

Slamf6 exacerbates colitis caused by a Gram- bacterium
Keywords: Citrobacter rodentium, IL-22, innate, mucosal immunology, Slamf6
Abstract
The homophilic cell surface receptors CD150 (Slamf1) and CD352 (Slamf6) are known to modulate adaptive immune responses. Although the Th17 response was enhanced in Slamf6 −/− C57BL/6 mice upon oral infection with Citrobacter rodentium, the pathologic consequences are indistinguishable from an infection of wild-type C57BL/6 mice. Using a reporter-based binding assay, we show that Slamf6 can engage structures on the outer cell membrane of several Gram− bacteria. Therefore, we examined whether Slamf6, like Slamf1, is also involved in innate responses to bacteria and regulates peripheral inflammation by assessing the outcome of C. rodentium infections in Rag −/− mice. Surprisingly, the pathology and immune responses in the lamina propria of C. rodentium-infected Slamf6 −/− Rag −/− mice were markedly reduced as compared with those of Rag −/− mice. Infiltration of inflammatory phagocytes into the lamina propria was consistently lower in Slamf6 −/− Rag −/− mice than in Rag −/− animals. Concomitant with the reduced systemic translocation of the bacteria was an enhanced production of IL-22, suggesting that Slamf6 suppresses a mucosal protective program. Furthermore, administering a mAb (330) that inhibits bacterial interactions with Slamf6 to Rag −/− mice ameliorated the infection compared with a control antibody. We conclude that Slamf6-mediated interactions of colonic innate immune cells with specific Gram− bacteria reduce mucosal protection and enhance inflammation, contributing to lethal colitis that is caused by C. rodentium infections in Rag −/− mice.
Introduction
Most hematopoietic cells express Slamf6, which is one the of nine signaling lymphocyte activating molecule (SLAM) family receptors. Slamf6 expression is highest in activated T cells and B cells, whereas expression in dendritic cells (DCs) and macrophages can be induced by inflammatory signals (1). Although Slamf6 signaling is extensively studied in adaptive immunity (2, 3), less is known about the function of Slamf6 in innate responses.
Several Slam family glycoproteins serve as receptors for certain pathogens (Table 1). For instance, human SLAMF1 is one of the receptors of Measles virus (4). Slamf2 is a receptor for the lectin present on pili of certain enterobacteriaceae (5). Recently, we found that Slamf1 can recognize the outer membrane porins OmpC and OmpF of Eschericia coli (6, 7). Furthermore, most of the Slamf receptors modulate mechanisms that protect against microbial challenges mediated by signals that are induced by Slamf–Slamf homophilic ligation. For example in T cells and B cells, Slamf6 recruits SH-2-containing signal-transducing molecules to its intracellular intracellular tyrosine-based switch motives (ITSM) domains following homophilic ligation and receptor clustering, which is critically involved in germinal center reactions (3, 8). Table 1 summarizes the susceptibility of Slam receptor-deficient mice to various infectious agents that have been used in to study Slamf functions.
Table 1.
Slamf receptors and their adaptor SAP modulate susceptibility to microbes
| Expression | Receptor deficiency results in resistance to: | Receptor deficiency results in susceptibility to: | Slamf ligand | Microbial ligand | |
|---|---|---|---|---|---|
| Slamf1, SLAM, CD150 | T, B, mono, Mø, DC, plat, HSC | Trypanosoma cruzi | Gram− bacteria, Leishmania major | Slamf1 | Measles virus E. coli (OmpC/F+) S. typhimurium |
| Slamf2, CD48 | Pan-lymphocyte | S. aureus | FimH+ enterobacterae | Slamf4, CD2 | E. coli (FimH+) |
| Slamf3, Ly-9, CD229 | T, B, iCD8, NKT, mono, Mø, HSC | Slamf3 | — | ||
| Slamf4, 2B4, CD244 | NK, NKT, T, B, γδ, CD8, DC, eo | LCMV, γHV-68 | Slamf2 | — | |
| Slamf5, CD84 | Pan-lymphocyte, plat, mast, eo | Slamf5 | — | ||
| Slamf6, NTB-A, Ly-108 | NK, NKT, T, B, Mø, pDC, Neu | Leishmania mexicana, C. rodentium | S. typhimurium | Slamf6 | E. coli, C. rodentium |
| Slamf7, CRACC, CS1, CD319 | T, B, mono, DC, NK | Slamf7 | — | ||
| Slamf8, BLAME | iCD8, mono, DC, Mø, Neu, endo, FRC | Slamf8 | — | ||
| Slamf9, SF2001 | T, B, mono, DC | ??? | — | ||
| SAP | NK, NKT, T, (B?) | Mouse: γHV-68, LCMV, Influenza Human: EBV, some other viruses |
Slamf1, 3, 4, 5, 6 Human only: Slamf7 |
N/A |
γHV-68, murine gamma-herpes virus 68; EBV, Epstein–Barr virus; FimH, bacterial lectin; LCMV, lymphocytic choriomengitis virus; SAP, Sh2d1a, Slam-associated protein.
Here, we assess whether Slamf6, like Slamf1, might be a microbial sensor for bacteria, especially E. coli and Citrobacter rodentium, which both reside in the gastrointestinal tract. Furthermore, enterohemorrhagic E. coli and enteropathogenic E. coli are attaching bacteria that harbor a pathogenicity island that renders them capable of colonizing colonic epithelia and causing lesions resulting in a compromised mucosal barrier (9). They represent a major threat to global health, as they are responsible for a large number of cases of diarrhea that can be life threatening for infants and children. The closely related bacterium C. rodentium is a natural Gram− murine pathogen, and oral infection with this bacterium results in an infectious colitis characterized by local Th1 responses, neutrophil and macrophage recruitment and epithelial hyperplasia. T cells and B cells are necessary for sterilizing immunity to C. rodentium and mice that lack CD4+ T cells have systemic dissemination of bacteria (9–11).
