

PIKfyve modulates endosomes in TLR9-mediated induction of type I IFNs
Keywords: dendritic cells, innate immunity, intracellular trafficking, signaling, TLR9, type I IFNs
Abstract
Toll-like receptors (TLRs) traffic to distinct membranes for signaling. TLR7 and TLR9 recognize viral nucleic acids in the endosomes and induce robust anti-viral program. Signaling from these TLRs bifurcate at the level of distinct endosomal compartments, namely VAMP3+ and LAMP+ endosomes, to mediate the induction of cytokine and type I interferon (IFN) genes, respectively. The formation of the TLR9 endosome competent for IFNs induction requires AP-3. Phosphoinositides (PIs) mark distinct subcellular membranes and control membrane trafficking. However, their role in TLR trafficking and signaling in different dendritic cell (DC) subsets remains unclear. Here, we examined the role of phosphatidylinositol 3P 5-kinase, PIKfyve, in TLR9 trafficking and signaling. We demonstrate that inhibition of PIKfyve activity preferentially blocks TLR9 signaling for type I IFN induction in FLT3L-bone marrow-derived DCs. By confocal microscopy using RAW264.7 cells, we show that trafficking of both TLR9 and CpG to the LAMP1+ compartment was blocked by PIKfyve inhibitor treatment, whereas their trafficking to the VAMP3+ endosome remained intact. Further, AP-3 recruitment to TLR9 endosomes was impaired by PIKfyve inhibition. These data indicate that PIKfyve provides critical PIs necessary for the formation of endosome from which TLR9 signals to induce type I IFNs.
Introduction
Virus infection is sensed by multiple pattern recognition receptors. Cell types such as B cells, macrophages, plasmacytoid dendritic cells (pDCs) and conventional DCs (cDCs) use Toll-like receptor (TLR) 9 and TLR7 to recognize viral DNA and RNA within the endosomes (1). TLR9 and TLR7 signals bifurcate downstream of MyD88 to induce pro-inflammatory cytokines and type I interferon (IFN) genes. The first pathway leads to the transcription of pro-inflammatory cytokine genes through activation of NF-κB, IRF5 and MAPK (2). The second pathway leads to the activation of type I IFN genes through phosphorylation of IRF7 (3). While both pathways depend on MyD88 (4), IRAK4 (5), UNC93B (6) and TRAF6, the latter pathway also requires additional molecules including IRAK1 (7), TRAF3 (8, 9), IKKα (10), osteopontin (11), DOCK2 (12), PDC TREM (13), viperin (14) and AP-3 (15). These two distinct signaling complexes have been termed the cytoplasmic transductional-transcriptional processors for IFN and pro-inflammatory cytokines (16). While the molecules involved in the two signaling pathways are being identified, the cell biological mechanisms by which these two types of signals are mediated through TLR7/9 remain less clear. Recent evidence indicates that TLR signaling heavily depends on its trafficking inside the cell (17, 18). Specifically, certain adaptor proteins (APs) mediate trafficking of TLR7 and TLR9 to appropriate endosomal compartments. The endosomes from which TLR7/9 signals through NF-κB or IRF7 are the ‘NF-κB endosome’ and the ‘IRF7 endosome’ (15, 19–21). TLR9 trafficking to the NF-κB endosome requires UNC93B-mediated recruitment of AP-2 and involves trafficking via the plasma membrane. In contrast, TLR7 trafficking from the Golgi to the NF-κB endosome occurs directly and relies on direct AP-4 binding to the cytoplasmic tail of TLR7 (22). AP-3 is required for trafficking of TLR9 from the NF-κB endosome to the IRF7 endosome in pDCs (15). However, another study found that AP-3 is required for signaling of TLR9 in general in pDCs but not in cDCs (23).
Phosphoinositides (PIs) are a key component of cell membranes. They can undergo rapid phosphorylation/dephosphorylation cycles at positions 3′, 4′ and 5′ of the inositol head group to generate a variety of phosphatidylinositol lipid species. Every organelle is equipped with a distinct array of PI kinases and PI phosphatases, which produces a unique set of PI species providing distinct properties to the membrane (24). Previous studies indicate the critical role of PI species in controlling TLR localization and signaling (25, 26). However, the role of PI in control of TLR7 and TLR9 trafficking and signaling remains less clear. In human pDCs, an inhibitor of phosphatidylinositol 3 kinase (PI3K) p100δ but not p100γ was shown to diminish IFN production without affecting tumor necrosis factor (TNF)-α, suggesting the involvement of PI(3,4,5)P3 in TLR9 signaling for activation of IFN genes (27).
PI(3,5)P2 is a member of the PIs that comprises 0.1% of the total cellular PI pool (28). It is found specifically in early endosomes (EEs), late endosomes (LEs) and the multivesicular body (MVB) compartment (29). In mammalian cells, the enzyme that phosphorylates PI(3)P on the fifth position of the inositol head group is PI(3)P 5-kinase known as PIPkIII or PIKfyve (29). Past studies have revealed the role of this enzyme in post early endosomal traffic to the MVB/LEs and endosome-to-trans-Golgi network membrane transport in mammalian cells by utilizing PIKfyve knockdown, dominant negative mutants and inhibitor treatment (30). While PIKfyve knockout mice are embryonically lethal (31), PIKfyve hypomorphic mice with ~10% of PIKfyve protein survive for a few days after birth (32). PIKfyve hypomorphic mice revealed that this enzyme is responsible for generating the PI(3,5)P2 pool, which serves as a major precursor of PI(5)P (32). Recent studies demonstrate that PI(5)P serves as a second messenger to trigger IRF3-dependent type I IFN induction downstream of TLRs and RIG-I-like receptors (RLRs) (33). Another study demonstrated that inhibition of PIKfyve results in decreased colocalization of CpG in LEs in the RAW264.7 macrophage cell line (34). Inhibition of PIKfyve in human pDCs by apilimod induces expression of the transcription repressor ATF3, which suppresses type I IFN transcription without affecting ther expression of other cytokines (35). Therefore, PIKfyve appears to play an important role in TLR9 trafficking and signaling in cell types including pDCs, cDCs and macrophages.
