

Abstract
Aims
The physiological determinants of RV diastolic function remain poorly understood. We aimed to quantify the contribution of elastic recoil to RV filling and determine its sensitivity to interventricular interaction.
Methods and results
High-fidelity pressure–volume loops and simultaneous 3-dimensional ultrasound sequences were obtained in 13 pigs undergoing inotropic modulation, volume overload, and acute pressure overload induced by endotoxin infusion. Using a validated method, we isolated elastic restoring forces from ongoing relaxation using conventional pressure–volume data. The RV contracted below the equilibrium volume in >75% of the data sets. Consequently, elastic recoil generated strong sub-atmospheric passive pressure at the onset of diastole [−3 (−4 to −2) mmHg at baseline]. Stronger restoring suction pressure was related to a shorter isovolumic relaxation period, a higher rapid filling fraction, and lower atrial pressures (all P < 0.05). Restoring forces were mostly determined by the position of operating volumes around the equilibrium volume. By this mechanism, the negative inotropic effect of beta-blockade reduced and sometimes abolished restoring forces. During acute pressure overload, restoring forces initially decreased, but recovered at advanced stages. This biphasic response was related to alterations of septal curvature induced by changes in the diastolic LV–RV pressure balance. The constant of elastic recoil was closely related to the constant of passive stiffness (R = 0.69).
Conclusion
The RV works as a suction pump, exploiting contraction energy to facilitate filling by means of strong elastic recoil. Restoring forces are influenced by the inotropic state and RV conformational changes mediated by direct ventricular interdependence.
Keywords: Diastolic function, Right ventricle, Hemodynamics, Relaxation, Diastolic stiffness
1. Introduction
Right-ventricular (RV) function is a powerful determinant of outcome in a number of lung and cardiovascular diseases.1,2 Although most clinical research has focused on systolic function, normal RV filling is also essential to maintain exercise activity and adapt to acute and chronic overload.3 However, the physiological foundations of RV filling are only beginning to be understood.4–7
One of the most extensively studied index of diastolic function is myocardial relaxation (lusotropy).2 At the cellular level, relaxation is produced by the detachment of strong actin-myosin bounds. Lusotropy determines the rate by which the ventricle lowers its pressure from end-systolic to unstressed baseline values. Moreover, this rate of pressure decay is also conditioned by the recovery of elastic energy generated by compression of myocardial elements during systole.8 Hence, relaxation and elastic restoration synergistically facilitate chamber pressure decay and early filling.9 Although never quantified, an important role of restoring forces in the RV can be anticipated. Because it is not possible for relaxation to generate sub-atmospheric pressure,10 strong elastic restoring forces are the only potential source for the negative intraventricular pressure frequently measured in the RV.11
In the isolated cardiomyocyte, restoring forces are generated when the sarcomere contracts below its slack length. Attached in parallel to the contractile apparatus, sarcomeric titin senses and modulates time-variable cellular stresses. Thus, restoring forces are driven by passive deformation of this protein, predominantly in its Ig-domain regions.12 In the myocardium, restoring forces are further conditioned by the amount, type, and degree of crosslinking of collagen fibres in the extracellular matrix.
In the intact ventricle, restoring forces are generated as the chamber contracts below its equilibrium volume—the volume of the fully relaxed chamber at zero transmural pressure.8 Whenever volume falls below this equilibrium value during systole, a fraction of contraction energy is used to compress elastic myocardial materials. Once mechanical systole ends, potential energy is rapidly released as a spring-like mechanism facilitating chamber depressurization. Detorsion of large-scale myocardial fibres complements microscopic conformational changes to enhance elastic recoil in the intact ventricle.13
In the whole heart, an additional large-scale mechanism may also modulate restoring forces. RV geometry and volume are determined by septal shape which, in turn, is conditioned by the pressure balance of direct RV–LV interdependence.14 We hypothesized that ventricular interdependence may also modulate RV elastic recoil during early filling by this mechanism.
Measuring elastic restoring forces is methodologically challenging because passive forces need to be detached from simultaneous active relaxation. Very complex surgical volume-clamp experiments have been designed for this purpose in the LV.8,13,15 However, a simpler algorithm, able to uncouple active and passive diastolic pressures from conventional pressure–volume (PV) data, has been recently proposed.16 This method is particularly well suited to assess RV elastic restoring forces.16
The present study was designed to quantify the role of RV elastic restoring forces in the intact heart. Additionally, we assessed whether direct ventricular interdependence could impact elastic restoring forces by means of regional variations of septal shape. For this purpose, we combined a new method of analysis of high fidelity RV pressure–volume data with custom-developed chamber geometry analyses of 3D echocardiographic datasets.
2. Methods
The experimental protocol was approved by the local Institute Animal Care Committee. All animal procedures were in accordance with guidelines from Directive 2010/63/EU.
2.1. Physical model and design rationale
For the purpose of this study, we conceived myocardial mechanics as the resultant of ‘active’ and ‘passive’ forces acting in parallel; instantaneous measured pressure are the net sum of active and passive components. ‘Active’ pressure (energy consuming) is caused by actin–myosin cross-bridge cycling. Active forces are therefore responsible for the time-varying elastance that causes contraction and relaxation. ‘Passive’ pressure (non-energy consuming, although modulated by post-transcriptional energy consuming processes) is generated by deformation of myocardial elastic structures. As in a spring, passive forces attempt to take the RV wall to its unstressed resting state. Deformation of passive elements can take place either by compression or by stretching. When ‘compressed’, myocardial elastic elements push the chamber wall outwards, generating negative pressure. These forces are the ‘elastic restoring forces’. When ‘stretched’, myocardial passive elastic elements push the ventricular wall inwards, generating positive pressure. These forces are responsible for myocardial diastolic ‘stiffness’.
Restoring forces and stiffness at the chamber level are best characterized in terms of the full passive PV curve.17 The negative-pressure part governs restoring forces, whereas the positive-pressure part of the PV curve governs stiffness (Figures 1 and 2). The slope of a particular RV curve is determined by a few constitutive parameters, establishing the amount of passive pressure generated at a given volume (discussed in what follows). A particularly relevant parameter is the position of the passive PV curve on the horizontal axis. This zero-pressure volume (V0) is analogous to the slack length of a spring and is critical because it defines the transition between stiffness and restoring forces; only when the ventricle operates beyond this value (ESV < V0) it will develop restoring forces.
Figure 1.