To study the role of Slamf6 in innate immune responses, we employ Slamf6 −/− Rag −/− and Rag −/− mice, which solely rely on innate mechanisms to combat C. rodentium infections, because they lack T cells and B cells. The absence of T cells and B cells renders Rag −/− mice unable to mount an effective immune response to C. rodentium and the infection results in severe and ultimately fatal colitis (12).
Roles for a range of innate cells have been implicated in the immunity against C. rodentium. Lamina propria CX3CR1+ macrophages are key in the early detection of the bacterium and specific depletion of these macrophages leads to enhanced pathologic inflammation and more systemic translocation (13, 14). Upon activation, CX3CR1+ macrophages produce IL-1β, IL-12 and IL-23 as well as other inflammatory mediators (14, 15). The role of innate lymphocytes manifests predominantly through their production of IL-22, which contributes to mucosal protection by inducing the production of anti-microbial peptides and accelerating mucosal healing (16, 17).
Here, we report that, surprisingly, Slamf6-deficiency renders Rag −/− mice resistant to C. rodentium-induced colitis, which suggests that Slamf6 negatively affects mucosal protection during colonic C. rodentium infection.
Methods
Mice
Slamf6 −/− C57BL/6J mice were described previously (18). These mice were interbred with Rag1 −/− mice that were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Age- and sex-matched wild-type (WT) and Rag1 −/− C57BL/6J mice were bred in-house and originally purchased from Jackson Laboratory. Mice were co-housed for at least 10 days prior to experimental use. All animals were maintained under specific pathogen-free conditions at the Center for Life Science animal facility of the Beth Israel Deaconess Medical Center (BIDMC) and were used at 8–13 weeks of age. The experiments were performed according to the guidelines of the Institutional Animal Care and Use Committee at BIDMC.
Evaluating bacterial binding by the IL-2 luciferase assay
Chimeric constructs, consisting of extracellular Ig domains of the Slamf6 receptor fused with the signaling competent cytoplasmic domain of CD3ζ, together with an IL-2 promoter-driven Firefly luciferase gene and Renilla luciferase under a mammalian promoter, are transfected into the Jurkat human T-cell line (6). Six hours after transfection, heat-inactivated bacteria are added as stimulation and incubated overnight. Cells are washed and lysed according to the manufacturer’s protocol (Dual Luciferase Reporter Assay, Promega, Madison, WI, USA). Substrates for Firefly luciferase and subsequently Renilla luciferase are added to 10 µl of the cell lysates and luminescence is measured using a standard Glomax luminometer (Promega). Values represent the ratio of Firefly and Renilla luminescence.
In vivo bacterial infection by oral gavage
Citrobacter rodentium (DBS100) (ATCC#51459) was cultured in LB-broth for 4h and washed in PBS prior to inoculation via oral gavage. Bacteria (2×109) were resuspended in 200 µl PBS, which was used for the inoculation of one mouse. The mice were monitored daily for morbidity and their weight was recorded every 3 or 4 days. Mice were sacrificed for analysis when their weight dropped below 80% of their starting weight or at the end of the experiment. Fresh stool pellets were collected in Eppendorf tubes for serial dilution in PBS and plating on MacConkey agar plates for quantification of colony forming units (CFU).
Treatment with an antibody directed against Slamf6
Rag −/− mice were intra-peritoneally injected with 100 µg anti-Slamf6 mAb (330) 1 day prior to infection and subsequently every seventh day after the first injection. Control mice were injected with 100 µg mouse IgG2a in the same regimen.
Adoptive CD45RBhi CD4+ T-cell transfer colitis
Adoptive transfer of CD45RBhi CD4+ T cells into Rag −/− Slamf6 −/− Rag −/− recipients was described previously (19).
Histology
Fresh colon tissue was harvested and fixed in paraformaldehyde (10%). Hematoxylin & eosin (H&E) staining was performed on slices of the proximal, medial and distal part of the colon. Scoring was performed by an independent pathologist (A.K.B.) assessing: (i) mononuclear cell infiltration, (ii) epithelial integrity, (iii) hyperplasia and (iv) edema.
Flow cytometry
Macrophages and DCs were incubated with anti-CD16/32 antibody to block Fc receptors at 4°C for 20min. All samples were stained with relevant antibodies on ice for 30min. Dead cells were excluded using 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Roche, Indianapolis, IN, USA). The cells were acquired on a BD LSRII flow cytometer and the data analysis was performed using the FlowJo analysis package (Tree Star Inc., Ashland, OR, USA).
Cytokine analysis
Colons were harvested, washed in PBS with gentamycin (100 µg ml−1) and 100mg tissue was cultured in 1ml complete DMEM for 24h. The amount of cytokines in the supernatant was analyzed using LEGENDplex reagents (Biolegend, San Diego, CA, USA) or CBA reagents (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions.
Taqman quantitative PCR
Fresh colon tissue was collected, washed in PBS and homogenized by sonication in TRIzol reagent (Life Technologies, Grand Island, NY, USA). The vendor’s protocol was followed for RNA purification. FAM/MGB 16sRNA probes were used as control housekeeping genes. Commercially available FAM/MGB probes (Mm04203745_mH and Hs01011518_m1) were used for Iltifb (IL-22) and Il12b [IL-12(p70)], respectively (Life Technologies). Analysis was performed using the 7500 FAST Real Time PCR.
Statistical analysis
The Prism 5.0 software (GraphPad, San Diego, CA, USA) was used for results analysis. Results are reported as mean ± SEM. Most of the statistical comparisons were performed using the two-tailed Student’s t-test. Values of P < 0.05 are considered to be statistically significant.
Results
Slamf6 engages in cognate interactions with several Gram− bacteria
To investigate a possible role of Slamf6 in innate immune responses during bacterial infections, we first assessed potential interactions between Slamf6 and Gram− E. coli, Salmonella typhimurium (SseB-), C. rodentium and the Gram+ bacterium Staphylococcus aureus. To this end, we used a cell-based reporter assay based on co-transfection of a DNA segment encoding a chimeric Slamf6/CD3ζ together with an IL-2 promoter-driven luciferase gene into Jurkat cells (6). Cognate interactions between the Ig domain of Slamf6 and bacterial surface entities result in increased luciferase activity. Using this assay, specific interactions between Slamf6 and E. coli and S. typhimurium (SseB-), but not between Slamf6 or S. aureus, were detected (Fig. 1A). Citrobacter rodentium displayed binding to the ectodomain of Slamf6 (Fig. 1B), albeit to a far lesser extent. Mutated E. coli that lack OmpC, OmpF or both lost the ability to interact with the ectodomain of Slamf6, suggesting that these bacterial proteins contain both the Slamf6- and Slamf1-interacting structures (Fig. 1C) (6). Further analysis of the interaction between Slamf6 and E. coli revealed that anti-Slamf6 mAb blocked this interaction (Fig. 1D).