Our previous study showed that TLR9 requires AP-3 for its LE localization in bone marrow (BM) macrophages (BMMs), and FLT3L-cultured BM cells from AP-3 deficient mice failed to secrete type I IFNs while maintaining the ability to secrete IL-12p40 and TNF-α (15). Moreover, assays using an engineered fusion protein in which the PH domain of centaurin-β2 with PI(3,5)P2 binding specificity (36) is fused to TRAF3 show that the production of IFNs downstream of TLR9 in AP-3−/− BMMs is restored by targeting TRAF3 to the NF-κB endosome. These data indicated that PI(3,5)P2 may mark the AP-3 independent endosome from which TLR9 can signal to induce cytokines. Here, to further understand the role of PI in TLR signaling and trafficking, we used inhibitors of PI kinases and examined the ability of FLT3L-cultured BM pDC and cDCs to respond to TLR agonists. Further, we investigated intracellular trafficking of TLR9 and its ligand in RAW264.7 cells. Our data reveal a critical role for PIKfyve activity in TLR9 trafficking to the IRF7 endosome, and subsequent IFN production downstream of TLR9.
Methods
Mice and reagents
Age- and sex-matched C57BL/6 (WT) mice from the National Cancer Institute (Frederick, MD, USA) and AP-3β−/− mice, and age- and sex-matched C57BL/6 mice purchased from Jackson Laboratories (Bar Harbor, ME, USA) were used for isolating BM cells. All procedures used in this study complied with federal guidelines and institutional policies of the Yale Animal Care and Use Committee. CpG 2216 (CpG type-A) was purchased from TriLink BioTechnologies (San Diego, CA, USA), and Poly I:C and R848 were purchased from Invivogen (San Diego, CA, USA), respectively. DOTAP-CpG-A stimulation was performed as described previously (37) using DOTAP obtained from Roche. YM201636 was purchased from Symansis and SantaCruz. PI(3)K inhibitors were purchased from Calbiochem. Water-soluble C8-PtdIns were obtained from Echelon Biosciences.
Cell lines and primary cell culture
RAW264.7 cells were purchased from ATCC and cultured in RPMI supplemented with 10% FCS, penicillin/streptomycin, 10mM HEPES (Invitrogen) and 2-mercaptoethanol (Sigma). TLR9-GFP and AP3µ3A-Flag co-expressing cells were prepared by transduction with MSCV2.2 retrovirus and GFP+ cells were sorted by a Sony SY3200 at the Yale Cell Sorting Core Facility. FLT3L-cultured BM-derived pDCs and cDCs (38) and GM-CSF-cultured BM-derived DCs (39) were prepared as previously described. To obtain FACS-sorted DCs, FLT3L-cultured cells or primary BM cells were stained with anti-B220-FITC (BioLegend) and anti-CD11c-PECy7 (BioLegend) for 15min at 4°C. pDCs (CD11c+ B220+) and cDCs (CD11c+ B220−) were sorted by a FACS Aria (BD Biosciences, Mountain View, CA, USA).
Plasmids
TLR9-HA plasmid is a kind gift from Dr Ruslan Medzhitov. Cherry-IRF7 was subcloned by means of PCR. Retrovirus vectors containing TLR9-HA and Cherry-IRF7 were generated by subcloning the genes into pMSCV2.2. The retrovirus vector containing AP3µ3A-Flag was generated as previously described (15).
Microscopy
Transduced RAW264.7 were settled onto Alcian Blue-coated coverslips overnight. Cells were pretreated with YM201636 (SantaCruz) for 1h, then stimulated with 3 µM CpG-A or 2 µM Cy-5-labeled CpG-A (synthesized by Sigma) together with DOTAP (Roche, Indianapolis, IN, USA) in the presence of YM201636 for 6h, washed with PBS and fixed in 2% paraformaldehyde/PBS. Cells were permeabilized and blocked with PBS-saponin (PS) solution (1% BSA, 0.05% saponin in PBS) for 15min. Primary antibodies were diluted in PS as follows: mouse anti-HA mouse 1:2000 (Sigma), rabbit anti-LAMP1 1:1000 (Abcam) and rabbit anti-VAMP3 1:2000 (Synaptic Systems). In 20 µl volumes, cells were stained with the antibody solution for 60min. After washing with PS, secondary antibodies Alexa Fluor 488 anti-mouse (Invitrogen) and Cy3 anti-rabbit (Jackson ImmunoResearch) were diluted at 1:400 in PS and cells were stained with the solution for 30min. Subsequently, cells were washed with PS and then PBS. Coverslips were mounted on slides with ProLong Gold (Invitrogen) and imaged by a Leica TCS SP8 confocal microscope using a ×64 objective lens. A minimum of 15–20 cells were examined per slide.
The calculation method for (%) colocalization is as follows:
(%) Colocalization = (The number of puncta positive for all three markers)/(The number of total TLR9+ puncta) (Figures 3 and 4) or (The number of puncta positive for all three markers)/(The number of total CpG+ puncta) (Figure 5).
Mean (%) colocalization = Average of (%) colocalization.
ELISA
The protein level of IFN-α present in the cell culture supernatant and serum was measured by ELISA as previously described (4). The protein level of IL-12p40 and TNF-α present in cell culture supernatants and serum was measured according to the manufacturer’s instructions with antibodies purchased from BioLegend and eBioscience, respectively.
Quantitative PCR
Fetal liver and Flt3L-cultured pDCs were stimulated with CpG-A (1 and 3 µM) for indicated time periods. Total RNA was prepared using RNeasy kit (Qiagen), and reverse transcription was carried out with an oligo (dT) primer using an iScript cDNA synthesis Kit (Bio-Rad). Expression of the indicated genes following stimulation was measured by quantitative reverse transcription–PCR using a CFX Connect™ Real-Time PCR Detection System (Bio-Rad).
Statistical analysis
Normally distributed continuous variable comparisons were performed with Student’s t-test using Prism software (GraphPad). Data are presented as mean ± SEM. P <0.05 was considered statistically significant.
