

Abstract
Fibroblast growth factor receptor 1 (FGFR1) is one of the causative genes for Kallmann syndrome (KS), which is characterized by isolated hypogonadotropic hypogonadism with anosmia/hyposmia. The third immunoglobulin-like domain (D3) of FGFR1 has the isoforms FGFR1-IIIb and FGFR1-IIIc, which are generated by alternative splicing of exons 8A and 8B, respectively. To date, the only mutations to have been identified in D3 of FGFR1 are in exon 8B. We performed mutation analysis of FGFR1 in a 23-year-old female patient with KS and found a missense mutation (c.1072C>T) in exon 8A of FGFR1. The c.1072C>T mutation was not detected in her family members or in 220 normal Japanese and 100 Caucasian female controls. No mutation in other KS genes, KS 1, prokineticin-2, prokineticin receptor-2 and FGF-8 was detected in the affected patient or in her family members. Therefore, this is the first case of KS carrying a de novo missense mutation in FGFR1 exon 8A, suggesting that isoform FGFR1-IIIb, as well as isoform FGFR1-IIIc, plays a crucial role in the pathogenesis of KS.
Keywords: Kallmann syndrome, FGFR1b mutation, fibroblast growth factor receptor 1 isoform expression
Introduction
Kallmann syndrome (KS), which is characterized by isolated hypogonadotropic hypogonadism (IHH) and anosmia/hyposmia, is a clinically and genetically heterogeneous disorder. To date, five causative genes for KS have been reported: KS 1 (KAL1, GenBank accession M97 252), prokineticin-2 (PROK2, GenBank accession NM 021935), prokineticin receptor-2 (PROKR2, GenBank accession NM 144773), fibroblast growth factor-8 (FGF-8, GenBank accession NM 033163) and fibroblast growth factor receptor 1 (FGFR1, GenBank accession NM 023110.2).
Although sporadic cases of KS are more frequent, families with KS have been reported with X-linked recessive or autosomal dominant or recessive modes of inheritance. Mutations in KAL1 have been found in familial cases with X-linked recessive inheritance (Franco et al., 1991; Legouis et al., 1991). Mutations in PROK2 were detected in the heterozygous state, whereas PROKR2 mutations were found in the heterozygous, homozygous or compound heterozygous state (Dodé et al., 2006). PROKR2/PROK2 mutations with true pathogenic potential were found only in the homozygous state (Abreu et al., 2008), and any dominant-negative effect of PROKR2 mutations was ruled out (Monnier et al., 2009). Mutations in FGFR1 or FGF8 underlie an autosomal dominant form with incomplete penetrance. Therefore, KS families harbouring heterozygous FGFR1 or FGF8 mutations display variable olfactory phenotypes (Dodé et al., 2003; Falardeau et al., 2008), and a few cases with heterozygous FGFR1 mutations show a normosmic IHH (Pitteloud et al., 2006a). The FGFR1 gene, which is located on chromosome 8p12, comprises 18 exons (Ruta et al., 1989), and various mutations, including missense and protein truncation mutations, have been reported (Trarbach et al., 2007). The third immunoglobulin-like domain (D3) of FGFR1 has the isoforms FGFR1-IIIb and FGFR1-IIIc, which are generated by alternative splicing of exons 8A and 8B, respectively (Johnson et al., 1991). To date, mutations in D3 of FGFR1 have only been identified in exon 8B, which encodes immunoglobulin domain IIIc, suggesting that isoform FGFR1-IIIc plays a crucial role in the pathogenesis of KS (Pitteloud et al., 2006b; Trarbach et al., 2006; Dodé et al., 2007).
Here, we report for the first time a KS case carrying a de novo missense mutation in the alternatively spliced exon 8A of FGFR1-IIIb.
