity.
Loss of iPLA2-VIA promotes brain lipid peroxidation and reduces whole body triacylglycerol levels
Despite the absence of changes in oxidized cardiolipin, we hypothesized that loss of PLA2G6 may lead to abnormal and excessive oxidation of other mitochondrial membrane lipids, which in turn would lead to the observed mitochondrial dysfunction. We therefore studied the effect of knocking out iPLA2-VIA on lipid peroxidation in Drosophila. Rates of lipid peroxidation were measured by live imaging of Day 25 iPLA2-VIA−/− and w1118 control dissected fly brains, using the fluorescent ratiometric oxidation-sensitive dye C11 BODIPY581/591, which shifts its fluorescence from red to green in a time-dependent manner, the rate being proportional to levels of lipid peroxidation. The changes in colour shift were detected by confocal microscopy. This demonstrated that levels of lipid peroxidation in flies lacking the iPLA2-VIA gene were 3.5-fold higher than in age-matched control fly brains (Fig. 5A).
Figure 5.

Flies lacking iPLA2-VIA have increased lipid peroxidation in their brains and reduced triacylglycerol (TAG) levels. (A) Lipid peroxidation was measured using C11-BODIPY581/591 and demonstrated that Day 25 iPLA2-VIA−/− fly brains have elevated levels of lipid peroxidation compared with age-matched w1118 controls (n = 6 fly brains per experiment; **P < 0.01). (B) Whole-body triacylglycerol levels are decreased in iPLA2-VIA−/− flies in an age-dependent manner compared with w1118 control flies. (***P = 0.005 Day 20 and *P = 0.01 Day 32). (C) Day 15 iPLA2-VIA−/− flies are more sensitive to starvation conditions than age-matched w1118 control flies (P = 0.001; n = 150 flies per genotype).
There is evidence that PLA2G6 may play a role in the regulation of triacylglycerol synthesis by providing endogenous fatty acids (Gubern et al., 2009). Triacylglycerol is the main form of lipid storage in the fly and therefore reflects the ability of flies to respond to starvation conditions (Ballard et al., 2008). Measurement of whole-body triacylglycerol levels in female flies revealed a significant age-dependent reduction in both control w1118 and iPLA2-VIA−/− flies. However, the reduction was significantly greater by Day 20 in the flies lacking iPLA2-VIA activity (Fig. 5B). In keeping with an age-dependent decrease in triacylglycerol levels, iPLA2-VIA−/− flies were not sensitive to starvation conditions at Day 7 but became sensitive at Day 15 compared with control w1118 flies (Fig. 5C).
Human mutant PLA2G6 fibroblasts display abnormal mitochondrial physiology
Given that loss of iPLA2-VIA led to increased lipid peroxidation and mitochondrial dysfunction in Drosophila, we examined fibroblasts taken from a patient with a known homozygous p.R747W mutation in PLA2G6 causing dystonia-parkinsonism (Paisan-Ruiz et al., 2009). Using TMRM fluorescence to probe for mitochondrial membrane potential, we found that there was a significant decrease in basal mitochondrial membrane potential in the mutant human fibroblasts (Fig. 6A and B), similar to that seen in fly brains lacking iPLA2-VIA.
Figure 6.

Human mutant PLA2G6 fibroblasts have reduced mitochondrial membrane potential and increased levels of mitochondrial and cytosolic reactive oxygen species levels. (A) The mitochondrial membrane potential of human p.R747W mutant PLA2G6 fibroblasts is reduced compared with control human fibroblasts (*P < 0.05 control 1 and **P < 0.002 control 2) as measured by TMRM fluorescence. (B) This is clearly seen as a reduction in red TMRM staining in the p.R747W mutant PLA2G6 human fibroblasts compared with control fibroblasts (Scale bars = 44 μm). (C) Mitochondrial reactive oxygen species production is increased 28% in human p.R747W mutant PLA2G6 fibroblasts compared to control fibroblasts (P < 0.001; n = 6). (D) Cytosolic reactive oxygen species production is also increased in human p.R747W mutant PLA2G6 fibroblasts compared with control fibroblasts (1.47-fold increase; P < 0.001; n = 6). (E) Rates of reactive oxygen species production in C and D are shown as a percentage. Het = dihydroethidium.