Results
Inhibitor of PIKfyve preferentially blocks TLR9 signaling for type I IFN synthesis in FLT3L-BM cells
PI(3,5)P2 synthesis initiates in the EE and PI(3,5)P2 decorates the membrane of the LE and the MVB compartment. On the basis of our previous findings that TLR9 trafficking to the LAMP2+ compartment is crucial for its signaling capacity to induce IFN genes (15), we hypothesized that PI(3,5)P2 may be important in the TLR9 signaling pathway. To test the role of PI(3,5)P2 in TLR9 trafficking and function, we used a specific inhibitor of PIKfyve, YM201636 (40). FLT3L-BM cells were stimulated with 1 µM of CpG-A in the presence of YM201636 at various concentrations. At concentrations above 1 µM, YM201636 completely inhibited IFN-α production while it had minimal effects on IL-12p40 production (Figure 1A). The differential effect of YM201636 on IFN-α and IL-12p40 was also observed in FLT3L-BM cells stimulated with CpG-A at 3 µM, where IFN-α production was significantly inhibited but there was little effect on IL-12p40 or TNF-α production (Figure 1B). Moreover, R848, a TLR7 ligand known to induce cytokines but not type I IFNs in FLT3L-DCs (41), induced secretion of IL-12p40 and TNF-α from stimulated FLT3L-BM cells that was not inhibited by YM201636 treatment suggesting that cytokine signaling downstream of TLR7 does not require PIKfyve activity (Figure 1C). The inhibition in IFN and cytokine protein secretion correlated with reduced levels of mRNA of the corresponding genes (Figure 1D). Moreover, the requirement of PI(3,5)P2 for signaling for IFN production is specific to TLR9, because when we inhibited PIKfyve in GM-CSF-cultured BM cells stimulated with Poly (I:C) (TLR3 ligand), or LPS (TLR4 ligand), the production of both type I IFN and cytokines remained intact (Supplementary Figure 1, available at International Immunology Online).
Fig. 1.
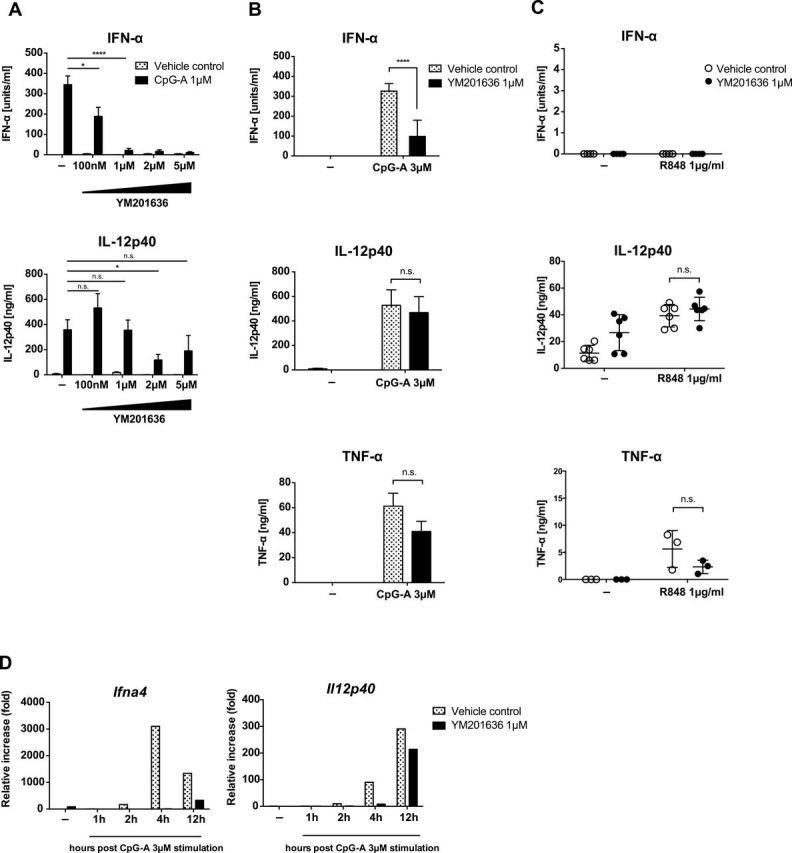
PIKfyve inhibition results in preferential reduction in IFN-α. Flt3L-cultured BM cells from B6 mice were stimulated with 1 µM CpG-A in the presence or absence of PIKfyve inhibitor, YM201636. After 24h, supernatant was collected and the amount of secreted IFN-α, IL-12p40 and TNF-α was determined by ELISA (A). Flt3L-BM cells from B6 mice were stimulated with 3 µM of CpG-A (B) or 1 µg ml−1 of R848 (C) for 24h. Levels of secreted IFN-α, IL-12p40 and TNF-α was determined by ELISA. IFN-α and IL-12p40 mRNA expression levels were assessed by means of quantitative reverse transcription–PCR at the indicated time points following CpG-A stimulation (D). n = 3–6, mean ± SEM, *P < 0.05, ****P < 0.0001.
These data indicated that signals downstream of TLR9 for IFN synthesis are more sensitive to the PIKfyve requirement compared to signals for IL-12p40 or TNF-α.
Exogenously added synthetic phosphoinositol lipids are not sufficient to enhance TLR9 signaling
A study reported that PIKfyve-generated PI5P enhances IFN production downstream of TLR and RLR stimulation through activation of IRF3 (33). In contrast to most cell types, pDCs use IRF7, and not IRF3, to activate transcription of IFN genes downstream of TLR7 and TLR9 (5, 16, 42). To determine whether supplementation of PI5P or PI(3,5)P2 could restore IFN synthesis in PIKfyve-inhibited cells, YM201636-treated FLT3L-BM cells were stimulated with CpG in the presence of synthetic water-soluble C8-PIs at various concentrations (33). In the absence of CpG-A, the C8-phosphoinosides were insufficient to induce expression of IFN-α or IL-12p40 in FLT3L-BM cells (below detection, data not shown). Further, exogenously added PIs were unable to restore IFN synthesis in YM201636 inhibitor-treated FLT3L-BM cells stimulated with CpG-A (Supplementary Figure 2A and B, available at International Immunology Online). In contrast, lipid addition to GM-CSF-cultured BM cells resulted in some induction of RANTES and IP-10 mRNA expression (Supplementary Figure 2C and D, available at International Immunology Online) as reported previously (33), although other PI species also showed similar effects. Thus, these data indicated that exogenously added PIs were insufficient to stimulate FLT3L-BM cells and restore IFN-α expression under YM201636 treatment.