Materials and Methods
Patient and family
Patient (Subject II-2) was a 23-year-old Japanese woman. When she was 18 years old, she was treated at Nagasaki University Hospital because of primary amenorrhea with anosmia. Her height was 159.2 cm and her weight was 72.0 kg. Her serum levels of luteinizing hormone (LH), follicle-stimulating hormone (FSH) and estradiol (E2) were less than 0.5 and 1.5m IU/ml and 10 pg/ml, respectively. Her LH frequent sampling study (sampling performed every 15 min) showed a low-amplitude pattern of LH pulsation (Fig. 1). Her brain magnetic resonance imaging (MRI) examination was negative for tumors and showed no anatomical abnormalities of the hypothalamic–pituitary region and olfactory bulbs. A scratch-and-sniff test (UPSIT, Sensonics, Haddon Hts, NJ, USA) (Doty et al., 1985), which determines ability to smell, indicated anosmia. She was diagnosed as having KS and received hormone replacement therapy for 5 years. Her mother (Subject I-1) was normosmic and had normal puberty and regular menstrual cycles. Her father (Subject I-2), elder brother (Subject II-1) and younger brother (Subject II-3) were also normosmic and had normal puberty (Fig. 2).
Figure 1.

LH pulsation pattern in a case of KS, assayed using an LH frequent sampling study. LH frequent sampling was performed every 15 min. *Low-amplitude pattern of LH pulse.
Figure 2.
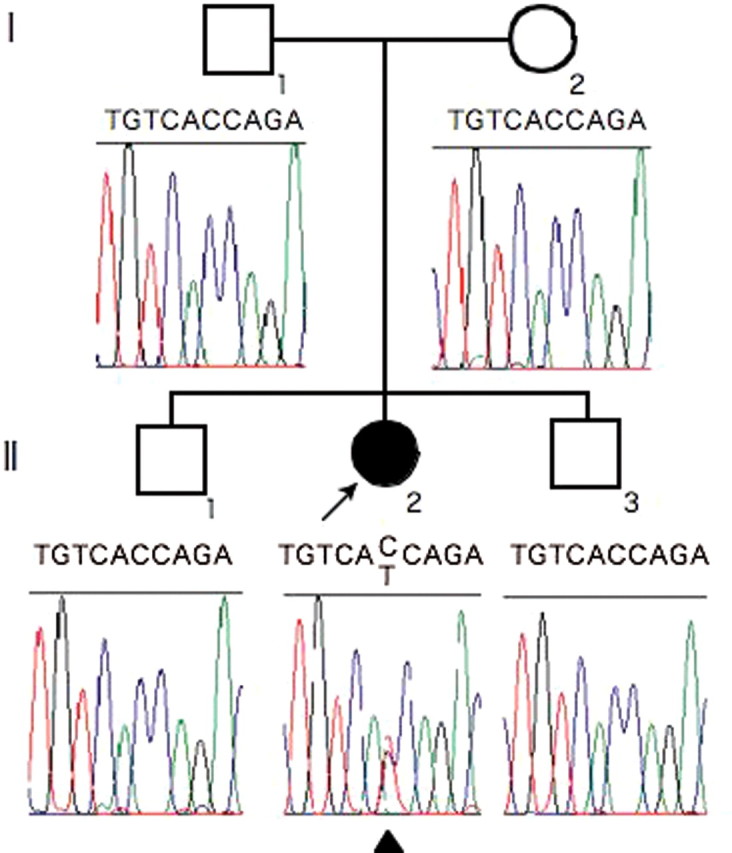
Pedigree of patient's family and the results of fibroblast growth factor receptor 1 (FGFR1) sequence analysis. II-2 is a 23-year-old woman with KS. The patient had a mutation in FGFR1 (C>T) but the other family members did not. The mutation was de novo because parentage was assured. Arrowhead under the electropherogram indicates the mutation site.
Molecular analysis
DNA extraction
Whole blood samples were obtained from the KS patient and from her mother, father, elder and younger brothers. All samples were collected after obtaining written informed consent and the study protocol was approved by the Institutional Review Board of Nagasaki University. Genomic DNA from lymphocytes was extracted using a QIAamp DNA blood mini kit (Qiagen, Düsseldorf, Germany), according to the manufacturer's instructions.