Fig. 1.
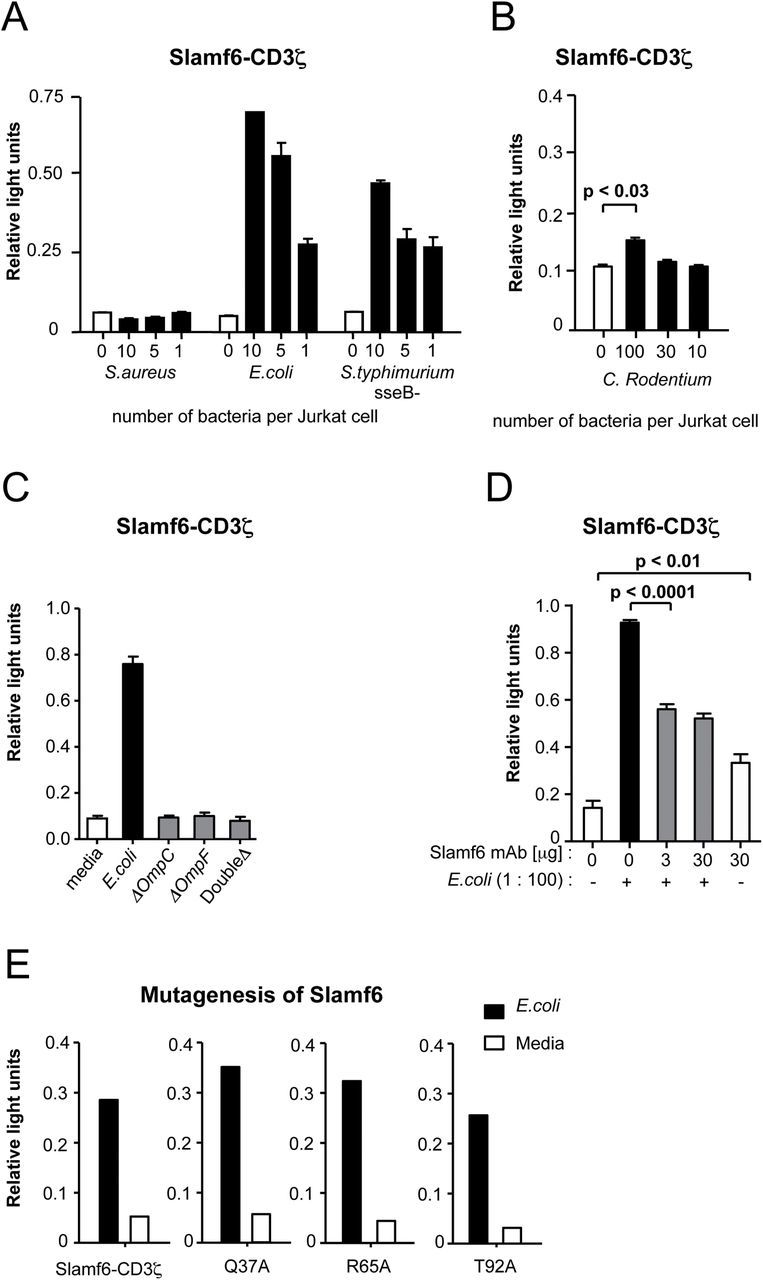
Slamf6 interacts with E. coli and C. rodentium. Relative luminescence measured in Jurkat cells that were transfected with a fusion construct of Slamf6 and CD3ζ and a Renilla luciferase reporter. (A) The luciferase activity of Jurkat cells after o/n stimulation with serial dilutions of heat-inactivated S. aureus, E. coli (F18) or S. typhimurium and (B) C. rodentium (DS100). (C) The luciferase activity of Jurkat cells after o/n stimulation with E. coli, E. coli ΔOmpC, E. coli ΔOmpF or double-deficient E. coli. (D) The luciferase activity of Jurkat cells after o/n stimulation in the absence and presence of anti-Slamf6 mAb (330). (E) The luciferase activity of Jurkat cells in which single mutations were made, after o/n stimulation.
Structural studies that assessed the formation of human SLAMF6–SLAMF6 homodimers revealed 13 amino acid residues that are critical for this interaction (20). To evaluate whether these residues are also involved in bacterial interactions, we constructed three mutant Slamf6–CD3ζ chimeras at positions that affect homodimer formation (Fig. 1E). These mutant Slamf6 molecules were capable of inducing a signal upon exposure to E. coli, suggesting that bacterial ligation does not involve amino acid residues that are essential in homodimer formation. Similar conclusions were made based upon the same type of analyses with mutants of human SLAMF6 and mouse Slamf1 (data not shown). Thus, the amino acids in the ectodomain of Slamf6 and Slamf1 that interact with structures in the outer cell wall of several Gram− bacteria are different from the amino acid residues, which are requisite for formation of the homodimers.
Stronger Th17 response in Slamf6−/− mice during C. rodentium infection
To evaluate the effect of Slamf6 on immune responses to bacteria, we selected C. rodentium, which is a well-studied model organism that induces intestinal inflammation. First, Slamf6 −/− and WT C57BL/6 mice were orally infected with 2×109 C. rodentium bacteria and analyzed for progression of colitis. Upon infection, both colonic macrophages and a set of CD11c− cells express Slamf6 (Supplementary Figure 1A and B, available at International Immunology Online). Slamf6 −/− mice, like WT mice, showed a slight reduction in their body weight on day 12 post-infection (Fig. 2A) with a fecal C. rodentium burden of ~107 CFU per mg feces. The fecal burden steadily declined after day 16 and both WT and Slamf6 −/− mice appeared to recover from the infection (Fig. 2B and C). Thus, Slamf6-deficiency has little impact on the pathogenesis of C. rodentium-induced colitis in the presence of an adaptive immune system.