Inhibitor of PIKfyve blocks TLR9 and CpG transport to the lysosome-related organelle
To understand whether the defect in type I IFN production in FLT3L-BM cells treated with PIKfyve inhibitor is due to mis-trafficking of TLR9 and/or its ligand CpG to the IRF7 endosomes, we compared intracellular localization of TLR9 and CpG in VAMP3+ [an NF-κB endosome marker (15)] and LAMP+ [an IRF7 endosome marker (15)] compartments in RAW264.7 cells transduced with TLR9-HA (15). To mimic trafficking of CpG and TLR9 in pDCs in RAW264.7 cells, CpG-A was delivered in complex with DOTAP (37). First, we tested whether RAW264.7 cells exhibit a similar phenotype as we had observed in the FLT3L-BM cells. As shown in Supplementary Figure 3, available at International Immunology Online, gene expression of IFN-β was selectively suppressed while there was no effect on IL-12p40 or TNF-α genes in PIKfyve suppressed RAW264.7 cells. Subsequent confocal microscopy experiments showed that in RAW264.7 cells stimulated with DOTAP CpG-A, TLR9 was mostly found in VAMP3+ or LAMP1+ endosomes with little staining found in the endoplasmic reticulum (Figures 2 and 3; Supplementary Figure 4, available at International Immunology Online). Moreover, PIKfyve inhibition resulted in vacuolization in cells (Figures 2 and 3) as reported previously (40). In the absence of PIKfyve inhibitor, CpG-A entered VAMP3+ endosomes (Figure 2C) and colocalized with TLR9 in LAMP1+ endosomes (Figure 3C), the latter associated with signaling for IFNs (15). In contrast, in the presence of PIKfyve inhibitor, both TLR9 and CpG were excluded from LAMP1+ endosomes (Figure 3D). CpG and TLR9 were still found to be associated with VAMP3+ endosomes (Figure 2D). Therefore, these data are consistent with a recent study that demonstrated exclusion of CpG-B from LAMP1+ endosomes in RAW264.7 cells (34) and support the notion that blockade of PIKfyve activity interferes with both TLR9 and CpG trafficking to the IRF7 endosome.
Fig. 2.
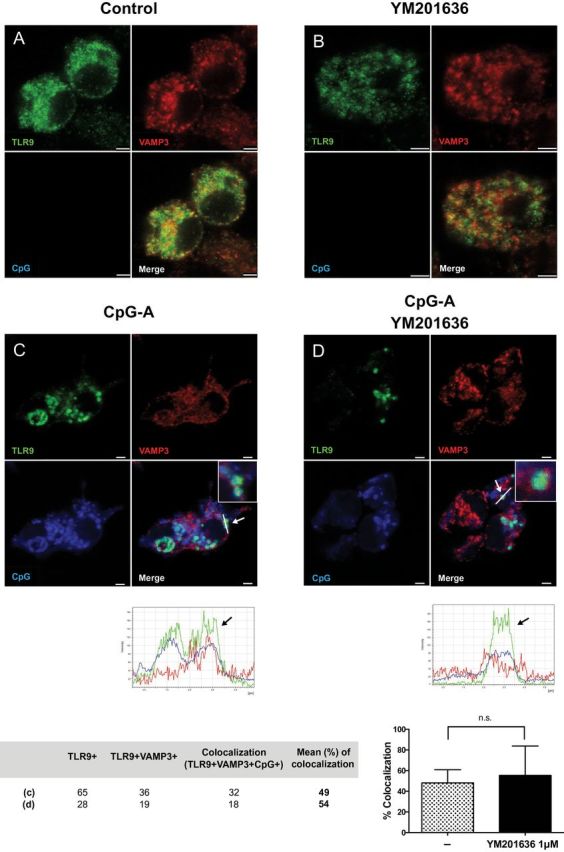
PIKfyve inhibitor does not affect TLR9 localization to the VAMP3+ compartment upon CpG stimulation. RAW 264.7 cells stably transduced with TLR9-HA were stimulated with DOTAP-CpG-A-Cy5 for 6 h in the absence (C) or presence (D) of YM201636 (1 μM) given 1 h prior to CpG-A stimulation. Subsequently, cells were stained and analyzed by confocal microscopy to detect TLR9-HA (green), VAMP3 (red) and CpG (blue). Scale bars, 2.5 μm. No-stimulation controls in the absence or presence of inhibitor are shown as (A), (B), respectively.
Fig. 3.
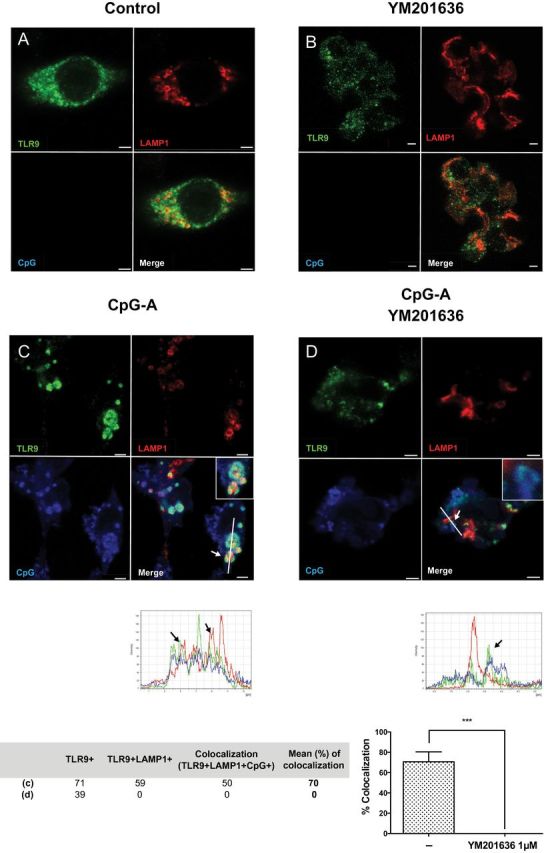
PIKfyve inhibitor blocks TLR9 localization to the LAMP1+ compartment upon CpG stimulation. RAW 264.7 cells stably transduced with TLR9-HA were stimulated with DOTAP-CpG-A-Cy5 for 6 h in the absence (C) or presence (D) of YM201636 (1 μM) given 1 h prior to CpG-A stimulation. Subsequently, cells were stained and analyzed by confocal microscopy to detect TLR9-HA (green), LAMP1 (red) and CpG (blue). Colocalization of TLR9 and LAMP1 or TLR9, LAMP1 and CpG was defined by structures in which TLR9/CpG signals were surrounded by LAMP1 staining. Scale bars, 2.5 μm. No-stimulation controls in the absence or presence of inhibitor are shown as (A), (B), respectively. ***P < 0.001.