Abnormal respiration and low mitochondrial membrane potential can induce excessive reactive oxygen species production in the matrix of mitochondria. Indeed the rate of mitochondrial reactive oxygen species production, as assessed using MitoSOX fluorescence, was increased by 28% relative to control human fibroblasts (Fig. 6C and E). Cytosolic reactive oxygen species were also significantly higher in the fibroblasts with a PLA2G6 mutation (Fig. 6D and E).
Mutations in PLA2G6 are associated with elevated mitochondrial lipid peroxidation levels in human fibroblasts and are reversed by treatment with deuterated polyunsaturated fatty acids
Following the demonstration that there is an increase in lipid peroxidation in the brains of flies lacking iPLA2-VIA compared with controls, we also assessed lipid peroxidation in two human PLA2G6 fibroblast lines; one taken from a patient with dystonia-parkinsonism with a p.R747W mutation (Paisan-Ruiz et al., 2009) and the other from a child with infantile neuroaxonal dystrophy. We assessed lipid peroxidation levels using the fluorescent ratiometric oxidation-sensitive C11 BODIPY581/591 dye. This demonstrated that PLA2G6 mutant fibroblasts had an increased level of lipid peroxidation compared to controls (Fig. 7A), suggesting therefore that lipid peroxides may be a potential therapeutic target.
Figure 7.

Elevated lipid peroxidation is a possible therapeutic target in PLA2G6-associated disease. (A) Lipid peroxidation levels in intact human fibroblasts taken from two patients with PLA2G6 mutations (mutant 1 fibroblasts were taken from a patient with a p.R747W PLA2G6 mutation and mutant 2 fibroblasts were from a patient with a PLA2G6 mutation and infantile neuroaxonal dystrophy) are increased compared with fibroblasts from healthy controls. In addition, pretreatment with D-PUFAs (D4-linoleic acid) for 24 h significantly rescues the elevated lipid peroxidation rates in the human PLA2G6 mutant fibroblasts (***P < 0.0001; n.s = non-significant). (B) The mitochondrial membrane potential of human mutant PLA2G6 fibroblasts is considerably reduced compared with control fibroblasts and is significantly recovered below control levels following 24 h pre-treatment with D4-linoleic acid (***P < 0.0001). (C) Lipid peroxidation levels are also increased in permeabilized mutant p.R747W PLA2G6 fibroblasts compared with controls in the absence (***P < 0.05 for control 1 and control 2) and presence of rotenone (***P < 0.05 for controls 1 and 2), suggesting that it is generated from a mitochondrial source. (D) Treatment of iPLA2-VIA−/− flies with 10 μM D2-linoleic acid partially rescues the climbing deficits in an age-dependent manner compared with untreated controls (n = 45 flies per group, three repeats per group; one-way ANOVA with Bonferroni correction ***P < 0.003). There is a trend for 100 μM D2-linoleic acid to rescue the climbing deficits of iPLA2-VIA−/− flies but this is not statistically significant. (E) Schematic diagram showing how loss of normal PLA2G6 activity might lead to mitochondrial dysfunction. A mutation in PLA2G6 results in loss of PLA2G6 activity. In turn this results in elevated lipid peroxidation of mitochondria, with the resultant decrease in mitochondrial membrane potential, decrease in ATP synthesis and mitochondrial reactive oxygen species production. The mitochondria eventually degenerate and become swollen in appearance. All of these abnormalities result in mitochondrial dysfunction and subsequent cellular dysfunction with consequent neuronal death. NBIA = neurodegeneration with brain iron accumulation; ROS = reactive oxygen species.