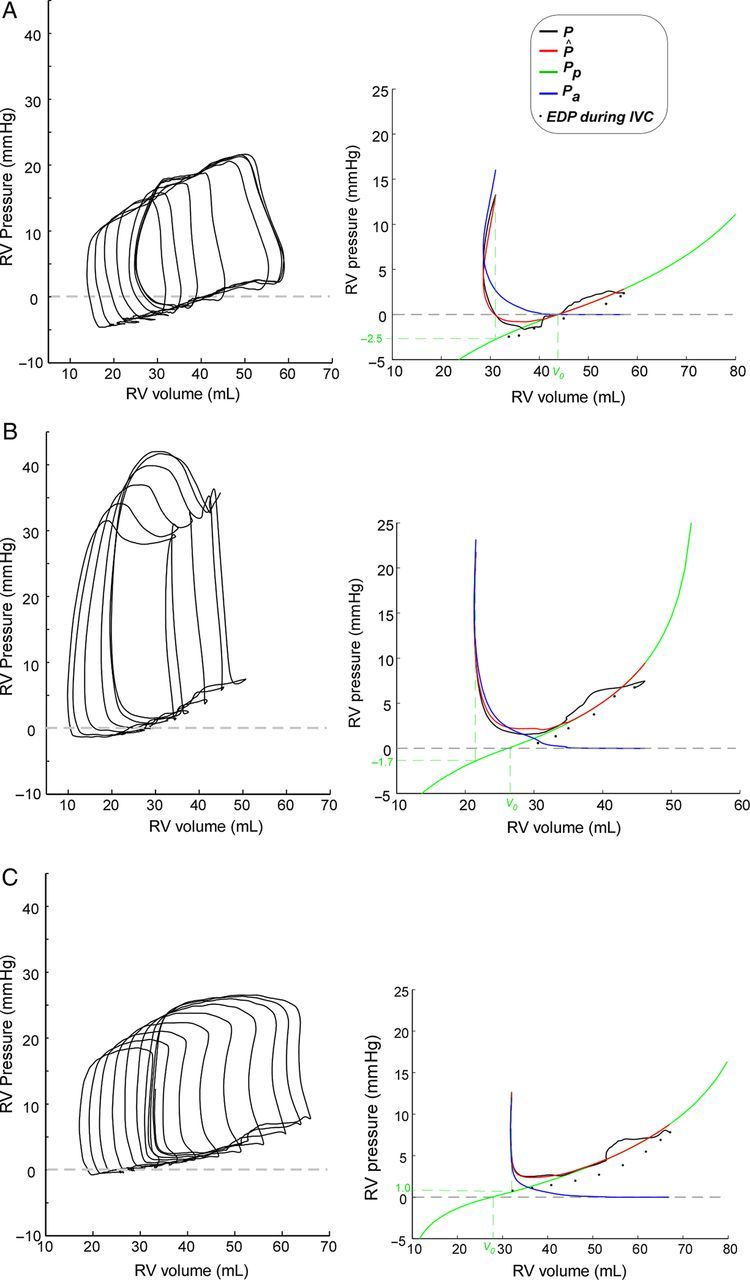
Representative examples of elastic restoring forces in the RV. Unprocessed PV-loops are shown in left panels. Right panels show the results of pressure decomposition, fitting the passive (green) and active (blue) pressures curves, as well as measured (black), and total fitted (red) pressure tracings ‘for the first beat (pre IVC-occlusion) in the data set’; black dots represent measured end-diastolic pressures and volumes for all beats. V0 is the zero-pressure volume. Values of passive pressure at pulmonary valve closure are shown in the vertical axis. Three patterns of the behaviour of restoring forces on the first beat are illustrated. (A) Negative pressure is measured because contraction is far below V0 from the first run, generating strong restoring forces. (B) Also contraction below V0 generates restoring forces. However, despite restoring forces are increasing the pressure gradient, measured pressure is positive because active pressure is higher. (C) The ventricle does not contract below V0 so restoring forces are not generated. The ventricle is exclusively filled by atrial emptying.
Figure 2.

Passive pressure curves of a representative animal during all haemodynamic interventions. Operational volumes, from end-systole to end-diastole are overlaid as solid lines on the full PV passive curve (dotted lines). During late endotoxin infusion, V0 increases, shifting the curve to the right. By this mechanism, the RV preserves restoring forces and lowers its stiffness, and reaches lower end-diastolic pressure than if operating volumes were projected on the baseline or the early endotoxin passive pressure curves.
On these foundations, we designed the acute haemodynamic interventions for our study. We used acute inotropic modulation (esmolol and dobutamine) and volume overload to modify operating volumes and thereby the position of ESV with respect to V0. We used acute pressure overload to induce overt RV geometrical changes that could impact V0. This allowed us to test our working hypothesis that elastic restoring forces are related to RV geometry.
2.2. Experimental preparation and protocol
We studied 13 adult minipigs (43 ± 8 kg) in a closed-chest instrumentation setup. After anaesthetic induction with intravenous propofol (1.5 mg/kg) and fentanyl (5 µg/kg), the animals were endotracheally intubated and mechanically ventilated without end-expiratory positive pressure. Complete anaesthesia and relaxation were maintained by propofol infusion (0.2 mg/kg/min), as well as repetitive boluses of fentanyl (0.05 mg i.v.) and atracurium (0.3 mg/kg/h). The abolition of eye reflexes, blood pressure, and heart rate to ensure deep anaesthesia was systematically monitored and verified before the administration of the neuromuscular blocker. Through the right jugular vein, we placed a 7F pigtail PV catheter (CD-Leycom) in the RV apex, and connected it to a dual-field conductance processor (Sigma 5DF, CD-Leycom). Additional 5F micromanometers (Millar Instruments Inc.) were placed in the right atrium (RA) and the LV. We placed an occlusion balloon (PTS404, NuMED Inc.) in the RA-inferior vena cava (IVC) junction through a femoral vein.
We performed a sequential data acquisition protocol which included baseline (n = 13), esmolol (75–200 µg/kg/min; n = 12), and dobutamine (2.5 µg/kg/min; n = 11) infusions, acute volume overload (1000–1500 mL saline isotonic solution in 5–10 min; n = 11), and endotoxic RV failure [lipopolysaccharide from Escherichia coli serotype 0127:B8 (Sigma Chemical); 0.5 mg/kg over 30 min infusion].18 Endotoxin infusion triggers the release of mediators by lung macrophages inducing an early phase of predominantly acute pulmonary hypertension, and a late phase of overt RV failure.18,19 Hence, we focused on early (30 min; n = 11) and late (4 h) endotoxin effects (n = 12). We waited for a recovering period ≥20′ before starting dobutamine, volume, and endotoxin. Animals were euthanized at the end of the experiments with intravenous sodium pentobarbital (100 mg/kg).
2.3. Data acquisition and analysis
Pressure micromanometres were carefully balanced and checked for drift as previously reported.18,20 Digital signals were recorded at 1000 Hz on a dedicated computer with custom-built amplifiers, a 16-channel analogue-to-digital converter board, and virtual instrumentation software. PV data were acquired during transient IVC occlusion, three times for each haemodynamic state. We obtained pulsed-wave Doppler and 3D RV volumetric recordings using either a Vivid-7 or a Vivid-9 ultrasound system (General Electric Healthcare) and broadband transducers from parasternal and apical (subxifoid abdominal incision) views. Data were collected at atmospheric (zero) intrathoracic pressure by disconnecting the endotracheal tube.