Inhibitor of PIKfyve blocks AP-3 recruitment to TLR9
Because AP-3 has been previously shown to bind to PI(3,5)P2 (43), we reasoned that reduced recruitment of AP-3 to TLR9 endosome resulted in altered lysosome-related organelle (LRO) formation by AP-3, which then led to the selective impairment of IFN signaling. To probe this hypothesis, we next tested whether the recruitment of AP-3 is impaired after PIKfyve inhibition. RAW264.7 cells expressing TLR9 were stimulated with CpG-A DOTAP for 1h in the presence or absence of PIKfyve inhibitor and AP-3 recruitment to the TLR9+ vesicles was detected by confocal microscopy. As expected, AP-3 was recruited to the TLR9+ vesicles after CpG stimulation (Figure 4C). However, AP-3 recruitment to the TLR9+ vesicles was lost in the presence of PIKfyve inhibitor (Figure 4D), indicating that PIKfyve activity is required for AP-3 recruitment to the TLR9+ compartment after CpG stimulation.
Fig. 4.
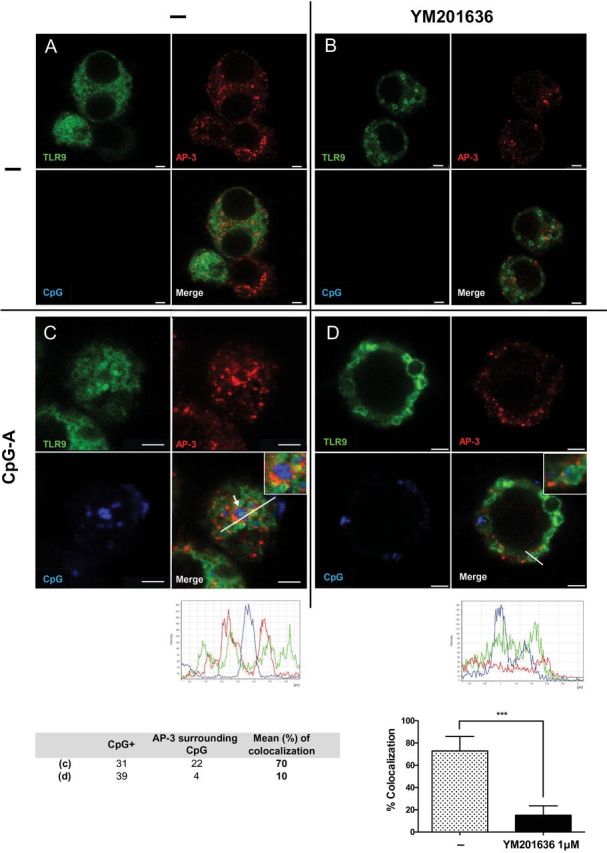
AP-3 fails to be recruited to TLR9+ endosomes after CpG stimulation in the presence of PIKfyve inhibitor. RAW 264.7 cells stably transduced with TLR9-GFP and AP-3μ3A-Flag were stimulated with DOTAP-CpG-A-Cy5 for 1 h in the absence (C) or presence (D) of YM201636 (1 μM) given 1 h prior to CpG-A stimulation. Subsequently, cells were stained and analyzed by confocal microscopy to detect TLR9-GFP (green), AP-3 (red) and CpG (blue). Scale bars, 2.5 μm. No-stimulation controls in the absence or presence of inhibitor are shown as (A), (B), respectively. ***P < 0.001.
Inhibitor of PIKfyve blocks TLR9 signaling in pDCs while AP-3 deficiency selectively targets type I IFN production downstream of TLR9 in cDCs
Because FLT3L-cultured BM cells contain CD11c+ B220+ pDCs as well as CD11c+ B220- cDCs, data obtained from the mixed culture could reflect differential contributions of pDCs and cDCs. In order to determine the importance of PIKfyve in pDC and cDCs, mixed cells from FLT3L-BM culture were FACS sorted and pure populations were obtained for subsequent assays. As in mixed FLT3L-BM culture (Figure 1), PIKfyve inhibitor blocked secretion of IFN-α in both sorted pDCs and cDCs (Figure 5). In contrast, production of IL-12p40 was blocked in pDCs but not in cDCs (Figure 5). Data from GM-CSF-cultured cDC further confirmed that only the production of IFN-β, but not IL-12p40, was inhibited in YM201636-treated cells (Supplementary Figure 1B, available at International Immunology Online). Altogether, these data indicated that PIKfyve is selectively required for TLR9 signaling for type I IFN in cDCs whereas PIKfyve inhibition blocks TLR9 signaling in general in pDCs.
Fig. 5.

PIKfyve inhibition results in differential effects in the production of IFN-α and IL-12p40 in sorted pDCs and cDCs differently. Flt3L-cultured BM cells from B6 mice were sorted into CD11cintB220+ pDCs (A) and CD11c+B220− cDCs (B), then stimulated with indicated concentrations of CpG-A in the presence or absence of PIKfyve inhibitor, YM201636. After 24h, supernatant was collected and the levels of secreted IFN-α and IL-12p40 were determined by ELISA. n = 3, mean ± SEM, *P < 0.05, ****P < 0.0001.