Sequence analysis
FGFR1 consists of 18 coding exons. Intragenic mutations were investigated by PCR amplification and sequence analysis using 14 pairs of primers, as previously described (Dodé et al., 2003; Sato et al., 2004). Genomic DNA was PCR amplified using conditions of 95°C for 12 min followed by 95°C for 30 s, 59°C for 30 s and 72°C for 60 s for 35 cycles and a final cycle of 72°C for 10 min. PCR products were analyzed by agarose gel electrophoresis, purified with ExoSAP-IT and subjected to sequencing reactions. Sequencing reactions were performed using the BigDye terminator v.3.1 kit and analyzed with an ABI PRISM 3100 Genetic Analyzer™ (Applied Biosystems). The KS patient carrying a mutation in FGFR1 and her family members were also screened for mutations in the other genes known to be involved in KS [KAL1, PROK2, PROKR2 and FGF8]. Whether the mutation leads to a change in the protein structure and function was predicted bioinformatically using the ExPASy proteomics server (http://au.expasy.org/) and PolyPhen (http://genetics.bwh.harvard.edu/pph/).
Confirmation of the alternatively spliced exon
Isolation of a full-length murine Fgfr1-IIIb showed that Fgfr1-IIIb was a transmembrane receptor (Beer et al., 2000). Although the mRNA encoding exon IIIb has been found in human (Johnson et al., 1991), the presence of sequences encoding the intracellular domain has not yet been demonstrated. Therefore, to determine the splice site of exon 8A and to detect FGFR1-IIIb mRNA encoding the intracellular domain, we performed RT–PCR using specific primers to amplify the splice isoform containing exon 8A. Kal23 is designed to span exons 7 and 8A for specific annealing to the FGFR1-IIIb isoform, which is spliced from exon 7 to exon 8A (Fig. 3A). Kal5 is designed within exon 8B for specific annealing to the FGFR1-IIIc isoform, which is spliced from exon 7 to exon 8B. Kal2 and Kal6 are designed within exons 7 and 9, respectively, for annealing to the D3 isoforms of FGFR1. Primer sequences were as follows: kal2: 5′-GACAGAAGGTCGGTTATGTC-3′, Kal23: 5′-CAGATCTTGAAGCATTCGGG-3′, Kal5: 5′-GGTGGTATTAACTCCAGCAG-3′ and Kal6: 5′-GTACAGGGGCGAGGTCATCA-3′. The BD multiple tissue complementary DNA (cDNA, MTC) panels Human I and Human II (BD Biosciences Clontech, Mountain View, CA, USA) were used to detect the expression of each isoform of FGFR1. PCR amplification was performed on cDNAs as follows: 94°C, 30 s; 62°C, 30 s; 72°C, 1 min; 40 cycles. PCR products were analyzed by agarose gel electrophoresis and sequenced using then ABI PRISM 3100 Genetic Analyzer™.
Figure 3.

Genomic organization of FGFR1 around exon 8A. (A) The numbers with bp indicate exon length (over the line) and intron length (under the line). The mutation is located near the end of exon 8A. Horizontal arrows indicate the locations of primers used to perform RT–PCR amplification of the isoform containing exons 7 and 8A. Primer Kal23 is designed to span exons 7 and 8A for specific annealing to the FGFR1-IIIb isoform, which is spliced from exon 7 to exon 8A (Fig. 3A). Kal5 is designed within exon 8B for specific annealing to the FGFR1-IIIc isoform, which is spliced from exon 7 to exon 8B. Kal2 and Kal6 are designed within exons 7 and 9, respectively, for annealing to the D3 isoforms of FGFR1. Vertical arrow indicates mutation site. (B) Composition of mRNA isoforms and of putative protein structures. FGFR1-IIIb: membrane-bound form of FGFR1 with immunoglobulin-like domain IIIb encoded by exon 8A, FGFR1-IIIc: membrane-bound form of FGFR1 with immunoglobulin-like domain IIIc encoded by exon 8B, FGFR1-secr: a secreted form of FGFR1.