The conductance signal was calibrated using end-systolic and end-diastolic volumes (ESV and EDVs) measured from 3D-echocardiographic data sets before IVC occlusion.21 We used synchronized18,20 Doppler spectrograms at the RV outflow tract during IVC occlusion to manually identify pulmonary valve closing (PVC). Tricuspid valve opening (TVO) was taken at the first diastolic crossover point of RV and RA pressures, and the isovolumic relaxation time (IVRT) was defined as the period between PVC and TVO. End-diastole (ED) was defined as the peak of R-wave slope, and confirmed beat-by-beat inspecting of the bottom-right corner of the PV loop diagram. Maximum time varying elastance (Emax) and preload recruitable stroke work (PRSW) were calculated using the iterative method.20,22 Pulmonary arterial elastance (Ea) was measured from the PV data of the last pre-occlusion beat and used as a surrogate of RV arterial load. Emax/Ea was used as a measure of ventricular–arterial coupling.23
To process the diastolic PV data, we adapted an already validated numerical algorithm based on global optimization (see Supplemental material online and Figure 1).16 This method makes use of all diastolic PV data to characterize main diastolic chamber properties. Measured diastolic pressure results from the combination of active (Pa) and passive (Pp) pressure terms throughout the full diastolic period, from PVC to ED. We used an exponential function to characterize Pa by means of the time-constant of relaxation (τ),16 and a piece-wise logarithmic fitting to characterize Pp below and above V0.24 We defined V0 as RV volume at Pp = 0, as in previous studies performed in intact hearts.8,16 Upon this conception, elastic restoring pressure (Pp(V < V0)) is generated when V < V0 , and is characterized by the constant of diastolic recoil (S−), and the RV dead volume asymptote (Vd). Likewise, positive passive pressure (Pp(V > V0)) is defined by the stiffness constant (S+) and the maximal achievable volume asymptote (Vm). Because the exponential fitting has known limitations to characterize Pa, we additionally recalculated all indices of diastolic function using a logistic fitting of τ.25 Remarkably, measured indices of elastic recoil and restoring suction pressure values were highly insensitive to the model used to characterize relaxation (see Supplementary material online, Table S2).
Instantaneous Pp and Pa values were retrieved at PVC, TVO, minimal pressure (Pmin), and ED. We defined ‘restoring suction pressure’ as the negative pressure generated by passive restoring forces when V < V0, such as coined by volume-clamp studies.13 We report restoring suction pressure as well as all physiological data for the last baseline beat ‘prior’ to IVC occlusion. The full PV data set during preload manipulation was used to characterize τ, V0, Vd, S−, Vm, and S+. The global optimization method accurately fitted diastolic pressures in all data sets (n = 244; Ric > 0.90 for PVC, TVO, Pmin, and ED) and showed good agreement with conventional methods (see Supplementary material online). Based on the uncoupling of active and passive pressures, we also measured the impact of restoring forces on the work performed by the cardiac chambers to generate rapid RV filling (see Supplementary material online).
2.4. RV geometry analysis
3D echocardiography RV images were analysed off-line using commercial software (4D-RV function; TomTec Imaging Systems). From the instantaneous RV inner-surface meshes, we calculated the RV shape-index (SI), a dimensionless metric that describes the local shape of the surface and which is not directly affected by volume (see Supplementary material online). Values of SI of +1 account for a convex spherical geometry bending towards the RV, whereas values of −1 account for a symmetric spherical concave geometry bending towards the LV.26 The SI and the transeptal pressure difference are reported at the time of Pmin, the closest time to V0 (see Results).
2.5. Statistical analysis
Experimental data were analysed using linear mixed-effects models, χ2, and paired t-tests where appropriate. The effects of interventions on haemodynamic indices were calculated as the fixed-effect estimates and their 95% confidence intervals. Differences among phases were tested using Dunnett's contrasts against baseline measurements. The association between quantitative physiological variables was also assessed by linear mixed-effects models as well as within-subject correlation coefficients accounting for repeated measures (Rrm). The intraclass regression coefficient (Ric) and its 95% confidence interval were used to assess agreement. Variables are described as mean ± standard deviation. Statistical analysis was performed using R v.3.0 (R Development Core Team, packages: lme4, rms, ggplot2, irr, multcomp) and P-values < 0.05 were considered significant.
3. Results
3.1. Global haemodynamics
Esmolol significantly lowered heart-rate, cardiac output, and dP/dt, whereas dobutamine produced the opposite effects (Table 1). Importantly, both drugs modified RV operating volumes as demonstrated by significant changes in ESV [from 22 (95% CI: 18–27) mL at baseline to 25 (20–29) mL, during esmolol, and to 19 (14–23) mL during dobutamine, P < 0.05 for both]. Acute volume infusion raised ESV up to 26 (21–30) mL (P < 0.05 vs. baseline). Endotoxin infusion led to a rapid increase in RV afterload as demonstrated by a progressive increase in Ea from baseline values of 1.0 (0.7–1.2) mmHg/mL to 1.8 (1.5–2.1) mmHg/mL during early infusion and 2.4 (2.2–2.7) mmHg/mL during the late phase (P < 0.05 for both vs. baseline). This acute pressure overload also resulted in RV dilatation and larger ESVs [29 (24–33) and 40 (35–45) mL during early and late endotoxin phases, respectively; P < 0.05 for both vs. baseline]. Both mean RAP and RVEDP significantly increased during volume and endotoxin infusions (P < 0.05 for all). Negative values of Pmin were found in 36% of data sets at baseline, 13% during esmolol infusion, 41% during dobutamine, never during acute volume overload, and in 15 and 11% of the data sets during early and late endotoxin infusion, respectively (P < 0.001).
Table 1.