Previous studies showed conflicting results for whether AP-3 is required for TLR9 signaling in FLT3L-BM cells for cytokine responses (15, 23). While both studies found the requirement for AP-3 in type I IFN induction upon TLR9 stimulation, one study found no AP-3 requirement for cytokine induction (15) while another did (23). In order to examine whether AP-3 is also differentially involved in TLR9 signaling, WT and AP-3−/− FLT3L-BM cells were FACS sorted into pDC and cDCs, and production of type I IFN and IL-12 after CpG stimulation was determined. Consistent with the previous study (15), we observed a requirement for AP-3 for TLR9 signaling for type I IFN induction but not IL-12 in purified pDCs (Figure 6A). Similarly, AP-3 deficiency resulted in reduced type I IFN induction but not cytokine production by CpG-stimulated cDCs (Figure 6B). Together, these results indicate that in both pDCs and cDCs, both PIKfyve and AP-3 are required for TLR9 signaling for type I IFN induction. In cDCs, PIKfyve and AP-3 are both dispensable for TLR9 signaling for IL-12 production, while in pDCs, PIKfyve, but not AP-3, is required for signaling for IL-12 production downstream of TLR9.
Fig. 6.

AP-3 is required for the production of IFN-α but not IL-12p40 in sorted pDCs and cDCs. Flt3L-cultured BM cells from WT or AP-3−/− B6 mice were sorted into CD11cintB220+ pDCs (A) and CD11c+B220- cDCs (B), then stimulated with indicated concentration of CpG-A. After 24h, supernatant was collected and the levels of secreted IFN-α and IL-12p40 were determined by ELISA. n = 3, mean ± SEM, *P < 0.05, **P < 0.01, ****P < 0.0001.
Discussion
Recent studies have revealed the mechanisms by which intracellular trafficking of TLR9 and TLR7 is coordinated in cDCs, pDCs and macrophages to generate signaling platforms capable of activating distinct sets of genes. Our current study highlights another pathway by which TLR9 trafficking and signaling is regulated, namely, through the action of PIKfyve. Through the use of PIKfyve inhibitor, YM201636, we showed that in FLT3L-cultured BM cells, TLR9 signals leading to IFN induction are preferentially blocked compared to those leading to NF-κB-dependent cytokines. Using RAW264.7 cells expressing TLR9, we showed the inability of TLR9 and CpG-A to traffic to the LAMP1+ IRF7 endosomes in the presence of PIKfyve inhibitor. In contrast, PIKfyve inhibition did not significantly impair TLR9 trafficking to the VAMP3+ NF-κB endosome upon CpG-A stimulation. Using sorted pDCs and cDCs from FLT3L-BM culture, we further demonstrated the requirement for PIKfyve and AP-3 in TLR9 signaling for type I IFN induction. Thus, our data indicate that PIKfyve activity is essential in TLR9 trafficking to the IRF7 endosome and signals leading to type I IFN induction in pDCs and cDCs.
Our data are consistent with a recent report demonstrating that PIKfyve inhibition by YM 201636 resulted in decreased colocalization of CpG-B DNA with LAMP1 and TLR9, with no obvious defect in TLR9 colocalization to EEs in RAW264.7 cells (34). However, it differs from another study (35) that showed that CpG-induced TLR9 trafficking to CD63+ endosomes is intact in the presence of another PIKfyve inhibitor, apilimod (44). Notably, apilimod is a compound that specifically blocks TLR-mediated IL-12/IL-23 production that has entered clinical trials (45). Apilimod selectively inhibits type I IFN and IL-12 production but not TNF-α or IL-8 production downstream of TLR9 (35, 44). The differences in whether a PIKfyve inhibitor interferes with TLR9 trafficking to LAMP+ endosomes, and which cytokines are blocked downstream, may reflect the different mode of action of apilimod and YM201636 on PIKfyve inhibition.
AP-3 is required for the generation of the IRF7 endosome, known as IFN LRO, which is critical for triggering IFN signaling, and mediates the divergence of NF-κB endosomes and IRF7 endosomes (15). Our data showed that PIKfyve inhibition blocks the recruitment of AP-3 to the TLR9 endosome after CpG stimulation as early as 1h post-stimulation suggesting that PIKfyve inhibitor blocks the formation of IFN LRO, resulting in the selective impairment in IRF7, but not NF-κB, signaling. Our findings support the idea that LRO generation is abolished upon PIKfyve inhibition, leading to the inhibition of IFN signaling and reduced type I IFN production in the cells after CpG stimulation. Further, our data provide a definitive demonstration that AP-3 is required in both pDCs and cDCs for signaling for IFN-α but not IL-12p40 downstream of TLR9.
PIKfyve is known to generate PI(3,5)P2 from PI(3)P, and the product PI(3,5)P2 serves as a major precursor to PI(5)P (32). A recent study showed that YM201636 inhibits PI(5)P synthesis twice more effectively compared with PI(3,5)P2 synthesis at 160nM (46). However, at 100nM, we did not observe any impairment of IL-12p40 secretion in response to CpG in FLT3L-BM cells. In addition, we were unable to rescue the defective TLR9 signaling for type I IFNs in YM201636-treated FLT3L-BM cells with exogenous C8-PI(5)P or C8-PI(3,5)P2. While these data indicate that endogenous products generated by PIKfyve are required, future studies are needed to delineate the specific contributions of PI(3,5)P2 and PI(5)P in TLR9 signaling.
On the basis of the data presented in this study, we hypothesize that TLR9 trafficking is coordinated by PIKfyve in the following manner (Figure 7). PIKfyve is recruited to PI(3)P on the VAMP3+ NF-κB endosome through its FYVE domain, where it catalyzes the synthesis of PI(3,5)P2. On the basis of our results, we hypothesize that the formation of PI(3,5)P2 promotes AP-3 recruitment. This hypothesis is supported by the fact that a comprehensive analysis of the PI(3,5)P2 PI interactome revealed AP-3δ and AP-3σ subunits as binding partners of PI(3,5)P2 (43). AP recruitment generally involves concurrent binding to multiple low-affinity sites comprising adenosine diphosphate ribosylation factor (Arf)–GTP, cargo sorting signals (such as the YXXØ motif) and PIs (47). For AP-3, Arf-1–GTP and Arf-3–GTP have been shown to regulate its recruitment to membranes, as well as YXXØ, [DE]XXXL[LI] sorting signals on transmembrane protein cargo (47). Our data support the hypothesis that PI(3,5)P2 could serve as the PI species that acts as the third signal to recruit AP-3 to TLR9-containing vesicles.