Natural polyunsaturated fatty acids (PUFAs) such as linoleic acid undergo autoxidation by reactive oxygen species, resulting in the production of toxic reactive carbonyl compounds that can mediate DNA damage, carcinogenesis and inflammation. However, replacement of the bis-allylic hydrogen atoms with deuterium atoms (termed site-specific isotope-reinforcement) arrests PUFA autoxidation due to the isotope effect. Indeed, even small amounts of isotope-reinforced D-PUFA are able to protect yeast from lipid autoxidation-mediated cell killing (Hill et al., 2011, 2012). These benefits have also been confirmed in vivo with the demonstration that D-PUFAs reduce nigrostriatal degeneration in a mouse model of Parkinson’s disease (Shchepinov et al., 2011). We therefore pretreated human PLA2G6 mutant fibroblasts for 24 h with D-PUFA (D4-linoleic acid) and found that this decreased lipid peroxidation in the cells to below control levels (Fig. 7A). This was also associated with a rescue in the reduced mitochondrial membrane potential (Fig 7B). Moreover, assessment specifically of lipid peroxidation levels in digitonin-permeabilized cells, which destroys all cell membranes except mitochondrial ones, also demonstrated that lipid peroxidation was substantially increased. In addition, when rotenone was added to the fibroblasts there was no further significant increase in the lipid peroxidation level, indicating that the lipid peroxidation was predominantly produced by mitochondria in the cell (Fig. 7C).
Deuterated linoleic acid partially rescued the locomotor abnormalities of flies lacking iPLA2-VIA
Finally, we tested the effect of inhibiting lipid peroxidation with D-PUFAs on the neurodegenerative locomotor phenotype seen in flies lacking iPLA2-VIA. Crosses for iPLA2-VIA−/− and w1118 control flies were set up on food containing vehicle (H20), 10 or 100 μM D2-linoleic acid. Eclosed offspring were also kept on standard food containing either vehicle or D2-linoleic acid and aged while their climbing ability was assessed. iPLA2-VIA−/− flies treated with 10 μM D2-linoleic acid had significantly improved motor performance in old age compared to flies treated with control food (Fig. 7D). There was a trend towards improved climbing ability at older ages when iPLA2-VIA−/− flies were treated with 100 μM D2-linoleic acid, but this was not statistically significant. The rescuing effect of D2-linoleic acid on climbing ability was specific to iPLA2-VIA−/− flies, as the climbing ability of control w1118 flies treated with 10 μM D2-linoleic acid was indistinguishable from controls (Supplementary Fig. 5B). The effect of D2-linoleic acid on the survival of iPLA2-VIA−/− flies was also assessed. There was no significant effect of 10 μM D2-linoleic acid on the lifespan of flies lacking iPLA2-VIA. Indeed, at the higher concentration of 100 μM D2-linoleic, the lifespan of the iPLA2-VIA−/− flies was reduced compared to control w1118 flies demonstrating that D2-linoleic acid is toxic to flies at higher concentrations (Supplementary Fig. 5B). Despite the lack of lifespan-prolonging effects, the fact that deuterated linoleic acid significantly improved the locomotor abnormalities of iPLA2-VIA flies suggests that low concentrations of D-PUFAs, or similar compounds, may act as potential therapeutic compounds for the treatment of PLA2G6-associated neurodegeneration.
Discussion
As the energy factories of cells, mitochondria play an essential role in neurons, in which oxidative phosphorylation is the main source of ATP. A previous study in a mouse model demonstrated that knockout of Pla2g6 results in abnormal mitochondrial membrane morphology (Beck et al., 2011). These findings lead to the question as to how loss of normal PLA2G6 gene function leads to abnormal mitochondrial morphology, and whether mitochondrial dysfunction is an early feature of PLA2G6-associated neurodegeneration.
Loss of normal phospholipase A2 activity leads to mitochondrial dysfunction, which precedes mitochondrial membrane defects
In this study, we demonstrated that loss of normal phospholipase A2 activity in the fruit fly and in fibroblasts taken from patients harbouring mutations in PLA2G6, results in striking abnormalities in both mitochondrial function and morphology. Furthermore, we demonstrate that mitochondrial dysfunction precedes the mitochondrial morphological abnormalities that are seen at the ultrastructural level. Deficits in mitochondrial oxidative respiration are seen in iPLA2-VIA knockout flies as young as 2 days of age, when there are no associated mitochondrial membrane abnormalities present on electron microscopy.