Results
Sequence analysis of the entire coding region of FGFR1, including exon–intron boundary regions, showed that the KS patient had a mutation (c.1072C>T) in exon 8A of FGFR1-IIIb, while the other family members did not (Fig. 2). However, the full-length FGFR1 mRNA that includes exon 8A is not deposited in the full-length cDNA database (GenBank accession no. NM 023110.2). RT–PCR analysis indicated that most transcripts containing exon 8A were spliced to exon 8B in all adult tissues except bone marrow (data not shown). We wished to demonstrate the existence of an alternative transcript, exon 8A which was spliced to exon 9 encoding the transmembrane helix; therefore, RT–PCR products from human fetal brain were cloned and sequenced. In 1 of 27 clones exon 8A was spliced to exon 9 (designated here ‘FGFR1-IIIb’, GenBank accession FJ809917, see Fig. 3B), while in the other clones exon 8A was spliced to exon 8B (designated here ‘FGFR1-secr’, GenBank accession FJ809916, see Fig. 3B). The exact acceptor and donor sites of exon 8A in ‘FGFR1-IIIb’ mRNA, which produces a membrane-bound FGFR1-containing D3, were determined by sequence analysis of splice isoforms, ‘FGFR1-IIIb’ and ‘FGFR1-secr’, (Fig. 3). As most full-length FGFR1 cDNAs in the database were transcripts containing exon 7–exon 8B–exon 9 (designated here ‘FGFR1-IIIc’, GenBank accession NM 023110.2, see Fig. 2B) without exon 8A, ‘FGFR1-IIIc’ is likely to be the most abundantly expressed human isoform. The EST, CA488712.1, was the only isoform in the EST database corresponding to ‘FGFR1-IIIb’. Although both ‘FGFR1-IIIb’ and ‘FGFR1-IIIc’ encode membrane-bound FGFR1, ‘FGFR1-secr’ encodes a secreted form of FGFR1 because of a sequence frameshift and a termination codon in exon 9.
The exons 8A and 8B of the human FGFR1 isoforms shared the amino acid sequence at 354–357; WLTV. However, exon 8A ends with six extra amino acids at 358–363, TRPVAK, whereas exon 8B ends with only two, LE. These sequences are identical in the mouse Fgfr1 isoforms (Beer et al., 2000). The mutation in exon 8A of FGFR1-IIIb (GenBank accession no. FJ809917 bankit1193625) is c.1072C>T at the cDNA level and p.T358I at the amino acid level. Bioinformatic analysis shows the mutated amino acid residue to be conserved between human and mouse and to be located in D3 of FGFR1-IIIb, which is a critical region for FGF ligand binding. However, the mutation was not predicted to produce a change in the human protein structure. The c.1072C>T mutation was not detected in 220 normal Japanese women or in 100 normal Caucasian women. The patient had no mutation in any of the other four KS genes.
Discussion
Mouse Fgfr1-IIIb has a low level of expression in a wide variety of adult tissues, but a high level of expression in skin and brain, indicating the existence of specific splicing factors in skin and brain that recognize the relatively weak Fgfr1-IIIb splice site (Beer et al., 2000). Consistent with the expression pattern of mouse Fgfr1-IIIb, we could isolate human FGFR1-IIIb from a fetal brain cDNA library but not from adult tissues. Most full-length FGFR1 cDNAs in the database represent FGFR1-IIIc. FGFR1-IIIc is expressed at high levels, but FGFR1-IIIb is expressed at very low levels (Johnson et al., 1991). We can amplify only a tiny amount of FGFR1-IIIb that has exon 8A spliced to exon 9 by RT–PCR using the primers Kal23 and Kal6. Most of the sequenced RT–PCR products corresponded to FGFR1-secr, suggesting that the expression level of the three isoforms is ‘FGFR1-IIIc’ ≫ ‘FGFR1-IIIsecr’ ≫ ‘FGFR1-IIIb’ (Fig. 3B).