Fig. 7.

Model for PIKfyve-dependent TLR9 trafficking and signaling in DCs. Activated TLR9 recruits the adaptor MyD88 and transduces signals through the recruitment of additional signaling molecules such as IRAK4 and TRAF6. This signal results in the activation of a distinct transcription factor such as NF-κB, which results in activation of pro-inflammatory genes. Furthermore, PIKfyve is recruited to the NF-κB endosome via its FYVE domain (red), which converts PI(3)P to PI(3,5)P2. AP-3 is recruited to the endosome via binding to PI(3,5)P2 and mediates the formation of the IRF7 endosome, which subsequently promotes TLR9 signaling for type I IFNs.
Taking our results together, we speculate that the AP-3 complex is recruited to PI(3,5)P2, and assembly of this coat drives membrane curvature. Subsequently, the membrane containing PI(3,5)P2, AP-3 and TLR9 buds out of the NF-κB endosome to generate the LAMP1+ IRF7 endosome, from which TLR9 triggers signaling for type I IFN gene transcription (Figure 7). This paradigm is applicable to FLT3L-cultured BM cDCs, GM-CSF-cultured BM cDCs and the RAW264.7 cell line. However, in pDCs, PIKfyve inhibitor impaired TLR9 signals in general. This likely reflects the inability of the sorted pDCs to maintain viability and to tolerate YM201636 treatment (48, 49). Future studies are needed to determine the detailed molecular pathways involved in the selective ability of TLR9 to recruit and activate IRF7 from this particular endosome.
In humans, mutations in the PIKfyve gene have been found in fleck corneal dystrophy (50–52). Those mutations result from non-sense or frameshift heterozygous mutations that lead to the production of truncated or dysfunctional protein, characterized by abnormal swollen keratinocytes with enlarged vesicles in the cornea. Moreover, the importance of PI(3)P to PI(3,5)P2 conversion in human disease is underscored by the strong association between mutations in the gene encoding the crucial component of PIKfyve complex (PAS complex), Sac3 and Charcot-Marie-Tooth type 4J neuropathy (CMT4J) (53). Whether or not the alteration in PIKfyve function results in abnormality in other organs is unknown. On the basis of our analysis, we speculate the possibility that PIKfyve mutations result in impaired IFN responses following viral infections, which leads to the increased susceptibility to infectious diseases.
Supplementary data
Supplementary data are available at International Immunology Online.
Funding
National Institutes of Health (NIH) (R01 AI064705; R01 AI081884 to A.I.). A.I. is an investigator of the Howard Hughes Medical Institute.
Supplementary Material
Acknowledgements
We thank Melissa Linehan and Huiping Dong for technical support. K.H. is a recipient of a Nakajima Foundation Predoctoral Fellowship, and M.S. is a recipient of fellowships from the Japan Society for the Promotion of Science, the Uehara Memorial Foundation, the Kanae Foundation for the Promotion of Medical Science and the Takeda Science Foundation. The authors have no conflicting financial interests.
References
- 1. Kawai T., Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131. [DOI] [PubMed] [Google Scholar]
- 2. Takaoka A., Yanai H., Kondo S., et al. 2005. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434:243. [DOI] [PubMed] [Google Scholar]
- 3. Honda K., Yanai H., Negishi H., et al. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772. [DOI] [PubMed] [Google Scholar]
- 4. Lund J., Sato A., Akira S., Medzhitov R., Iwasaki A. 2003. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198:513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Honda K., Yanai H., Mizutani T., et al. 2004. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl Acad. Sci. USA 101:15416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tabeta K., Hoebe K., Janssen E. M., et al. 2006. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 7:156. [DOI] [PubMed] [Google Scholar]
- 7. Uematsu S., Sato S., Yamamoto M., et al. 2005. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J. Exp. Med. 201:915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Häcker H., Redecke V., Blagoev B., et al. 2006. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439:204. [DOI] [PubMed] [Google Scholar]
- 9. Oganesyan G., Saha S. K., Guo B., et al. 2006. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439:208. [DOI] [PubMed] [Google Scholar]
- 10. Hoshino K., Sugiyama T., Matsumoto M., et al. 2006. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 440:949. [DOI] [PubMed] [Google Scholar]
- 11. Shinohara M. L., Lu L., Bu J., et al. 2006. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat. Immunol. 7:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gotoh K., Tanaka Y., Nishikimi A., et al. 2010. Selective control of type I IFN induction by the Rac activator DOCK2 during TLR-mediated plasmacytoid dendritic cell activation. J. Exp. Med. 207:721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Watarai H., Sekine E., Inoue S., Nakagawa R., Kaisho T., Taniguchi M. 2008. PDC-TREM, a plasmacytoid dendritic cell-specific receptor, is responsible for augmented production of type I interferon. Proc. Natl Acad. Sci. USA 105:2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saitoh T., Satoh T., Yamamoto N., et al. 2011. Antiviral protein Viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity 34:352. [DOI] [PubMed] [Google Scholar]
- 15. Sasai M., Linehan M. M., Iwasaki A. 2010. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science 329:1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Honda K., Taniguchi T. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6:644. [DOI] [PubMed] [Google Scholar]
- 17. Barton G. M., Kagan J. C. 2009. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat. Rev. Immunol. 9:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kagan J. C., Iwasaki A. 2012. Phagosome as the organelle linking innate and adaptive immunity. Traffic 13:1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang X., Chen Z. J. 2011. Viperin links lipid bodies to immune defense. Immunity 34:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Engel A., Barton G. M. 2010. Compartment-specific control of signaling from a DNA-sensing immune receptor. Sci. Signal. 3:pe45. [DOI] [PubMed] [Google Scholar]