Fig. 2.
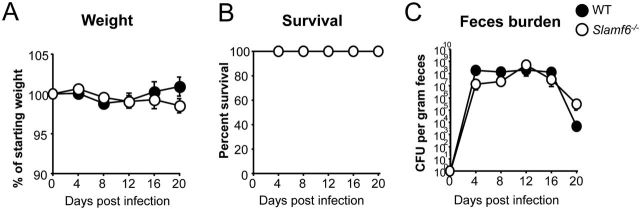
Slamf6 −/− mice are equally resistant to C. rodentium-induced colitis as WT mice. WT and Slamf6 −/− mice were infected by oral gavage of 2×109 C. rodentium bacteria. The animals were checked daily and sacrificed when their weight dropped below 80% of their starting weight. (A) The weight of individual mice was measured every 4 days and represented as a percentage of their weight on the day of infection. (B) Survival of infected WT and Slamf6 −/− mice. (C) C. rodentium counts of fecal pellets that were obtained directly from mice every 3 days. Serial dilutions were made in PBS and plated on MacConkey plates. Counted bacterial colonies are represented as CFU per g feces.
As both Th1 and Th17 cells have been implicated in inflammation and mucosal protection during C. rodentium infections (21, 22), isolated T cells from infected WT and Slamf6 −/− mice were assessed for the production of IFNγ and IL-17A. Intracellular staining of these cells revealed that Th17 cells are more prevalent in the lamina propria of Slamf6 −/− mice, compared with WT mice (Fig. 3A). However, an equal percentage of CD4+ IFNγ+ T cells were found in the colonic lamina propria of WT and Slamf6 −/− mice (Fig. 3B and C), suggesting that Th1 development is unaffected by Slamf6. To assess whether the enhanced percentage of Th17 cells in the lamina propria of infected Slamf6 −/− mice is a T-cell intrinsic phenomenon, WT and Slamf6 −/− T cells were stimulated in vitro. The culture supernatant of Slamf6 −/− T cells contained significantly more IL-17A upon stimulation with αCD3 mAb compared with WT culture supernatant, while IFNγ production was similar (Fig. 3D and E). Experiments in which colitis-inducing CD45RBhi CD4+ T cells were transferred to Rag −/− mice also showed that Slamf6 −/− T cells are more prone to IL-17A production (Fig. 3F) (23), as the percentage of IL17A+ lymphocytes was significantly higher in Rag −/− mice that had received Slamf6 −/− T cells, compared with WT T cells. Again, equal percentages of Th1 cells were detected in the lamina propria of Rag −/− mice 6 weeks after transfer of WT or Slamf6 −/− T cells (Fig. 3G and H). Taken together, the data suggest that Slamf6 inhibits the production of IL-17A by CD4+ T cells.
Fig. 3.
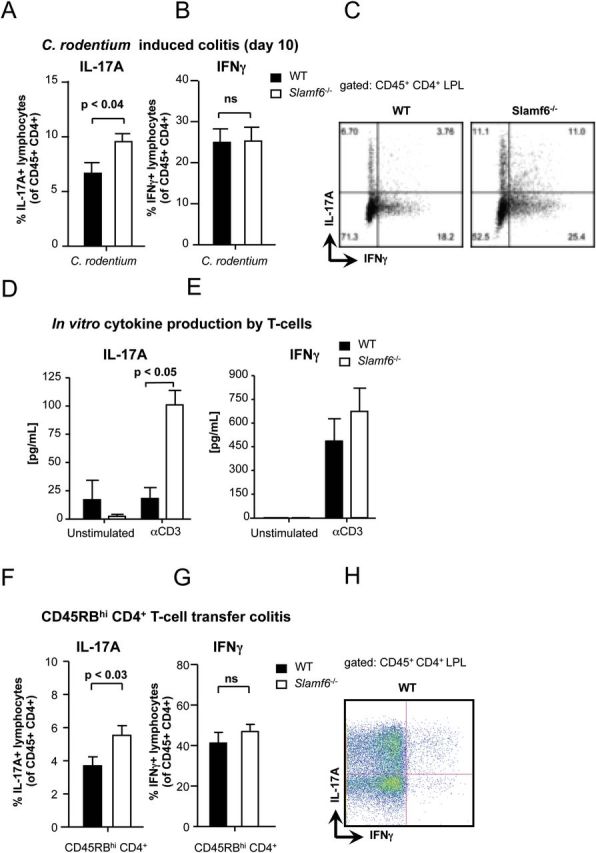
Enhanced Th17 response in Slamf6 −/− mice during C. rodentium-induced colitis. WT and Slamf6 −/− mice were infected by oral gavage of 2×109 C. rodentium bacteria. At day 10 post-infection, leukocytes were isolated from the colon lamina propria. The percentage of (A) IL-17A+ and (B) IFNγ+ lymphocytes is represented as a percentage of total CD45+ CD4+ lymphocytes. (C) Representative dot plots of intracellular staining for IL-17A and IFNγ obtained from WT and Slamf6 −/− mice. Isolated CD4+ splenocytes were cultured o/n in the presence of plate-bound anti-CD3 antibody. The amount of (D) IL-17A and (E) IFNγ cytokines in the supernatant of these cultures is represented. WT and Slamf6 −/− CD45RBhi CD4+ splenocytes were transferred into Rag −/− mice. The percentage of (F) IL-17A+ and (G) IFNγ+ lymphocytes is represented as a percentage of total CD45+ CD4+ lymphocytes. (H) Representative dot plot of intracellular staining for IL-17A and IFNγ obtained from Rag −/− mice in which WT T cells were transferred.