Haemodynamic data and RV systolic chamber function
| Baseline | Esmolol | Dobutamine | Volume | Endotoxin (early) | Endotoxin (late) | |
|---|---|---|---|---|---|---|
| Number of animals | 13 | 12 | 11 | 11 | 11 | 12 |
| Number of runs | 47 | 45 | 32 | 25 | 39 | 36 |
| Heart rate (b.p.m.) | 92 (86–99) | 85 (78–91)* | 118 (111–125)* | 99 (92–106) | 89 (82–96) | 114 (107–121)* |
| Cardiac output (L/min) | 2.3 (2.0–2.5) | 1.9 (1.6–2.2)* | 2.6 (2.4–2.9)* | 2.9 (2.6–3.2)* | 2.2 (1.9–2.4) | 2.3 (2.1–2.6) |
| RV end-diastolic volume (mL) | 47 (41–53) | 47 (41–54) | 42 (35–48)* | 56 (49–62)* | 53 (47–59)* | 61 (55–67)* |
| RV end-systolic volume (mL) | 22 (18–27) | 25 (20–29)* | 19 (14–23)* | 26 (21–30)* | 29 (24–33)* | 40 (35–45)* |
| RV ejection fraction | 0.53 (0.49–0.57) | 0.49 (0.45–0.53)* | 0.56 (0.52–0.6) | 0.55 (0.51–0.6) | 0.46 (0.42–0.5)* | 0.36 (0.32–0.4)* |
| Pressures | ||||||
| LV Pmax (mmHg) | 114 (108–120) | 103 (96–109)* | 138 (131–144)* | 103 (96–110)* | 102 (96–109)* | 76 (69–83)* |
| Mean RAP (mmHg) | 2.7 (1.3–4) | 3.3 (2–4.7) | 1.6 (0.2–3) | 10.3 (8.8–11.8)* | 5.5 (4.2–6.9)* | 3.9 (2.5–5.3)* |
| RV Pmin (mmHg) | 0.9 (0–1.8) | 1.4 (0.4–2.3) | −0.1 (−1.1–0.9) | 7.7 (6.6–8.7) | 3.4 (2.5–4.4) | 2.3 (1.3–3.3) |
| RVEDP (mmHg) | 5.4 (4.3–6.6) | 6.7 (5.6–7.9)* | 4.2 (2.9–5.4) | 14.9 (13.6–16.2)* | 9.2 (8–10.3)* | 6.8 (5.6–8.0)* |
| RV Pmax (mmHg) | 29.9 (25.6–34.1) | 28.4 (24.1–32.7) | 35.9 (31.4–40.4)* | 42.1 (37.4–46.7)* | 41.8 (37.4–46.2)* | 45.2 (40.8–49.7)* |
| RVESP (mmHg) | 23.4 (19.6–27.2) | 24 (20.2–27.8) | 29 (25.0–32.9)* | 34.9 (30.8–38.9)* | 39.7 (35.9–43.6)* | 44.8 (40.9–48.6)* |
| Pmin < 0 (% of data sets) | 36 | 13 | 41 | 0 | 15 | 11 |
| RV systolic chamber function | ||||||
| dP/dtmax (mmHg/s) | 617 (506–728) | 528 (416–639)* | 1187 (1070–1304)* | 798 (677–919)* | 671 (557–784) | 723 (608–837)* |
| Total RV SW (mmHg · mL) | 548 (453–643) | 482 (385–578) | 715 (616–814)* | 825 (719–931)* | 717 (618–816)* | 656 (556–756)* |
| Preload-recruitable SW (mmHg) | 12.3 (6.3–18.3) | 8.3 (2.3–14.2) | 21.9 (15.7–28.1)* | 14 (7.5–20.5) | 13.9 (7.3–20.5) | 18.3 (12.2–24.4)* |
| Emax (mmHg/mL) | 0.8 (0.6–1.0) | 0.9 (0.7–1.1) | 1.1 (0.8–1.3)** | 1.4 (1.1–1.6)* | 0.9 (0.7–1.2) | 1.1 (0.8–1.3)* |
| Ea (mmHg/mL) | 1.0 (0.7–1.2) | 1.1 (0.9–1.4) | 1.3 (1.0–1.6)* | 1.2 (0.9–1.5) | 1.8 (1.5–2.1)* | 2.4 (2.2–2.7)* |
| Emax /Ea | 0.8 (0.7–0.9) | 0.8 (0.7–0.9) | 0.9 (0.8–1.0) | 1.0 (0.9–1.1)* | 0.5 (0.4–0.7)* | 0.5 (0.4–0.6)* |
LV Pmax, LV peak pressure; RV Pmin, minimum diastolic RV pressure; RVEDP, RV end-diastolic pressure; RVESP, RV end-systolic pressure; dP/dtmax, peak time derivative of RV pressure; SW, stroke work; Emax, peak systolic elastance; Ea, pulmonary arterial elastance; Emax/Ea, RV ventriculo-arterial coupling.
*P < 0.05 vs. baseline. Early-endotoxin data of one animal undergoing the full endotoxin protocol was unreadable.
3.2. Diastolic properties
Dobutamine and volume overload significantly shortened and prolonged τ, respectively, whereas esmolol and endotoxin had no effect on τ (Table 2). The full passive PV curve below and above V0 was characterized in all phases (Figure 2). Indices of passive diastolic properties (V0, Vd, S−, Vm, and S+) were relatively stable among different phases, although slightly sensitive to volume overload (Table 2). Notice that late endotoxin infusion increased V0 by 50% [46 (40–51) mL vs. 30 (25–36) mL at baseline, P < 0.05). Correlation between S− and S+ was R = 0.69 (P < 0.001, baseline values only), and S−was in magnitude approximately two-thirds of S+ (Figure 3).
Table 2.
Indices of RV diastolic function
| Baseline | Esmolol | Dobutamine | Volume | Endotoxin (early) | Endotoxin (late) | |
|---|---|---|---|---|---|---|
| Diastolic haemodynamic data | ||||||
| Heart rate (b.p.m.) | 92 (86–99) | 85 (78–91)* | 118 (111–125)* | 99 (92–106) | 89 (82–96) | 114 (107–121)* |
| Filling time (ms) | 274 (244–303) | 319 (290–349)* | 215 (184–245)* | 208 (177–239)* | 261 (231–291) | 166 (136–196)* |
| IVRT (ms) | 37 (30–43) | 43 (36–49) | 35 (27–42) | 39 (31–46) | 57 (50–64)* | 57 (50–64)* |
| -dP/dtmin (mmHg/s) | −416 (−477 to −355) | −412 (−473 to −350) | −605 (−670 to −540)* | −470 (−538 to −403) | −554 (−617 to −492)* | −588 (−651 to −525)* |
| Diastolic properties | ||||||
| τ (ms) | 41 (35–47) | 38 (33–44) | 29 (23–35)* | 65 (58–71)* | 40 (34–46) | 46 (40–51) |
| V0 (mL) | 30 (25–36) | 29 (23–34) | 27 (22–33) | 31 (26–37) | 32 (26–37) | 45 (40–51)* |
| S− (mmHg) | 7.9 (5.6–10.2) | 8.4 (6.0–10.7) | 5.7 (3.1–8.3) | 10.5 (7.7–13.2) | 8.6 (6.1–11) | 13 (10.6–15.5)* |
| Vd (mL) | 2 (0–3) | 2 (0–3) | 3 (2–5) | 8 (6–10)* | 2 (1–4) | 3 (2–5) |
| S+ (mmHg) | 11.1 (8.9–13.3) | 14.2 (12.0–16.5)* | 9 (6.5–11.5) | 23.1 (20.4–25.9)* | 11 (8.7–13.4) | 8.1 (5.7–10.5) |
| Vm (mL) | 71 (60–81) | 75 (65–86) | 69 (58–79) | 87 (75–98)* | 73 (62–83) | 79 (68–89) |
| dP/dV at V0 (mmHg/mL) | 0.29 (0.22–0.35) | 0.32 (0.25–0.39) | 0.24 (0.16–0.31) | 0.52 (0.44–0.60)* | 0.29 (0.22–0.36) | 0.28 (0.21–0.36) |
IVRT, isovolumic relaxation time; dP/dtmin, minimal derivative of RV pressure with respect to time; τ, time-constant of relaxation; V0, RV volume at zero passive pressure; S−, constant of diastolic elastic recoil; S+, constant of passive stiffness; Vm, RV maximal achievable volume; Vd, RV minimum dead volume; dP/dV at V0, derivative of RV pressure with respect to volume at V0.
*P < 0.05 vs. baseline.
Figure 3.