Moreover, our findings that mitochondrial dysfunction occurs early in PLA2G6-associated neurodegeneration, is consistent with the clinical features seen in infantile neuroaxonal dystrophy, neurodegeneration with brain iron accumulation and dystonia-parkinsonism, all of which display clear early-onset phenotypes, that have previously been associated with mitochondrial disease.
Loss of normal PLA2G6 activity leads to reactive oxygen species production
In addition to mitochondrial dysfunction, we also demonstrate that loss of PLA2G6 activity is associated with raised levels of reactive oxygen species. These species play important roles in cell signalling, but at abnormally high levels are detrimental to cells, ultimately leading to cell death (Martindale and Holbrook, 2002). Lipids are important targets of reactive oxygen species and therefore elevated reactive oxygen species levels will lead to increases in mitochondrial lipid peroxidation. Furthermore, it may also be that elevated lipid peroxidation arises in the absence of phospholipase A2 activity because some phospholipases preferentially cleave oxidized lipids to maintain redox homeostasis (Domijan et al., 2014). In turn, lipid peroxides are known to trigger a cascade of events that include a fall in mitochondrial membrane potential, mitochondrial swelling and loss of mitochondrial matrix components (Castilho et al., 1994). Furthermore, it has been shown that complex I, the largest complex of the respiratory chain, is susceptible to oxidative stress (Lin and Beal, 2006). Indeed we saw a decrease in oxidative respiration in flies lacking iPLA2-VIA when we used substrates specific for complex I of the respiratory chain. Furthermore, Lewy body disease has been shown to be associated with reduced levels of complex I activity (Gu et al., 1998), further implicating mitochondrial dysfunction in neurodegeneration, and more specifically in PLA2G6-associated neurodegeneration, which has been linked to parkinsonism and Lewy body pathology.
In addition, lipid peroxidation is strongly catalysed through the Fenton reaction by iron and iron complexes (Koster et al., 1980) and it is iron that is often deposited in the basal ganglia of the brains of patients affected with PLA2G6 mutations (Morgan et al., 2006). Thus, iron-mediated oxidative insults seem to have a crucial role in the function of PLA2G6 and in the progression of diseases associated with mutations in this gene.
PLA2G6-associated neurodegeneration is not likely mediated by oxidization of mitochondrial cardiolipin
Given the increase in levels of lipid peroxidation in flies lacking iPLA2-VIA, we looked for elevated levels of oxidized cardiolipin, one of the most common lipids in the inner mitochondrial membrane. It has previously been hypothesized that PLA2G6 is involved in the remodelling of cardiolipin's fatty acid side chains (Malhotra et al., 2009) and in removing oxidized fatty acid side chains. Oxidation of mitochondrial phospholipids such as cardiolipin leads to cytochrome c release and apoptosis (Kagan et al., 2005). To determine whether cardiolipin is oxidized in PLA2G6 loss-of-function, we analysed the cardiolipin composition of iPLA2-VIA−/− fly heads using mass spectrometry. We demonstrated that mitochondrial membrane disintegration and mitochondrial dysfunction occur in the absence of any significant differences in cardiolipin amount or side chain composition in fly brains lacking iPLA2-VIA. Furthermore, oxidized cardiolipin was not detected in significant amounts. This may be related to the types of fatty acids present in Drosophila cardiolipin. Palmitic acid is the major component of cardiolipin side chains in Drosophila and is more resistant to oxidation than linoleic acid, the main cardiolipin side chain in most mammalian tissues. However, it could also point towards redundancy in the cardiolipin remodelling enzymes. There are a number of enzymes involved in cardiolipin remodelling and it may be the case that fatty acid remodelling of the side chains, in particular removal of oxidized fatty acids, can be performed by other enzymes involved in cardiolipin remodelling such as tafazzin (Schlame, 2013).