Slamf6−/− Rag−/− mice are resistant to C. rodentium
To focus on the role of Slamf6 in the surface of innate immune cells, Slamf6 −/− Rag −/− and Rag −/− mice were infected with C. rodentium. Slamf6 −/− Rag −/− mice have a mucosal homeostasis that is similar to Rag −/− mice before and after co-housing. However, after oral infection with 2×109 C. rodentium, Slamf6 −/− Rag −/− mice are resistant to the typical lethal colitis that develops in Rag −/− mice (Fig. 4) (10, 17, 22). The loss in weight that is caused by a progressing colitis in infected Rag −/− mice, which starts 2 weeks after the oral gavage with C. rodentium, was not observed in Slamf6 −/− Rag −/− mice (Fig. 4A). Ultimately, Rag −/− mice are unable to manage the infection, leading to diarrhea and fatal colitis (~60% at day 20), whereas all of the Slamf6 −/− Rag −/− mice survived past day 24 (Fig. 4B).
Fig. 4.

Slamf6 −/− Rag −/− mice are resistant to C. rodentium-induced colitis, while Rag −/− mice are not. Rag −/− and Slamf6 −/− Rag −/− mice were infected by oral gavage of 2×109 C. rodentium bacteria. The animals were checked daily and sacrificed when their weight dropped below 80% of their starting weight. (A) The weight of individual mice was measured every 3 days and represented as a percentage of their weight on the day of infection. (B) Survival of infected Rag −/− and Slamf6 −/− Rag −/− mice. (C) C. rodentium counts of fecal pellets that were obtained directly from mice every 3 days. Serial dilutions were made in PBS and plated on MacConkey plates. Counted bacterial colonies are represented as CFU per g feces. (D) H&E staining of representative longitudinal sections of the medial colon of Rag −/− and Slamf6 −/− Rag −/− mice, 18 days after infection. (E) Histological score of proximal, medial and distal colon longitudinal sections, 18 days after infection. An independent pathologist performed scoring blind. Maximal scores were 13, judged by mononuclear cell infiltration (3), epithelial integrity (4), hyperplasia (4) and edema (2). (F) C. rodentium counts of spleen homogenates. Serial dilutions were made in PBS and plated on MacConkey plates. Counted bacterial colonies are represented as CFU per g spleen tissue.
Fecal cultures of infected mice reveal an expansion of the colonic bacterial burden over the first ~14 days after infection in both Rag −/− and Slamf6 −/− Rag −/− mice. As expected, Rag −/− mice were unable to control the C. rodentium burden after day 14, reaching >1010 CFU per mg feces (11). In contrast, Slamf6 −/− Rag −/− mice stabilize the bacterial burden at ~107 CFU per mg feces (Fig. 4C). Slamf6 −/− Rag −/− mice were incapable of sterile clearance as late as day 40 post-infection (data not shown). Thus, Slamf6 −/− Rag −/− mice can harbor C. rodentium without developing lethal colitis.
At day 18 post-infection when most Rag −/− mice were moribund, Slamf6 −/− Rag −/− mice showed a strongly reduced pathology of the colon (Fig. 4D and E). A loss in epithelial integrity as well as some inflammatory infiltration of mononuclear phagocytes was observed in Slamf6 −/− Rag −/− mice. In contrast, Rag −/− mice showed a strong reduction of epithelial integrity and more cellular infiltration, paired with submucosal edema (Fig. 4D). This indicates that mucosal barrier damage and inflammation as a consequence of C. rodentium-induced colitis is reduced in Slamf6 −/− Rag −/− mice.
Citrobacter rodentium translocates to the spleen of both Rag−/− and Slamf6−/− Rag−/− mice
One of the major differences in the early response to C. rodentium between Rag −/− and WT mice is the presence of mucosal CD4+ T cells. Th1 cells localize at sites of bacterial lesions, produce IFNγ and aid in an early inflammatory response. However, Th1 cells also contribute to immune pathology (10–12, 24). Th17 cells are required to contain C. rodentium and to prevent aberrant intestinal pathology (25). Overall, the lack of T cells causes Rag −/− mice to develop a systemic multi-bacterial infection past the second week after oral infection due to this loss of colon barrier integrity (12). Although pathologic features were significantly lower in Slamf6 −/− Rag −/− mice (Fig. 4D and E), C. rodentium was detected in splenic homogenates of these mice when cultured on MacConkey agar plates. However, more C. rodentium CFU were cultured from Rag −/− spleens (Fig. 4F). Citrobacter rodentium systemic translocation manifested in mice with both genetic backgrounds, albeit to a lesser extent in Slamf6 −/− Rag −/− mice. This indicates that the mucosal barrier is less compromised in Slamf6 −/− Rag −/− mice.
Reduced inflammation in Slamf6−/− Rag−/− mice during C. rodentium infection
Having established that the absence of Slamf6 in T-cell- and B-cell-deficient mice improves the health of infected animals, the immune activation in the colonic lamina propria was assessed. The colonic lamina propria of Rag −/− and Slamf6 −/− Rag −/− mice showed an influx of phagocytes in the early progressive stage of a C. rodentium infection. As predicted from our histopathology analysis (Fig. 4D and E), smaller numbers of CD11b+ CD103− macrophages and Ly6G+ neutrophils were detected in the lamina propria of Slamf6 −/− Rag −/− mice compared with Rag −/− mice 6 days post-infection (Fig. 5A and B).
Fig. 5.

Reduced lamina propria inflammation during C. rodentium-induced colitis of Slamf6 −/− Rag −/− mice. Rag −/− and Slamf6 −/− Rag −/− mice were infected by oral gavage of 2×109 C. rodentium bacteria. Uninfected Rag −/− mice are represented as controls. Animals were sacrificed on day 6 post-infection for analysis. (A) Quantification of flow cytometric analysis of isolated lamina propria macrophages. The total number of CD11chi MHC-II+ CD11b+ CD103− Ly6G− CD45+ cells and the percentage of CD11chi MHC-II+ CD11b+ CD103− cells relative to the total pool of Ly6G− CD45+ lamina propria cells are represented. (B) Quantification of flow cytometric analysis of isolated lamina propria neutrophils. The total number of CD11b+ Ly6G+ CD45+ cells and the percentage of CD11b+ Ly6G+ relative to the total pool of CD45+ lamina propria cells are represented. (C) Quantitative PCR relative expression of IL-12(p70) in colon homogenates of uninfected Rag −/− and Slamf6 −/− Rag −/− mice and 6 days after infection with C. rodentium.