Relative role of RV passive mechanical properties below (elastic restoring forces) and beyond (passive stiffness) V0. Data shows baseline values for each animal (n = 13). Individual data and boxplots are shown for the distribution of S− and S+ (A), the slope of the passive PV curve (dP/dV) at the instants of end-systole (@ ESV) and end-diastole (@ EDV) (B), and the relative operative deformation from V0 (C). See text for details.
3.3. Impact of elastic restoring forces on early filling
We quantified the effects of restoring forces on diastolic pressures of spontaneous beats prior to IVC occlusion. RV contraction below V0 was found in 96% of data sets at the baseline phase. V0 was reached close to the instant of Pmin (difference = 4 ± 52 ms, P = 0.2). This allowed restoring suction to facilitate chamber depressurization, by generating −2.7 (−3.8 to −1.6) mmHg of passive pressure in the baseline state at the time PVC (Figure 4 and Table 3). Small volumetric changes between PVC and TVO slightly lowered passive pressure to −2.2 (−3.3 to −1.2) mmHg by the onset of filling (baseline phase). This passive pressure facilitated entering 40 (30–50) % of stroke volume before Pmin (baseline phase; Table 3). Notice that the negative passive pressure most frequently did not translate to negative net (measured) pressure during early filling because of active relaxation pressure was predominant (Figure 4).
Figure 4.

Scatterplots and boxplots of active (red) and passive (blue) RV diastolic pressure components measured at the times of pulmonary valve closure, tricuspid valve opening, minimal RV pressure, and end-diastole at baseline (n = 13), esmolol (n = 12), dobutamine (n = 11), volume overload (n = 11), as well as early (n = 11) and late (n = 12) phases after endotoxin administration. Measured pressure values of RV (black) and RA (grey) pressure are overlaid. Values have been plotted at average time values for each of these events. *P < 0.05 vs. baseline values for the same time event.
Table 3.
Impact of passive diastolic forces on RV filling pressures
| Baseline | Esmolol | Dobutamine | Volume | Endotoxin (early) | Endotoxin (late) | |
|---|---|---|---|---|---|---|
| Elastic suction pressure present (% of runs) | 96 | 80 | 88 | 100 | 77 | 83** |
| Filling fraction while V < V0 | 0.4 (0.3–0.5) | 0.2 (0.1–0.4)* | 0.5 (0.3–0.6) | 0.2 (0.1–0.4)* | 0.2 (0.0–0.3)* | 0.4 (0.2–0.5) |
| Active diastolic pressure | ||||||
| At pulmonary valve closing (mmHg) | 18.8 (16.2–21.4) | 17.1 (14.5–19.8) | 18.0 (15.2–20.7) | 30.3 (27.4–33.3)* | 26.7 (23.9–29.4)* | 33.8 (31.0–36.5)* |
| At tricuspid valve opening (mmHg) | 6.3 (4.6–7.9) | 4.8 (3.2–6.5) | 5.6 (3.9–7.3) | 15.9 (14.1–17.7)* | 7.5 (5.8–9.2) | 8.1 (6.4–9.8)* |
| Passive diastolic pressure | ||||||
| At pulmonary valve closing (mmHg) | −2.7 (−3.8 to −1.6) | −1.0 (−2.1 to 0.1)* | −2.7 (−3.8 to −1.5) | −4.1 (−5.4 to −2.8) | −1.2 (−2.3 to 0.0)* | −2.4 (−3.6 to −1.2) |
| At tricuspid valve opening (mmHg) | −2.2 (−3.3 to −1.2) | −0.7 (−1.8 to 0.4)* | −1.8 (−3.0 to −0.6) | −3 (−4.3 to −1.8) | −0.1 (−1.2 to 1.0)* | −1.9 (−3.1 to −0.8) |
V0, RV volume at zero passive pressure.
*P < 0.05 vs. baseline.
**P < 0.05 among the six groups.
During interventions, changes in restoring suction pressure were mostly mediated by the position of operating volumes relative to V0. Thus, aforementioned increases in ESV caused by esmolol and early endotoxin infusions significantly lowered suction pressure at TVO (Table 3). However, during late endotoxin infusion, restoring forces reappeared, despite operating volumes were higher. This was possible because the passive PV curve shifted to the right due to a significant increase in V0 (Figure 2 and Table 3). Notice that the right-shift of the passive PV curve placed operating volumes on lower passive pressures than if the same volume values were to be extrapolated to the PV passive curves recorded at baseline or early-endotoxin; not only passive suction pressure was recovered but also EDP was much lower than if placed on the original curve. Restoring suction pressure closely correlated both with S− (Rrm = −0.68, P < 0.001) and with the relative volume difference between V0 and ESV (Rrm = −0.84, P < 0.001).
Stronger restoring suction pressure was related to a shorter IVRT (Rrm = 0.30, P < 0.001) and a lower mean RAP (Rrm = 0.56; P < 0.001). In fact, data sets with restoring suction showed 23% lower values of mean RAP than those without restoring suction (4.4 ± 3.7 vs. 5.4 ± 1.5 mmHg, respectively, P = 0.02; pooled data). Pmin was achieved 20 ms earlier in data sets with elastic suction (P < 0.001). Nevertheless, the fraction of stroke volume entering before Pmin strongly correlated with restoring suction pressure (Rrm = −0.71, P < 0.001, all phases pooled-data). This favourable effect of elastic recoil on early filling was corroborated by the work decomposition analysis (see Supplementary material online).
3.4. The effects of septal curvature and ventricular interdependence
To address the role of ventricular interpendence we first assessed whether changes in V0 were related to septal conformational changes. Because V0 is achieved close to the time of Pmin (discussed earlier), we compared values in this particular instant. Within animals, V0 moderately correlated with septal SI (Rrm = −0.67; P < 0.001). And secondly, we analysed whether interventricular pressures could justify changes in septal geometry. We observed a weak but significant correlation between transeptal pressure gradient and SI at Pmin (Rrm = 0.28; P = 0.003). Figure 5 shows a representative example of these relationships.
Figure 5.

Impact of ventricular interdependence on elastic restoring forces. (A and B) Example of shape index (SI) analysis at the instant of minimum diastolic RV pressure (Pmin) at baseline (A) and during late endotoxin infusion (B). Local 3-dimensional values of SI are shown for each vertex of the septum. A value of SI +1 accounts for a convex spherical geometry bending towards the RV, whereas a value of −1 accounts for a symmetric spherical concave geometry bending towards the LV. Changes in average SI values for the whole septum take place in parallel to changes in V0. (C) Simultaneous RV and LV pressures during the same two phases showing the inversion in the trans-septal pressure difference from baseline to the late endotoxin phase. Arrowheads indicate the time of Pmin. (D) The RV passive stiffness pressure curves obtained in the same animal following changes in V0.
4. Discussion
The major finding of our study is that strong elastic restoring forces contribute to RV filling. By means of elastic restoration acting as a spring-like mechanism, the RV makes use of contraction energy to facilitate chamber depressurization and to build-up a significant amount of the filling pressure gradient. Remarkably, we observed that the RV almost systematically operates below V0 to facilitate diastolic function. Inotropic modulation, volume, and acute pressure overload modified elastic restoring forces by shifting operating volumes relative to V0. V0 increased during RV failure induced by sustained pressure-overload, allowing the RV to recover elastic restoring forces despite severe dilatation. This was related to modifications in septal geometry as measured by 3-dimensional ultrasound. Hence, by its effect on septum conformation, the interventricular pressure gradient during early diastole conditions RV elastic restoring forces. This source of direct diastolic ventricular interdependence may be clinically relevant in extreme overload situations of either ventricle.