The abnormal lipid peroxidation seen in PLA2G6 knockout leads to a toxic cascade of downstream events
In this work we report that loss of normal PLA2G6 function is strongly associated with elevated mitochondrial lipid peroxidation. Furthermore lipid peroxidation is known to induce mitochondrial injury, leading to a decrease in oxidative respiratory chain function, reduced mitochondrial membrane potential, reduced cellular ATP levels and raised reactive oxygen species levels, all features we have demonstrated in flies and human fibroblasts lacking normal PLA2G6 gene activity (Fig. 7D). This in turn would be predicted to lead to cell death and neurodegeneration. As lipid peroxides are harmful mediators of oxidative stress, strategies focused on quenching, recycling or protecting such products could be highly effective in the treatment of the disease.
This work suggests that lipid peroxidation-lowering strategies may be beneficial in PLA2G6-associated neurodegeneration
In addition to showing that lipid peroxidation is increased in both fly brains lacking iPLA2-VIA and human PLA2G6 mutant fibroblasts, we have demonstrated that D-PUFAs reduce the rates of lipid peroxidation in two lines of PLA2G6 mutant fibroblasts and restore mitochondrial membrane potential. This suggests that D-PUFAs may have potential therapeutic roles in PLA2G6-associated neurodegeneration and its associated mitochondrial dysfunction.
Our data promote Drosophila as a useful model for studying PLA2G6-associated neurodegeneration, and for manipulating pathways and therapeutic targets in order to reverse or prevent the deleterious effects of lipid peroxides. Loss of iPLA2-VIA gene function results in reduced lifespan and locomotor ability, increased sensitivity to oxidative insults as well as hypoxia, osmotic and starvation stressors. Thus flies lacking iPLA2-VIA display phenotypes that can be used as markers in genetic modifying screens or in the screening of potential therapeutic compounds. To test the validity of the fly model for drug screening and to confirm the benefits of D-PUFAs in vivo in an organism lacking iPLA2-VIA, we examined the effects of D-PUFAs on the locomotor deficits of iPLA2-VIA−/− flies. This demonstrated that treatment with D2-linoleic acid partially rescues the climbing abnormalities in aged iPLA2-VIA knockout flies. Therefore this approach of using compounds, which act to reduce the autoxidation of lipids, may represent a future therapeutic strategy for patients with PLA2G6-associated neurodegeneration and warrants further investigation.
Supplementary Material
Acknowledgements
We would like to thank Drs Fiona Kerr, Teresa Niccoli, Nazif Alic and Ekin Bolukbasi for technical advice and useful discussions, and Filiz Senbabaoglu and Helena Cantwell for technical assistance. We also thank Kerrie Venner for help with electron microscopy and Dr Una Sheerin for help with patient fibroblast sample collection.
We also thank the TRiP at Harvard Medical School (NIH/NIGMS R01-GM084947) for providing a transgenic RNAi fly stock used in this study, and Dr Mikhail S. Shchepinov (Retrotope, Inc) for providing D-PUFAs.
Glossary
Abbreviations
- D/H-PUFA
deuterated/hydrogenated polyunsaturated fatty acids
- D2-linoleic acid
11-11-D2-linoleic acid
- D4-linoleic acid
11,11,14,14-D4-α-linolenic acid
- TMRM
tetramethylrhodamine methylester
Funding
K.J.K, L.P. and J.H. are funded by the Wellcome Trust. F.B. is supported by a fellowship from the Spanish Ministerio de Educacion through the FECYT and partially supported by an ALS Association Initiated award (ID 2109) and Motor Neuron Disease Association Small Grants (Hardy/Oct12/6603). L.P. is also supported by the Max Planck Society. This work was also supported by the Welcome Trust MRC strategic neurodegenerative disease initiative award (Ref. number WT089698). K.J.K. was the recipient of a post-doctoral Wellcome Trust fellowship (Ref. number 090541/Z/09/Z). Part of this work was undertaken at the University College of London Hospitals who received a proportion of their funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. K.B.B. was also funded by a Parkinson’s UK Grant (Ref. number G-1009). L.L. is also the recipient of a Parkinson's UK PhD studentship (Ref. number H-1105).