As judged by quantitative PCR of colon homogenates, a reduced production of the inflammatory cytokine IL-12(p70) was observed in the colons of Slamf6 −/− Rag −/− mice (Fig. 5C). This indicates that fewer inflammatory phagocytes migrate into the lamina propria of Slamf6 −/− Rag −/− mice. These inflammatory cells contribute to the clearance of the infection but can also drive mucosal damage. Without an effective adaptive response, these inflammatory infiltrates contribute to local immune pathology. Therefore, the reduced pathology in Slamf6 −/− Rag −/− mice could perhaps be explained by impaired monocyte and neutrophil recruitment.
The presence of Slamf6 reduces the production of IL-22 in the colon of infected Slamf6−/− Rag−/− mice
IL-22 and the cells that produce it are a key component of protective immunity to C. rodentium in the early stages of infection (16, 17). IL-22 production was assessed in infected colons of Rag −/− and Slamf6 −/− Rag −/− mice at days 0, 3 and 6 after infection. As judged by quantitative PCR of mRNA that was isolated from whole colon homogenates and by cytokine analysis of colon culture supernatant, the peak of IL-22 production in both Rag −/− and Slamf6 −/− Rag −/− mice is at day 3 post-infection. At this time, higher amounts of IL-22 were detected in the colon of Slamf6 −/− Rag −/− mice. This difference reduced to a non-significant level at day 6 post-infection, when IL-22 levels had dropped to 3-fold higher than those of uninfected mice (Fig. 6A and B). As IL-22 is one of the key protective cytokines during C. rodentium infections, this observation may contribute to the reduced pathology and lower number of translocated bacteria in Slamf6 −/− Rag −/− mice (Fig. 4F). As IL-22 production is driven primarily by IL-23, colon cultures were assessed for the production of this cytokine after C. rodentium infection. In contrast to the IL-12(p70) subunit of the IL-12 cytokine, Slamf6 −/− Rag −/− colons produced more IL-12/IL-23(p40) (Fig. 6C), suggesting an enhanced production of IL-23 in Slamf6 −/− Rag −/− mice. Interestingly, Slamf6 expression was detected in a small set of CD45.2+ CD90.2+ CD127+ RORγt+ (ILC3) cells, which represent the main producers of IL-22 in Rag −/− mice (Supplementary Figure 1C, available at International Immunology Online) (14, 17).
Fig. 6.
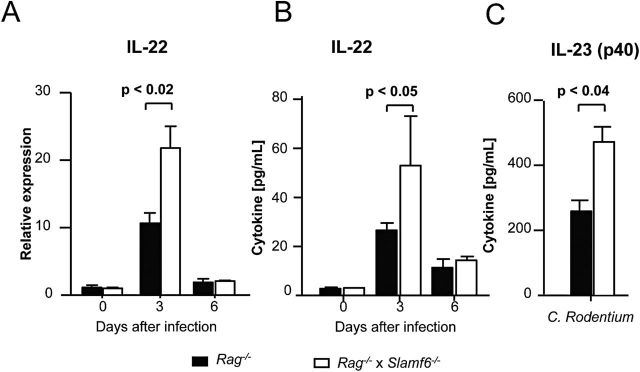
Enhanced IL-22 production in Slamf6 −/− Rag −/− mice during C. rodentium-induced colitis. Rag −/− and Slamf6 −/− Rag −/− mice were infected by oral gavage of 2×109 C. rodentium bacteria. Uninfected Rag −/− and Slamf6 −/− Rag −/− mice are represented as controls (day 0). Animals were sacrificed on day 3 or day 6 post-infection for analysis. (A) Quantitative PCR relative expression of IL-22 in colon homogenates. (B) IL-22 production of ex vivo-cultured colon tissue. (C) Ex vivo IL-23(p40) production by 3-day-infected colon tissue cultures.
Administering a mAb directed against Slamf6 reduces C. rodentium colitis in Rag−/− mice
In order to test whether the anti-Slamf6 mAb, which inhibits the cognate interactions of Slamf6 with bacteria in the recognition assay (Fig. 1), affects the bacterial colitis pathology, Rag −/− mice were intra-peritoneally injected with anti-Slamf6 or isotype (100 µg) 1 day before and every 7 days after C. rodentium infection (Fig. 7A). Rag −/− mice that received the anti-Slamf6 mAb showed a delay in their weight loss, a reduction of fecal C. rodentium burden at the peak of infection and an increased survival rate (Fig. 7B–D). These findings suggest that blocking the interaction of Slamf6 with bacteria reduces the pathology of C. rodentium-induced colitis.
Fig. 7.

Administering a mAb directed against Slamf6 ameliorates C. rodentium-induced colitis in Rag −/− mice. (A) Infection and treatment diagram for Rag −/− mice that were intra-peritoneally injected with 100 µg anti-Slamf6 mAb (330) 1 day prior to infection and subsequently every seventh day after the first injection. Control mice were injected with 100 µg mouse IgG2a in the same regimen. (B) The weight of individual mice was measured every 4 days and represented as a percentage of their weight on the day of infection. (C) Survival of infected anti-Slamf6 mAb-treated and isotype-treated Rag −/− mice. (D) C. rodentium counts in fecal pellets that were obtained directly from mice every 3 days. Serial dilutions were made in PBS and plated on MacConkey plates. Counted bacterial colonies are represented as CFU per g feces.
Discussion
In this report, we provide evidence that Slamf6 −/− Rag −/− mice are markedly less susceptible to C. rodentium-induced colitis than Rag −/− mice. Through the course of infection, Slamf6 −/− Rag −/− mice show a reduction in the inflammatory response in the lamina propria of the colon. Fewer bacteria translocate to the spleen of Slamf6 −/− Rag −/− mice and ultimately the infection is fatal in a significantly lower percentage of infected mice when compared with co-housed control Rag −/− mice. Moreover, anti-Slamf6 mAb treatment of infected Rag −/− mice improves the disease outcome associated with less weight loss, an improved survival rate and a reduction in the fecal C. rodentium burden.