4.1. Mechanisms of RV filling
Early filling is a complex phenomenon with distinct sources of force simultaneously interacting to generate the driving filling pressure gradient (RA–RV pressure).27 By decomposing RV pressure components, we clarified how these sources interact during early filling. Until relaxation is completed, any degree of residual ‘active’ pressure is always positive and lowers the filling pressure gradient. For this reason, rapid filling heavily depends on the rate of relaxation.27 Due to strong elastic restoring forces, ‘passive’ pressure is negative most of the time during rapid filling, therefore increasing the filling pressure gradient. Notice that only whenever passive pressure is larger (in absolute terms) than active pressure, total (measured) RV diastolic pressure will become negative.
The existence of negative RV pressures was described long time ago,28 and it is known not to be caused by negative intra-thoracic pressure.11 We demonstrate that, whenever present (36% of cases in the present study), negative RV pressures are the testimony of strong elastic restoring forces which are actively pulling flow from the RA.10 However, this a a very insensitive marker of restoring suction pressure since, as we also demonstrate, restoring forces most frequently act without reaching the level enough to generate negative total RV pressure.
We showed that once the tricuspid valve opens, restoring forces are a major source of the work developed by the heart during RV rapid filling. Thus, without the mechanism of elastic restoration, TVO is delayed until active pressure falls below atrial pressure and the RV fills exclusively by means of atrial emptying work. As shown in our study, this leads to longer IVRTs, smaller rapid filling fractions, and higher RA pressures. However, even in such cases, ongoing relaxation lowers RV pressure despite volume is increasing during rapid filling. Thus, a negative dP/dV does not prove the existence of elastic restoring forces. Instead, specific force uncoupling methods as used in the present study are required for this purpose.
4.2. Elastic restoration and stiffness
Interestingly, RV restoring suction pressures were considerably stronger than previously described in the LV, typically in the range of −1 to 0 mmHg.8,29 The reasons for this difference can be inferred by comparing constitutive parameters measured in both ventricles using identical methodology.16 Previously reported LV passive constants, S− and S+, were significantly lower than RV values measured in the present study; so was LV dP/dV at V0.16 Furthermore, operative volumes with respect to V0 seem to be different between both ventricles. At baseline we found that the LV V0 – ESV difference was close to 10% of V0,16 whereas in the present study this difference was measured close to 30% in the RV. Due to the nonlinearity of the passive PV relationship, such an augmented compression below V0 has a great impact on restoring forces. These comparisons suggest that, in the RV, passive mechanical properties are optimized to potentiate restoring forces and facilitate working as a suction pump.30 Conversely, in the LV they seem to be adjusted towards lowering stiffness and facilitating emptying.
Because wall thickness is a major determinant of chamber passive properties, higher stiffness constants found in the RV may seem paradoxical. However, recent studies have shown that passive mechanical properties of ventricular chambers in vivo are more complex than initially suggested. Particularly relevant is the effect of large scale 3D orientation of myocardial fibres and layers which may modulate chamber torsion and detorsion.13 Additionally, intraventricular fluid dynamics significantly modulate global chamber operative stiffness.31 Further studies are warranted to clarify the relative contribution of each of these properties on RV diastolic chamber mechanics.
4.3. Active relaxation vs. the isovolumic relaxation phase
At the molecular level, relaxation is triggered by removal of calcium from the cytosol and from troponin C. A vast number of factor are known to modulate troponin C switch-off kinetics.9 Among these, protein kinase A membrane and sarcomeric pathways mediate the lusotropic effects of acute beta-adrenergic receptor stimulation. At the chamber level, the rate of myocardial relaxation is typically measured by fitting the depressurization transient curve during the isovolumic phase. Because passive forces cannot be uncoupled using this method, elastic restoration is sometimes considered within the process of relaxation itself.9 Our study illustrates that new insight is provided when physical models are improved; in addition to its direct effect on calcium turnover, we found that beta-adrenergic receptor modulation impacts chamber diastolic function by its inotropic effect which conditions operating volumes. Further studies using refined computational models will probably facilitate translating the well-characterized molecular aspects of relaxation to the clinical setting.
4.4. Interdependence, geometry, and elastic recoil
Indirect interdependence designates the impact of LV haemodynamics on RV physiology mediated by the retrograde transmission of pressure across pulmonary vasculature. Direct interdependence classically accounts for the impact of volume-dependent conformational changes on the passive stiffness curves of either ventricles mediated by changes in septal bulging.32 This is a widely recognized source of modifications in LV and RV passive stiffness mediated by acute distension of the opposite ventricle. However, fitting the negative and positive parts of the PV curve, we found that V0 was the only parameter that significantly changed during RV extreme acute pressure overload. We showed that V0 depends on the degree of septal bulging induced by the interventricular pressure balance. Notice that this shift in the position of the PV curve cannot be detected by simpler mathematical models that do not include a zero-pressure volume intercept.33 But most importantly, changes in V0 explain the sensitivity of elastic recoil to direct interdependence, a mechanism that preserves restoring forces in a situation of extreme acute overload. One would expect severe ventricular dilatation to prevent contraction below V0. However, by displacing the septum towards the LV, the RV is ‘pre-stretched’ at zero passive pressure and its V0 increases. Making use of this mechanism, the RV re-adjusted itself to recover elastic recoil at advanced stages of acute overload. Interestingly, this mechanism also works in the opposite direction: acute LV dilatation may impair RV diastolic function by impairing restoring forces and not only by increasing stiffness.33
4.5. Clinical implications
The demonstration of strong restoring forces suggests that chamber mechanics have become optimized for the RV to work not only as a forward pump for the pulmonary circulation, but also as a suction pump that drives central venous return. Thus, deleterious effects take place when suction is impaired. Long-standing Fontan circulation is a paradigmatic scenario in which the lack of suction function leads to severe complications due to the chronic venous hypertension in the liver and splanchnic territories. Currently a promising field of pre-clinical research, the findings of our study encourage incorporating suction pressure on flow-assistance devices in the Fontan circulation.34
Our findings may have important clinical implications in right and left heart failure syndromes. Elastic restoring forces recover energy expended during contraction. Thus, when working under compression, passive structural elements (titin, extracellular matrix proteins, and large-scale fibre arrangements) impose a certain amount of tension to the myocyte. This ‘intrinsic’ load is functionally attached in parallel to the sarcomere, and sums to the ‘extrinsic’ (conventional, blood pressure-mediated) afterload connected in series. As inferred from our pressure decomposition results, intrinsic stresses accounts for ∼10% of total afterload of the RV. How intrinsic stresses are sensed and handled by the myocyte's molecular mechanisms is not well understood.12 Our observations under esmolol confirm that depression of myocardial contractility abolishes restoring forces by simply increasing end-systolic volume.13 Thus, acting as a source of systolic–diastolic interaction, ‘pure’ systolic RV dysfunction may induce disproportionately high RAP by this mechanism.