- 21. Henault J., Martinez J., Riggs J. M., et al. 2012. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 37:986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee B. L., Moon J. E., Shu J. H., et al. 2013. UNC93B1 mediates differential trafficking of endosomal TLRs. Elife 2:e00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blasius A. L., Arnold C. N., Georgel P., et al. 2010. Slc15a4, AP-3, and Hermansky-Pudlak syndrome proteins are required for Toll-like receptor signaling in plasmacytoid dendritic cells. Proc. Natl Acad. Sci. USA 107:19973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Matteis M. A., Godi A. 2004. PI-loting membrane traffic. Nat. Cell Biol. 6:487. [DOI] [PubMed] [Google Scholar]
- 25. Kagan J. C., Medzhitov R. 2006. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 125:943. [DOI] [PubMed] [Google Scholar]
- 26. Kagan J. C., Su T., Horng T., Chow A., Akira S., Medzhitov R. 2008. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 9:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guiducci C., Ghirelli C., Marloie-Provost M. A., et al. 2008. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J. Exp. Med. 205:315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ho C. Y., Alghamdi T. A., Botelho R. J. 2012. Phosphatidylinositol-3,5-bisphosphate: no longer the poor PIP2. Traffic 13:1. [DOI] [PubMed] [Google Scholar]
- 29. Shisheva A. 2008. PIKfyve: Partners, significance, debates and paradoxes. Cell Biol. Int. 32:591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ikonomov O. C., Sbrissa D., Shisheva A. 2001. Mammalian cell morphology and endocytic membrane homeostasis require enzymatically active phosphoinositide 5-kinase PIKfyve. J. Biol. Chem. 276:26141. [DOI] [PubMed] [Google Scholar]
- 31. Ikonomov O. C., Sbrissa D., Delvecchio K., et al. 2011. The phosphoinositide kinase PIKfyve is vital in early embryonic development: preimplantation lethality of PIKfyve-/- embryos but normality of PIKfyve+/- mice. J. Biol. Chem. 286:13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zolov S. N., Bridges D., Zhang Y., et al. 2012. In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc. Natl Acad. Sci. USA 109:17472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kawasaki T., Takemura N., Standley D. M., Akira S., Kawai T. 2013. The second messenger phosphatidylinositol-5-phosphate facilitates antiviral innate immune signaling. Cell Host Microbe 14:148. [DOI] [PubMed] [Google Scholar]
- 34. Hazeki K., Uehara M., Nigorikawa K., Hazeki O. 2013. PIKfyve regulates the endosomal localization of CpG oligodeoxynucleotides to elicit TLR9-dependent cellular responses. PLoS One 8:e73894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cai X., Xu Y., Kim Y. M., Loureiro J., Huang Q. 2014. PIKfyve, a class III lipid kinase, is required for TLR-induced type I IFN production via modulation of ATF3. J. Immunol. 192:3383. [DOI] [PubMed] [Google Scholar]
- 36. Dowler S., Currie R. A., Campbell D. G., et al. 2000. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem. J. 351(Pt 1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Honda K., Ohba Y., Yanai H., et al. 2005. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434:1035. [DOI] [PubMed] [Google Scholar]
- 38. D’Amico A., Wu L. 2003. The early progenitors of mouse dendritic cells and plasmacytoid predendritic cells are within the bone marrow hemopoietic precursors expressing Flt3. J. Exp. Med. 198:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sato A., Linehan M. M., Iwasaki A. 2006. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc. Natl Acad. Sci. USA 103:17343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jefferies H. B., Cooke F. T., Jat P., et al. 2008. A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 9:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Diebold S. S., Massacrier C., Akira S., Paturel C., Morel Y., Reis e Sousa C. 2006. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol. 36:3256. [DOI] [PubMed] [Google Scholar]
- 42. Kawai T., Sato S., Ishii K. J., et al. 2004. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 5:1061. [DOI] [PubMed] [Google Scholar]
- 43. Catimel B., Schieber C., Condron M., et al. 2008. The PI(3,5)P2 and PI(4,5)P2 interactomes. J. Proteome Res. 7:5295. [DOI] [PubMed] [Google Scholar]
- 44. Cai X., Xu Y., Cheung A. K., et al. 2013. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem. Biol. 20:912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wada Y., Cardinale I., Khatcherian A., et al. 2012. Apilimod inhibits the production of IL-12 and IL-23 and reduces dendritic cell infiltration in psoriasis. PLoS One 7:e35069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sbrissa D., Ikonomov O. C., Filios C., Delvecchio K., Shisheva A. 2012. Functional dissociation between PIKfyve-synthesized PtdIns5P and PtdIns(3,5)P2 by means of the PIKfyve inhibitor YM201636. Am. J. Physiol. Cell Physiol. 303:C436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robinson M. S. 2004. Adaptable adaptors for coated vesicles. Trends Cell Biol. 14:167. [DOI] [PubMed] [Google Scholar]
- 48. Magyarics Z., Csillag A., Pazmandi K., Rajnavolgyi E., Bacsi A. 2008. Identification of plasmacytoid pre-dendritic cells by one-color flow cytometry for phenotype screening. Cytometry A. 73:254. [DOI] [PubMed] [Google Scholar]
- 49. Weijer K., Uittenbogaart C. H., Voordouw A., et al. 2002. Intrathymic and extrathymic development of human plasmacytoid dendritic cell precursors in vivo . Blood 99:2752. [DOI] [PubMed] [Google Scholar]
- 50. Kotoulas A., Kokotas H., Kopsidas K., et al. 2011. A novel PIKFYVE mutation in fleck corneal dystrophy. Mol. Vis. 17:2776. [PMC free article] [PubMed] [Google Scholar]
- 51. Li S., Tiab L., Jiao X., et al. 2005. Mutations in PIP5K3 are associated with François-Neetens mouchetée fleck corneal dystrophy. Am. J. Hum. Genet. 77:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kawasaki S., Yamasaki K., Nakagawa H., et al. 2012. A novel mutation (p.Glu1389AspfsX16) of the phosphoinositide kinase, FYVE finger containing gene found in a Japanese patient with fleck corneal dystrophy. Mol. Vis. 18:2954. [PMC free article] [PubMed] [Google Scholar]
- 53. Chow C. Y., Zhang Y., Dowling J. J., et al. 2007. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.