Our in vitro observation that Slamf6 can interact with certain Gram− bacteria suggests that the reduced colitis displayed by Slamf6 −/− Rag −/− mice in the context of C. rodentium infections might be mediated by the absence of the binding of Slamf6 to this bacterium. The observations that anti-Slamf6 mAb inhibited bacterial binding and that in vivo administration of this antibody reduced the pathology of infected Rag −/− mice further corroborate this notion.
The balance between immune activation and mucosal protection is one of the defining factors in the pathogenic outcome of a C. rodentium infection. Whereas a range of key regulators of innate immune responses is indispensible for an effective response to C. rodentium, not all innate mechanisms are beneficial to the host. Several recent review articles discuss how innate receptors are involved in detection of C. rodentium (21, 26–28). Two major receptor families mediate inflammatory signaling in response to C. rodentium: Toll-like receptors and Nod-like receptors, both of which utilize the NF-κB pathway. Interestingly, TLR4 signaling does not appreciably contribute to host protection against C. rodentium but rather enhances mucosal pathologic immune responses (29). Contrary to this, TLR2 signaling promotes inflammatory responses as well as mucosal integrity (30, 31). The authors of these studies argue that TLR4-mediated immune activation drives immune pathology during C. rodentium infections, which can be reduced by the protective function of TLR2 (27). Along similar lines, an excess of IL-1β is detrimental to infected mice, whereas normal levels are required for an appropriate response to C. rodentium (32). From our data in Rag −/− mice, we conclude that Slamf6 also breaks tolerance and enhances inflammatory responses.
An interesting notion is that Slamf6 appears to affect the development of colitis only in Rag −/− mice. T cells and B cells play key roles in the development of an adequate immune response to C. rodentium. Mice that are impaired in their ability to mount a specific humoral response to the bacterium are unable to generate sterilizing immunity (11). However, the protective role of CD4+ T cells appears to be 2-fold: the direct protective effects of lesion-proximal T cells and eventually T-cell help to induce a sterilizing humoral immune response. µMT mice, which lack B cells, but have T cells, develop a milder colitis than Rag −/− mice (12). Additionally, Rag −/− mice in which CD4+ T cells were adoptively transferred show more inflammation, but also better mucosal protection (22). Both Th1 and Th17 cells have been implicated in the immune response to C. rodentium (21). The lesions at sites of C. rodentium infections are associated with IFNγ-producing Th1 clusters that contribute to the local response to the infection (10, 24). Th17 development contributes to a protective response and CD4+ T-cell depletion leads to an exacerbated pathology and increased loss of barrier function. Although Slamf6 negatively affects IL-17A production by CD4+ T cells, this suppressed Th17 response does not appear to significantly affect the pathology that is caused by C. rodentium infections. The observations that Slamf6 contributes to the pathology in Rag −/− mice, but not WT mice, are likely due to the lack of CD4+ T cells that play a protective role in mucosal integrity as differences in immune activation between Rag −/− and Slamf6 −/− Rag −/− mice precede the time when a humoral response would arise in WT mice.
Ample studies have addressed functions of Slam receptors in lymphocyte and phagocyte functions. However, little is known about their functions in mucosal phagocytes and innate lymphocytes. Here, we show that Slamf6 affects the cytokine production of these cells. Interactions of CD103− CX3CR1+ colonic macrophages with C. rodentium result in the phagocytosis of the bacterium (14, 15, 33). CX3CR1-deficient macrophages are impaired in their ability to probe the lumen of the intestine, which in WT as well as Rag −/− mice results in decreased IL-22 production and hence increased pathology of C. rodentium infection. Thus, interactions of colonic phagocytes with C. rodentium result in the production of IL-22 by phagocyte-proximal innate lymphocytes in Rag −/− mice (33). In another study, the role of IL-22 in the innate immunity to C. rodentium infection was clearly demonstrated utilizing an IL-22-specific antibody. Administration of this antibody to Rag −/− mice led to rapid weight loss, more severe colitis and expedited mortality (17). Conversely, vitamin D receptor-deficient mice have higher numbers of ILC3 and produce more IL-22. Consequently, these mice have a reduced bacterial burden and lower immune activation (34).
In this study, we report that Slamf6 interacts with several Gram− bacterial, including C. rodentium. Furthermore, Slamf6 −/− Rag −/− mice produce significantly higher amounts of IL-22 during C. rodentium infections. Higher levels of IL-22 result in stronger mucosal protection, thereby lessening epithelial damage (21). We therefore speculate that engagement of C. rodentium by Slamf6 negatively affects phagocyte-induced signals that promote IL-22 production. Longman et al. (14) have shown that IL-23 and IL-1β production by phagocytes plays a role in the induction of IL-22 production by ILC3 cells. We report that infected colons of Slamf6 −/− Rag −/− mice produce less IL-12(p70) and more IL-12/23(p40). Thus, Slamf6 has important implications in the development of a colonic innate immune response to pathogenic Gram− bacterial challenge. Slamf6-deficiency in innate cells renders the outcome of C. rodentium-mediated immune activation more inflammatory and less protective, which profoundly affects the pathology and survival of Slamf6 −/− Rag −/− mice.
Supplementary data
Supplementary data are available at International Immunology Online.
Funding
National Institutes of Health (RO1 NIH AI-15066 to C.T.).
Conflict of interest statement: The authors declared no conflict of interest.
Supplementary Material
Acknowledgements
We thank members of the Terhorst lab and Balthasar Heesters for thoughtful discussions.