The sensitivity of elastic recoil to septal geometry contributes to explain why septal curvature is a well-established prognostic indicator in acute pulmonary embolism.35 Because direct interdependence is bidirectional,33,36 RV dilatation may also impair LV diastolic function due to this mechanism. Thereby syndromes leading to LV dilatation are expected to aggravate systemic congestion by reducing RV recoil-mediated filling. We believe this mechanism contributes to the abnormalities in RV filling induced by isolated LV ischaemia.37 Future studies are warranted to clarify how chronic overload conditions of either ventricle modify these reciprocal interactions.
In the field of cardiac resynchronization therapy, it has been recently suggested that LV pacing may acutely improve LV diastolic function by increasing the left-to-right transseptal pressure gradient during early diastole.38 However, our results suggest that this potential benefit may cause opposite effects on diastolic function of the RV. Consequently, we believe the long-term clinical consequences of cardiac asynchrony and resynchronization on RV diastolic mechanics deserves further research. The methodology of the present study, combining conventional PV data with modern signal processing techniques, is applicable in the clinical setting16 and may be particularly useful for this purpose.
Pharmacological modulation of interstitial remodelling to reduce stiffness has been identified as potential therapeutic target for the treatment of heart failure.39,40 However, our study suggests that reducing chamber stiffness may have associated reciprocal effects on RV elastic recoil which need to be considered. The high correlation we found between S− and S+ demonstrates that chamber passive mechanics under compression and expansion are tightly related. Thus, steering the development of these drugs must take into account their effects both on stiffness and restoring forces.
4.6. Limitations
A complete understanding of direct ventricular interdependence requires complex modelling of the time-varying rigidity of the interventricular septum which was beyond the scope of this study. Instead, we only attempted to analyse the pre-stretch effect the interventricular pressure balance generated on septal geometry, its impact on V0, and consequently its implications on restoring forces. We also did not take into account additional potential confounders of RV diastolic function such as pericardial constraint, indirect ventricular interdependence, myocardial viscoelasticity, intraventricular flow-dynamics,31 and non-volume dependent untwisting deformation which may modify, to some degree, the PV relationship.
4.7. Conclusions
Strong elastic restoring forces provide the RV with a useful mechanism to forward contraction energy from systole to early filling. The magnitude of restoring forces is influenced by the inotropic state and RV geometrical changes caused by direct ventricular interdependence.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This study was supported by grants, PI12/02885, RD12/0042 (Red de Investigación Cardiovascular), BA11/00067 (to J.B.), CM12/00273 (to C.P.V.), and CM11/00221 (to T.M.) from the Instituto de Salud Carlos III, Spain and partially funded by project TEC-2010-21619-C04-03 from the Ministerio de Economía y Competitividad, Spain. C.P.V. and T.M. were partially supported by grants from the Fundación para Investigación Biomédica Gregorio Marañón, Spain. P.M.L. was partially supported by RD12/0042 and NIH grant 2R21 HL108268-01 (to J.C.A.).
References
- 1.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 2006;114:1883–1891. [DOI] [PubMed] [Google Scholar]
- 2.Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation 2008;117:1717–1731. [DOI] [PubMed] [Google Scholar]
- 3.McCabe C, White PA, Hoole SP, Axell RG, Priest AN, Gopalan D, Taboada D, Mackenzie Ross R, Morrell NW, Shapiro LM, Pepke-Zaba J. Right ventricular dysfunction in chronic thromboembolic obstruction of the pulmonary artery: a pressure-volume study using the conductance catheter. J Appl Physiol 2014;116:355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rain S, Handoko ML, Trip P, Gan CT, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CA, Marcus JT, Dorfmuller P, Guignabert C, Humbert M, Macdonald P, Dos Remedios C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk-Noordegraaf A, van der Velden J, de Man FS. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation 2013;128:2016–2025. [DOI] [PubMed] [Google Scholar]
- 5.Leeuwenburgh BP, Steendijk P, Helbing WA, Baan J. Indexes of diastolic RV function: load dependence and changes after chronic RV pressure overload in lambs. Am J Physiol Heart Circ Physiol 2002;282:H1350–H1358. [DOI] [PubMed] [Google Scholar]
- 6.Pasipoularides AD, Shu M, Shah A, Glower DD. Right ventricular diastolic relaxation in conscious dog models of pressure overload, volume overload, and ischemia. J Thorac Cardiovasc Surg 2002;124:964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu CM, Sanderson JE, Chan S, Yeung L, Hung YT, Woo KS. Right ventricular diastolic dysfunction in heart failure. Circulation 1996;93:1509–1514. [DOI] [PubMed] [Google Scholar]
- 8.Nikolic S, Yellin EL, Tamura K, Vetter H, Tamura T, Meisner JS, Frater RW. Passive properties of canine left ventricle: diastolic stiffness and restoring forces. Circ Res 1988;62:1210–1222. [DOI] [PubMed] [Google Scholar]
- 9.Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res 2004;94:1533–1542. [DOI] [PubMed] [Google Scholar]
- 10.Remme EW, Opdahl A, Smiseth OA. Mechanics of left ventricular relaxation, early diastolic lengthening, and suction investigated in a mathematical model. Am J Physiol Heart Circ Physiol 2011;300:H1678–H1687. [DOI] [PubMed] [Google Scholar]
- 11.Sabbah HN, Stein PD. Negative diastolic pressure in the intact canine right ventricle. Evidence of diastolic suction. Circ Res 1981;49:108–113. [DOI] [PubMed] [Google Scholar]
- 12.Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res 2008;77:637–648. [DOI] [PubMed] [Google Scholar]
- 13.Bell SP, Nyland L, Tischler MD, McNabb M, Granzier H, LeWinter MM. Alterations in the determinants of diastolic suction during pacing tachycardia. Circ Res 2000;87:235–240. [DOI] [PubMed] [Google Scholar]
- 14.Dong SJ, Smith ER, Tyberg JV. Changes in the radius of curvature of the ventricular septum at end diastole during pulmonary arterial and aortic constrictions in the dog. Circulation 1992;86:1280–1290. [DOI] [PubMed] [Google Scholar]