Several studies suggested that mutations in exon 8B of isoform FGFR1-IIIc are implicated in the pathogenesis of KS (Pitteloud et al., 2006b; Trarbach et al., 2006; Dodé et al., 2007). Mice homozygous for alleles with a stop codon in exon IIIc displayed phenotypes resembling those of embryos homozygous for null alleles, while mice carrying an in-frame stop codon in exon IIIb were viable and fertile (Partanen et al., 1998). Therefore, Fgfr1-IIIc is the dominant isoform that carries out the majority of the biological functions of the Fgfr1 gene, whereas Fgfr1-IIIb plays a minor and to some extent redundant role (Partanen et al., 1998). A receptor-binding analysis revealed no difference in the binding specificity between the endogenous Fgfr1-IIIb and an artificially created Fgfr1-IIIb, which had two different amino acids in the 3′-end of the unique IIIb exon (Beer et al., 2000), suggesting that the carboxyl terminus of D3 may not overtly influence binding specificity. However, being expressed at low levels does not imply that ‘FGFR1-IIIb’ has an unimportant role. The mutation we found, p.T358I, was located in exon 8A of FGFR1-IIIb. Therefore, p.T358I may affect ligand binding and cause the KS phenotype, although how this mutation affects the loss of function of FGFR1-IIIb is unknown. The spatio-temporal expression of any gene involved in development is key; therefore, the expression and functional involvement of FGFR1-IIIb may be important in the early embryonic brain, in particular during GnRH neuronal development. KS missense mutations in FGFR1 are distributed in the first, second and third immunoglobulin-like domains (D1–D3), in the tyrosine kinase domain and also in the intracellular domain (Dodé et al., 2003; Sato et al., 2004; Albuisson et al., 2005; Pitteloud et al., 2006a; Trarbach et al., 2006; Dodé et al., 2007); therefore, the membrane-bound form of FGFR1 is probably important for the KS phenotype. The membrane-bound form of FGFR1-IIIb could, therefore, be the critical isoform for the KS phenotype.
We present here, for the first time, a case of KS carrying a missense mutation in exon 8A of FGFR1, suggesting that the minor isoform ‘FGFR1-IIIb’ as well as the major isoform ‘FGFR1-IIIc’ has a crucial role in the pathogenesis of KS. Therefore, immunoglobulin-like domain IIIb may have an essential role in GnRH neuronal migration, which is initiated from the nasal placode and runs towards the forebrain following the olfactory sensory neuron axonal connection with the developing olfactory bulb. Further experiments are needed to show that the mutation in exon 8A causes KS, such as expression of the mutated isoform in transfected cells to analyze receptor stability and signaling efficiency. Although FGFR1 containing immunoglobulin-like domain IIIb has not been analyzed intensively, our mutation report should encourage researchers to analyze immunoglobulin-like domain IIIb function and the spatio-temporal expression of exon 8A in fetal brain development.
Funding
K.M. was supported, in part, by Seeds (No.15-B09) from the Japan Science and Technology Agency (JST), by grants from the Naito Foundation, by Grant-in-Aid for Young Scientists (B) (no. 21791567) from the Ministry of Education, Sports, Culture, Science and Technology of Japan, by a Grant for Child Health and Development (20C-1) from the Ministry of Health, Labor and Welfare, and by Grant-in-Aid for Scientific Research from Nagasaki University, Japan.
Acknowledgements
We thank Ms Yasuko Noguchi and Miho Ooga for their technical assistance and we thank Drs Nelly Pitteloud and William Crowley for their valuable contribution.