References
- 1. Calpe S., Wang N., Romero X., et al. 2008. The SLAM and SAP gene families control innate and adaptive immune responses. Adv. Immunol. 97:177. [DOI] [PubMed] [Google Scholar]
- 2. Kageyama R., Cannons J. L., Zhao F., et al. 2012. The receptor Ly108 functions as a SAP adaptor-dependent on-off switch for T cell help to B cells and NKT cell development. Immunity 36:986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Qi H., Cannons J. L., Klauschen F., Schwartzberg P. L., Germain R. N. 2008. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 455:764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tatsuo H., Ono N., Tanaka K., Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893. [DOI] [PubMed] [Google Scholar]
- 5. Malaviya R., Gao Z., Thankavel K., van der Merwe P. A., Abraham S. N. 1999. The mast cell tumor necrosis factor alpha response to FimH-expressing Escherichia coli is mediated by the glycosylphosphatidylinositol-anchored molecule CD48. Proc. Natl Acad. Sci. USA 96:8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berger S. B., Romero X., Ma C., et al. 2010. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nat. Immunol. 11:920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma C., Wang N., Detre C., Wang G., O’Keeffe M., Terhorst C. 2012. Receptor signaling lymphocyte-activation molecule family 1 (Slamf1) regulates membrane fusion and NADPH oxidase 2 (NOX2) activity by recruiting a Beclin-1/Vps34/ultraviolet radiation resistance-associated gene (UVRAG) complex. J. Biol. Chem. 287:18359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Engel P., Eck M. J., Terhorst C. 2003. The SAP and SLAM families in immune responses and X-linked lymphoproliferative disease. Nat. Rev. Immunol. 3:813. [DOI] [PubMed] [Google Scholar]
- 9. Guttman J. A., Li Y., Wickham M. E., Deng W., Vogl A. W., Finlay B. B. 2006. Attaching and effacing pathogen-induced tight junction disruption in vivo . Cell. Microbiol. 8:634. [DOI] [PubMed] [Google Scholar]
- 10. Chan J. M., Bhinder G., Sham H. P., et al. 2013. CD4+ T cells drive goblet cell depletion during Citrobacter rodentium infection. Infect. Immun. 81:4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vallance B. A., Deng W., Knodler L. A., Finlay B. B. 2002. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect. Immun. 70:2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simmons C. P., Clare S., Ghaem-Maghami M., et al. 2003. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium . Infect. Immun. 71:5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Niess J. H., Brand S., Gu X., et al. 2005. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307:254. [DOI] [PubMed] [Google Scholar]
- 14. Longman R. S., Diehl G. E., Victorio D. A., et al. 2014. CX₃CR1⁺ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 211:1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schreiber H. A., Loschko J., Karssemeijer R. A., et al. 2013. Intestinal monocytes and macrophages are required for T cell polarization in response to Citrobacter rodentium . J. Exp. Med. 210:2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ota N., Wong K., Valdez P. A., et al. 2011. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium . Nat. Immunol. 12:941. [DOI] [PubMed] [Google Scholar]
- 17. Zheng Y., Valdez P. A., Danilenko D. M., et al. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14:282. [DOI] [PubMed] [Google Scholar]
- 18. Keszei M., Detre C., Rietdijk S. T., et al. 2011. A novel isoform of the Ly108 gene ameliorates murine lupus. J. Exp. Med. 208:811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Powrie F., Leach M. W., Mauze S., Caddle L. B., Coffman R. L. 1993. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 5:1461. [DOI] [PubMed] [Google Scholar]
- 20. Cao E., Ramagopal U. A., Fedorov A., et al. 2006. NTB-A receptor crystal structure: insights into homophilic interactions in the signaling lymphocytic activation molecule receptor family. Immunity 25:559. [DOI] [PubMed] [Google Scholar]
- 21. Rubino S. J., Geddes K., Girardin S. E. 2012. Innate IL-17 and IL-22 responses to enteric bacterial pathogens. Trends Immunol. 33:112. [DOI] [PubMed] [Google Scholar]
- 22. Bry L., Brigl M., Brenner M. B. 2006. CD4+-T-cell effector functions and costimulatory requirements essential for surviving mucosal infection with Citrobacter rodentium . Infect. Immun. 74:673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Uhlig H. H., McKenzie B. S., Hue S., et al. 2006. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25:309. [DOI] [PubMed] [Google Scholar]
- 24. Shiomi H., Masuda A., Nishiumi S., et al. 2010. Gamma interferon produced by antigen-specific CD4+ T cells regulates the mucosal immune responses to Citrobacter rodentium infection. Infect. Immun. 78:2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishigame H., Kakuta S., Nagai T., et al. 2009. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30:108. [DOI] [PubMed] [Google Scholar]
- 26. Collins J. W., Keeney K. M., Crepin V. F., et al. 2014. Citrobacter rodentium: infection, inflammation and the microbiota. Nat. Rev. Microbiol. 12:612. [DOI] [PubMed] [Google Scholar]
- 27. Bergstrom K. S., Sham H. P., Zarepour M., Vallance B. A. 2012. Innate host responses to enteric bacterial pathogens: a balancing act between resistance and tolerance. Cell. Microbiol. 14:475. [DOI] [PubMed] [Google Scholar]
- 28. Liu Z., Zaki M. H., Vogel P., et al. 2012. Role of inflammasomes in host defense against Citrobacter rodentium infection. J. Biol. Chem. 287:16955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khan M. A., Ma C., Knodler L. A., et al. 2006. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect. Immun. 74:2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gibson D. L., Ma C., Rosenberger C. M., et al. 2008. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell. Microbiol. 10:388. [DOI] [PubMed] [Google Scholar]
- 31. Gibson D. L., Montero M., Ropeleski M. J., et al. 2010. Interleukin-11 reduces TLR4-induced colitis in TLR2-deficient mice and restores intestinal STAT3 signaling. Gastroenterology 139:1277. [DOI] [PubMed] [Google Scholar]
- 32. Alipour M., Lou Y., Zimmerman D., et al. 2013. A balanced IL-1β activity is required for host response to Citrobacter rodentium infection. PLoS One 8:e80656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Manta C., Heupel E., Radulovic K., et al. 2013. CX(3)CR1(+) macrophages support IL-22 production by innate lymphoid cells during infection with Citrobacter rodentium . Mucosal Immunol. 6:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen J., Waddell A., Lin Y. D., Cantorna M. T. 2014. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol. 3:618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.