Supplementary material
Supplementary material is available at Brain online.
References
- Ballard JW, Melvin RG, Simpson SJ. Starvation resistance is positively correlated with body lipid proportion in five wild caught Drosophila simulans populations. J Insect Physiol 2008; 54: 1371–6. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993; 118: 401–15. [DOI] [PubMed] [Google Scholar]
- Beck G, Sugiura Y, Shinzawa K, Kato S, Setou M, Tsujimoto Y, et al. Neuroaxonal dystrophy in calcium-independent phospholipase A2β deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J Neurosci 2011; 31: 11411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilho RF, Meinicke AR, Almeida AM, Hermes-Lima M, Vercesi AE. Oxidative damage of mitochondria induced by Fe (II) citrate is potentiated by Ca2+ and includes lipid peroxidation and alterations in membrane proteins. Arch Biochem Biophys 1994; 308: 158–63. [DOI] [PubMed] [Google Scholar]
- Chance B, Williams GR. A simple and rapid assay of oxidative phosphorylation. Nature 1955; 175: 1120–1. [DOI] [PubMed] [Google Scholar]
- Cheon Y, Kim HW, Igarashi M, Modi HR, Chang L, Ma K, et al. Disturbed brain phospholipid and docosahexaenoic acid metabolism in calcium-independent phospholipase A(2)-VIA (iPLA(2)β)-knockout mice. Biochim Biophys Acta 2012; 1821: 1278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006; 441: 1162–6. [DOI] [PubMed] [Google Scholar]
- Domijan AM, Kovac S, Abramov AY. Lipid peroxidation is essential for phospholipase C activity and the inositol-trisphosphate-related Ca2+ signal. J Cell Sci 2014; 127: 21–6. [DOI] [PubMed] [Google Scholar]
- Engel LA, Jing Z, O'Brien DE, Sun M, Kotzbauer PT. Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One 2010; 5: e12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd ME, Broekemeier KM, Crouser ED, Kumar J, Graff G, Pfeiffer DR. Mitochondrial iPLA2 activity modulates the release of cytochrome c from mitochondria and influences the permeability transition. J Biol Chem 2006; 281: 6931–9. [DOI] [PubMed] [Google Scholar]
- Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, et al. Neurodegeneration associated with genetic defects in phospholipase A(2). Neurology 2008; 71: 1402–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Owen AD, Toffa SE, Cooper JM, Dexter DT, Jenner P, et al. Mitochondrial function, GSH and iron in neurodegeneration and Lewy body diseases. J Neurol Sci 1998; 158: 24–9. [DOI] [PubMed] [Google Scholar]
- Gubern A, Barceló-Torns M, Casas J, Barneda D, Masgrau R, Picatoste F, et al. Lipid droplet biogenesis induced by stress involves triacylglycerol synthesis that depends on group VIA phospholipase A2. J Biol Chem 2009; 284: 5697–708. [DOI] [PubMed] [Google Scholar]
- Gui YX, Xu ZP, Wen-Lv, Liu HM, Zhao JJ, Hu XY. Four novel rare mutations of PLA2G6 in Chinese population with Parkinson's disease. Parkinsonism Related Disord 2013; 19: 21–6. [DOI] [PubMed] [Google Scholar]
- Hill S, Lamberson CR, Xu L, To R, Tsui HS, Shmanai VV, et al. Small amounts of isotope-reinforced polyunsaturated fatty acids suppress lipid autoxidation. Free Radic Biol Med 2012; 53: 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S, Hirano K, Shmanai VV, Marbois BN, Vidovic D, Bekish AV, et al. Isotope-reinforced polyunsaturated fatty acids protect yeast cells from oxidative stress. Free Radic Biol Med 2011; 50: 130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heather LC, Carr CA, Stuckey DJ, Pope S, Morten KJ, Carter EE, et al. Critical role of complex III in the early metabolic changes following myocardial infarction. Cardiovasc Res 2010; 85: 127–36. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 2005; 1: 223–32. [DOI] [PubMed] [Google Scholar]
- Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet 2006; 79: 942–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster JF, Slee RG. Lipid peroxidation of rat liver microsomes. Biochim Biophys Acta 1980; 620: 489–99. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006; 443: 787–95. [DOI] [PubMed] [Google Scholar]
- Ma Z, Turk J. The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog Nucleic Acid Res Mol Biol 2001; 67: 1–33. [DOI] [PubMed] [Google Scholar]
- Malhotra A, Edelman-Novemsky I, Xu Y, Plesken H, Ma J, Schlame M, et al. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc Natl Acad Sci USA 2009; 106: 2337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 2002; 192: 1–15. [DOI] [PubMed] [Google Scholar]
- Miwa S, St-Pierre J, Partridge L, Brand MD. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic Biol Med 2003; 35: 938–48. [DOI] [PubMed] [Google Scholar]
- Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 2006; 38: 752–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 2009; 65: 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging 2012; 33: 814–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006; 441: 1157–61. [DOI] [PubMed] [Google Scholar]
- Pope S, Land JM, Heales SJ. Oxidative stress and mitochondrial dysfunction in neurodegneration: cardiolpin a critical target? Biochim Biophys Acta 2008; 1777: 794–9. [DOI] [PubMed] [Google Scholar]
- Rival T, Soustelle L, Strambi C, Besson MT, Iche M, Birman S. Decreasing glutamate buffering capacity triggers oxidative stress and neuropil degeneration in the Drosophila brain. Curr Biol 2004; 14: 599–605. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Cuenca S, Cochemé HM, Logan A, Abakumova I, Prime TA, Rose C, et al. Consequences of long-term oral administration of the mitochondria-targeted antioxidant Mito Q to wild-type mice. Free Radic Biol Med 2010; 48: 161–72. [DOI] [PubMed] [Google Scholar]
- Schaeffer EL, Gattaz WF. Cholinergic and glutamatergic alterations beginning at the early stages of Alzheimer disease: participation of the phospholipase A2 enzyme. Psychopharmacology (Berl) 2008; 198: 1–27. [DOI] [PubMed] [Google Scholar]
- Shchepinov MS, Chou VP, Pollock E, Langston JW, Cantor CR, Molinari RJ, et al. Isotopic reinforcement of essential polyunsaturated fatty acids diminishes nigrostriatal degeneration in a mouse model of Parkinson's disease. Toxicol Lett 2011; 207: 97–103. [DOI] [PubMed] [Google Scholar]
- Schlame M. Cardiolipin remodeling and the function of tafazzin. Biochim Biophys Acta 2013; 1831: 582–8. [DOI] [PubMed] [Google Scholar]
- Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J Biol Chem 2006; 281: 22275–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strokin M, Seburn KL, Cox GA, Martens KA, Reiser G. Severe disturbance in the Ca2+ signaling in astrocytes from mouse models of human infantile neuroaxonal dystrophy with mutated Pla2g6. Hum Mol Genet 2012; 21: 2807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaarmann A, Gandhi S, Abramov AY. Dopamine induces Ca2+ signaling in astrocytes through reactive oxygen species generated by monoamine oxidase. J Biol Chem 2010; 285: 25018–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MJ, Gross RW. Expression, purification, and kinetic characterization of a recombinant 80-kDa intracellular calcium-independent phospholipase A2. J Biol Chem 1996; 271: 30879–85. [DOI] [PubMed] [Google Scholar]
- Xu C, Warsh JJ, Wang KS, Mao CX, Kennedy JL. Association of the iPLA2β gene with bipolar disorder and assessment of its interaction with TRPM2 gene polymorphisms. Psychiatr Genet 2013; 23: 86–9. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Wang J, Zhao C, Bi W, Yue Z, Ma ZA. Genetic ablation of PLA2G6 in mice leads to cerebellar atrophy characterized by Purkinje cell loss and glial cell activation. PLoS One 2011; 6: e26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.