- 15.Yellin EL, Hori M, Yoran C, Sonnenblick EH, Gabbay S, Frater RW. Left ventricular relaxation in the filling and nonfilling intact canine heart. Am J Physiol 1986;250:H620–H629. [DOI] [PubMed] [Google Scholar]
- 16.Bermejo J, Yotti R, Perez del Villar C, del Alamo JC, Rodriguez-Perez D, Martinez-Legazpi P, Benito Y, Antoranz JC, Desco MM, Gonzalez-Mansilla A, Barrio A, Elizaga J, Fernandez-Aviles F. Diastolic chamber properties of the left ventricle assessed by global fitting of pressure-volume data: improving the gold standard of diastolic function. J Appl Physiol 2013;115:556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burkhoff D, Mirsky I, Suga H. Assessment of systolic and diastolic ventricular properties via pressure-volume analysis: a guide for clinical, translational, and basic researchers. Am J Physiol Heart Circ Physiol 2005;289:H501–H512. [DOI] [PubMed] [Google Scholar]
- 18.Cortina C, Bermejo J, Yotti R, Desco MM, Rodriguez-Perez D, Antoranz JC, Rojo-Alvarez JL, Garcia D, Garcia-Fernandez MA, Fernandez-Aviles F. Noninvasive assessment of the right ventricular filling pressure gradient. Circulation 2007;116:1015–1023. [DOI] [PubMed] [Google Scholar]
- 19.Schmidhammer R, Wassermann E, Germann P, Redl H, Ullrich R. Infusion of increasing doses of endotoxin induces progressive acute lung injury but prevents early pulmonary hypertension in pigs. Shock 2006;25:389–394. [DOI] [PubMed] [Google Scholar]
- 20.Yotti R, Bermejo J, Desco MM, Antoranz JC, Rojo-Alvarez JL, Cortina C, Allue C, Rodriguez-Abella H, Moreno M, Garcia-Fernandez MA. Doppler-derived ejection intraventricular pressure gradients provide a reliable assessment of left ventricular systolic chamber function. Circulation 2005;112:1771–1779. [DOI] [PubMed] [Google Scholar]
- 21.Zhang QB, Sun JP, Gao RF, Lee AP, Feng YL, Liu XR, Sheng W, Liu F, Yang XS, Fang F, Yu CM. Feasibility of single-beat full-volume capture real-time three-dimensional echocardiography for quantification of right ventricular volume: validation by cardiac magnetic resonance imaging. Int J Cardiol 2013;168:3991–3995. [DOI] [PubMed] [Google Scholar]
- 22.Yotti R, Bermejo J, Benito Y, Sanz-Ruiz R, Ripoll C, Martinez-Legazpi P, Perez del Villar C, Elizaga J, Gonzalez-Mansilla A, Barrio A, Banares R, Fernandez-Aviles F. Validation of noninvasive indices of global systolic function in patients with normal and abnormal loading conditions: a simultaneous echocardiography pressure-volume catheterization study. Circ Cardiovasc Imaging 2014;7:164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brimioulle S, Wauthy P, Ewalenko P, Rondelet B, Vermeulen F, Kerbaul F, Naeije R. Single-beat estimation of right ventricular end-systolic pressure-volume relationship. Am J Physiol Heart Circ Physiol 2003;284:H1625–H1630. [DOI] [PubMed] [Google Scholar]
- 24.Yellin EL, Meisner JS. Physiology of diastolic function and transmitral pressure-flow relations. Cardiol Clin 2000;18:411–433. [DOI] [PubMed] [Google Scholar]
- 25.Matsubara H, Takaki M, Yasuhara S, Araki J, Suga H. Logistic time constant of isovolumic relaxation pressure-time curve in the canine left ventricle. Better alternative to exponential time constant. Circulation 1995;92:2318–2326. [DOI] [PubMed] [Google Scholar]
- 26.Koenderink JJ, Van Doorn AJ. Surface shape and curvature scales. Image Vision Comput 1992;10:557–564. [Google Scholar]
- 27.Sun Y, Belenkie I, Wang JJ, Tyberg JV. Assessment of right ventricular diastolic suction in dogs with the use of wave intensity analysis. Am J Physiol Heart Circ Physiol 2006;291:H3114–H3121. [DOI] [PubMed] [Google Scholar]
- 28.Bloom WL, Ferris EB. Elastic recoil of the heart as a factor in diastolic filling. Trans Assoc Am Physician 1956;69:200–206. [PubMed] [Google Scholar]
- 29.Woodard JC, Chow E, Farrar DJ. Isolated ventricular systolic interaction during transient reductions in left ventricular pressure. Circ Res 1992;70:944–951. [DOI] [PubMed] [Google Scholar]
- 30.Robinson TF, Factor SM, Sonnenblick EH. The heart as a suction pump. Sci Am 1986;254:84–91. [DOI] [PubMed] [Google Scholar]
- 31.Martinez-Legazpi P, Bermejo J, Benito Y, Yotti R, Perez Del Villar C, Gonzalez-Mansilla A, Barrio A, Villacorta E, Sanchez PL, Fernandez-Aviles F, Del Alamo JC. Contribution of the diastolic vortex ring to left ventricular filling. J Am Coll Cardiol 2014;64:1711–1721. [DOI] [PubMed] [Google Scholar]
- 32.Santamore WP, Dell'Italia LJ. Ventricular interdependence: significant left ventricular contributions to right ventricular systolic function. Prog Cardiovasc Dis 1998;40:289–308. [DOI] [PubMed] [Google Scholar]
- 33.Weber KT, Janicki JS, Shroff S, Fishman AP. Contractile mechanics and interaction of the right and left ventricles. Am J Cardiol 1981;47:686–695. [DOI] [PubMed] [Google Scholar]
- 34.Derk G, Laks H, Biniwale R, Patel S, De LaCruz K, Mazor E, Williams R, Valdovinos J, Levi DS, Reardon L, Aboulhosn J. Novel techniques of mechanical circulatory support for the right heart and Fontan circulation. Int J Cardiol 2014;176:828–832. [DOI] [PubMed] [Google Scholar]
- 35.Jardin F, Dubourg O, Gueret P, Delorme G, Bourdarias JP. Quantitative two-dimensional echocardiography in massive pulmonary embolism: emphasis on ventricular interdependence and leftward septal displacement. J Am Coll Cardiol 1987;10:1201–1206. [DOI] [PubMed] [Google Scholar]
- 36.Baker AE, Dani R, Smith ER, Tyberg JV, Belenkie I. Quantitative assessment of independent contributions of pericardium and septum to direct ventricular interaction. Am J Physiol 1998;275:H476–H483. [DOI] [PubMed] [Google Scholar]
- 37.Fabbiocchi F, Galli C, Sganzerla P, Montorsi P, Loaldi A, de Cesare N, Bartorelli AL. Changes in right ventricular filling dynamics during left anterior descending, left circumflex and right coronary artery balloon occlusion. Eur Heart J 1997;18:1432–1437. [DOI] [PubMed] [Google Scholar]
- 38.Boe E, Russell K, Remme EW, Gjesdal O, Smiseth OA, Skulstad H. Cardiac responses to left ventricular pacing in hearts with normal electrical conduction: beneficial effect of improved filling is counteracted by dyssynchrony. Am J Physiol Heart Circ Physiol 2014;307:H370–H378. [DOI] [PubMed] [Google Scholar]
- 39.Tarone G, Balligand JL, Bauersachs J, Clerk A, De Windt L, Heymans S, Hilfiker-Kleiner D, Hirsch E, Iaccarino G, Knoll R, Leite-Moreira AF, Lourenco AP, Mayr M, Thum T, Tocchetti CG. Targeting myocardial remodelling to develop novel therapies for heart failure: A position paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur J Heart Fail 2014;16:494–508. [DOI] [PubMed] [Google Scholar]
- 40.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008;456:980–984. [DOI] [PubMed] [Google Scholar]