References
- Abreu AP, Trarbach EB, de Castro M, Frade Costa EM, Versiani B, Matias Baptista MT, Garmes HM, Mendonca BB, Latronico AC. Loss-of-function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive Kallmann syndrome. J Clin Endocrinol Metab. 2008;93:4113–4118. doi: 10.1210/jc.2008-0958. [DOI] [PubMed] [Google Scholar]
- Albuisson J, Pêcheux C, Carel JC, Lacombe D, Leheup B, Lapuzina P, Bouchard P, Legius E, Matthijs G, Wasniewska M, et al. Kallmann syndrome: 14 novel mutations in KAL1 and FGFR1 (KAL2) Hum Mutat. 2005;25:98–99. doi: 10.1002/humu.9298. [DOI] [PubMed] [Google Scholar]
- Beer HD, Vindevoghel L, Gait MJ, Revest JM, Duan DR, Mason I, Dickson C, Werner S. Fibroblast growth factor (FGF) receptor 1-IIIb is a naturally occurring functional receptor for FGFs that is preferentially expressed in the skin and the brain. J Biol Chem. 2000;275:16091–16097. doi: 10.1074/jbc.275.21.16091. [DOI] [PubMed] [Google Scholar]
- Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Dû N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, et al. Loss-of-function mutation in FGFR1 cause autosomal dominant Kallmann syndrome. Nature Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, Lespinasse J, Lienhardt-Roussie A, Mathieu M, Moerman A, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 prokineticin receptor-2. PLoS Genet. 2006;2:e175. doi: 10.1371/journal.pgen.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodé C, Fouveaut C, Mortier G, Janssens S, Bertherat J, Mahoudeau J, Kottler ML, Chabrolle C, Gancel A, François I, et al. Novel FGFR1 sequence variants in Kallmann syndrome, and genetic evidence that the FGFR1c isoform is required in olfactory bulb and palate morphogenesis. Hum Mutat. 2007;28:97–98. doi: 10.1002/humu.9470. [DOI] [PubMed] [Google Scholar]
- Doty RL, Applebaum S, Zusho H, Settle RG. Sex differences in odor identification ability: a cross-cultural analysis. Neuropsychologia. 1985;23:667–672. doi: 10.1016/0028-3932(85)90067-3. [DOI] [PubMed] [Google Scholar]
- Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118:2822–2831. doi: 10.1172/JCI34538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B, Tonlorenzi R, Carrozzo R, Maestrini E, Pieretti M, Taillon-Miller P, et al. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529–536. doi: 10.1038/353529a0. [DOI] [PubMed] [Google Scholar]
- Johnson DE, Lu J, Chen H, Werner A, Williams LT. The human fibroblast growth factor receptor genes: a common structure arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol Cell Biol. 1991;11:4627–4634. doi: 10.1128/mcb.11.9.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legouis R, Hardelin JP, Levilliers J, Claverie JM, Compain S, Wunderle V, Millasseau P, Le Paslier D, Cohen D, Caterina D, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. 1991;67:423–435. doi: 10.1016/0092-8674(91)90193-3. [DOI] [PubMed] [Google Scholar]
- Monnier C, Dodé C, Fabre L, Teixeira L, Labesse G, Pin JP, Hardelin JP, Rondard P. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum Mol Genet. 2009;18:75–81. doi: 10.1093/hmg/ddn318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen J, Schwartz L, Rossant J. Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev. 1998;12:2332–2344. doi: 10.1101/gad.12.15.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteloud N, Acierno JS, Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA. 2006;103:6281–6286. doi: 10.1073/pnas.0600962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteloud N, Meysing A, Quinton R, Acierno JS, Jr, Dwyer AA, Plummer L, Fliers E, Boepple P, Hayes F, Seminara S, et al. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. 2006;254–255:60–69. doi: 10.1016/j.mce.2006.04.021. [DOI] [PubMed] [Google Scholar]
- Ruta M, Burgess W, Givol D, Epstein J, Neiger N, Kaplow J, Crumley G, Dionne C, Jaye M, Schlessinger J. Receptor for acidic fibroblast growth factor is related to the tyrosine kinase encoded by the fms-like gene (FLG) Proc Natl Acad Sci USA. 1989;86:8722–8726. doi: 10.1073/pnas.86.22.8722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Katsumata N, Kagami M, Hasegawa T, Hori N, Kawakita S, Minowada S, Shimotsuka A, Shishiba Y, Yokozawa M, et al. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endorinol Matab. 2004;89:1079–1088. doi: 10.1210/jc.2003-030476. [DOI] [PubMed] [Google Scholar]
- Trarbach EB, Costa EM, Versiani B, de Castro M, Baptista MT, Garmes HM, de Mendonca BB, Latronico AC. Novel fibroblast growth factor receptor 1 mutations in patients with congenital hypogonadotropic hypogonadism with and without anosmia. J Clin Endocrinol Metab. 2006;91:4006–4012. doi: 10.1210/jc.2005-2793. [DOI] [PubMed] [Google Scholar]
- Trarbach EB, Silveira LG, Latronico AC. Genetic insights into human isolated gonadotropin deficiency. Pituitary. 2007;10:381–391. doi: 10.1007/s11102-007-0061-7. [DOI] [PubMed] [Google Scholar]