

Abstract
Aims
Immune system activation is a common feature of hypertension pathogenesis. However, the mechanisms that initiate this activation are not well understood. Innate immune system recognition and response to danger are becoming apparent in many cardiovascular diseases. Danger signals can arise from not only pathogens, but also damage-associated molecular patterns (DAMPs). Our first hypothesis was that the DAMP, mitochondrial DNA (mtDNA), which is recognized by Toll-like receptor 9 (TLR9), is elevated in the circulation of spontaneously hypertensive rats (SHR), and that the deoxyribonuclease enzymes responsible for its degradation have decreased activity in SHR. Based on these novel SHR phenotypes, we further hypothesized that (i) treatment of SHR with an inhibitory oligodinucleotide for TLR9 (ODN2088) would lower blood pressure and that (ii) treatment of normotensive rats with a TLR9-specific CpG oligonucleotide (ODN2395) would cause endothelial dysfunction and increase blood pressure.
Methods and results
We observed that SHR have elevated circulating mtDNA and diminished deoxyribonuclease I and II activity. Additionally, treatment of SHR with ODN2088 lowered systolic blood pressure. On the other hand, treatment of normotensive rats with ODN2395 increased systolic blood pressure and rendered their arteries less sensitive to acetylcholine-induced relaxation and more sensitive to norepinephrine-induced contraction. This dysfunctional vasoreactivity was due to increased cyclooxygenase and p38 mitogen-activated protein kinase activation, increased reactive oxygen species generation, and reduced nitric oxide bioavailability.
Conclusion
Circulating mtDNA and impaired deoxyribonuclease activity may lead to the activation of the innate immune system, via TLR9, and contribute to elevated arterial pressure and vascular dysfunction in SHR.
Keywords: Innate immunity, Mitochondrial DNA, Toll-like receptor 9, Vascular dysfunction, Hypertension
1. Introduction
Immune system activation is a hallmark characteristic of hypertension;1,2 however, the exact mechanisms that initiate this pathophysiological response are not well understood. In fact, immune system activation and inflammation have been proposed as a unifying mechanism linking the three major organ systems involved in the development of high blood pressure—the kidneys, the cardiovascular system, and the autonomic nervous system.1,2
One of the functions of the innate immune system is to recognize and respond to danger.3,4 Danger signals can arise not only from pathogens, but also from endogenous damage-associated molecular patterns (DAMPs), which can either be secreted from cells or be released unintentionally following cell injury and death.5,6 DAMPs are a broad classification for a wide range of endogenous molecules. These endogenous molecules can include extracellular matrix, cytosolic compounds, and fragments of damaged organelles.2 In the circulation, DAMPs are free to activate pattern recognition receptors on immune and non-immune cells and lead to a pro-inflammatory response.5
The toll-like receptor (TLR) family is a class of innate immune system pattern recognition receptors that recognize and respond to DAMPs.5 In naïve cells, TLR9 is localized to the endoplasmic reticulum (ER), and upon cellular recognition of its ligand in early endosomes, it traffics first to early endosomes and subsequently to tubular lysosomal compartments.7 Toll-like receptor 9 has specific affinity for unmethylated cytosine and guanine nucleotides separated by a phosphate-backbone (CpG). CpG dinucleotides are common to prokaryotic DNA but not vertebrate DNA,8 and this specificity is important for preventing TLR9-dependent autoimmunity.9
Unmethylated CpG dinucleotides are also seen in mitochondrial DNA (mtDNA), as mitochondria evolved from saprophytic bacteria and became intracellular organelles (endosymbiosis).10 Due to this bacterial ancestry, mitochondria hold many evolutionarily conserved molecular signatures, including DNA with unmethylated CpG dinucleotide repeats. Therefore, when mtDNA is released into the circulation due to cellular injury or death, it is able to elicit a sterile immune response.6,11,12 Normally, the autophagy system degrades damaged mitochondria (termed ‘mitophagy’), and this involves the sequestration of cytoplasmic contents, or endocytosis of extracellular constituents, into double-membrane vacuoles known as the endosomes. Subsequently, endosomes fuse with lysosomes, which contain deoxyribonuclease (DNase) and other catabolic enzymes, to form an autophagolysosome and degrade the sequestered content.13
Traditionally, activation of TLR9 by unmethylated CpG-DNA involves an intracytoplasmic signalling cascade that proceeds through myeloid differentiation primary response protein (MyD88), IL-1-receptor-activated kinase (IRAK), and TNF receptor associated factor (TRAF).6 This signalling leads to the up-regulation of pro-inflammatory transcription factors, including nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) and mitogen-activated protein kinase (MAPK) stimulation of activator protein 1 (AP-1), collectively promoting a Th1 immune response.14 However, recently it was also observed that TLR9 participates in an alternative non-canonical stress tolerance signalling cascade in non-immune cells (cardiomyocytes and neurons specifically).15,16 In this novel signalling pathway, TLR9 temporarily reduces energy substrates to induce cellular protection from stress. Specifically, TLR9 stimulation reduces sarco/endoplasmic reticulum Ca2+ ATPase pump 2 (SERCA2) activity, modulating calcium homeostasis and its handling between the ER and mitochondria, which leads to a decrease in mitochondrial ATP levels and the activation of cell survival protein 5′ AMP-activated protein kinase (AMPKα).
As hypertension has exaggerated levels of cell injury and death,17–19 hypertensive patients have elevated levels of cell-free CpG-DNA,20 and TLRs have been implicated in the pathophysiology of hypertension,21 we wanted to know whether the mtDNA/TLR9 signalling axis could be a novel contributing mechanism to hypertension in spontaneously hypertensive rats (SHR). We first hypothesized that the DAMP mtDNA would be elevated in the circulation of SHR and the activity of enzymes responsible for nucleic acid and mtDNA degradation (DNase I and II) would be decreased. Confirmation of these novel SHR phenotypes led us to our second hypothesis that TLR9 inhibition in SHR would lower blood pressure and that TLR9 activation in normotensive rats would cause endothelial dysfunction and increases in arterial pressure. We observed that an inhibitory oligodinucleotide for TLR9 (ODN2088) lowered blood pressure in SHR and that a CpG oligonucleotide specific for TLR9 (ODN2395) caused a significant increase in blood pressure and endothelial dysfunction in previously normotensive rats. This investigation provides a new mechanism by which activation of the innate immune system, via mtDNA and TLR9, contributes to the pathogenesis of hypertension.
2. Methods
2.1. Animals
Male and female Wistar Kyoto (WKY) rats, spontaneously hypertensive rats (SHR), and Sprague Dawley (SD) rats, 12–15 weeks old, were used for this investigation (Harlan Laboratories, Indianapolis, IN, USA). The sample size indicated per experiment (see figure legends) is the number of independent rats used, respective of strain and treatment group. All rats were maintained on a 12:12 h light–dark cycle with both rat chow and water ad libitium. All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH) and were reviewed and approved by the Institutional Animal Care and Use Committee of Georgia Regents University. All surgical procedures were undertaken on rats under isoflurane anaesthesia, administered via nose cone (5% in 100% O2). Rats were then euthanized by thoracotomy and exsanguination via cardiac puncture.
2.2. Treatments
2.2.1. Anti-hypertensive treatment
Beginning at 6 weeks of age, male and female SHR were randomly assigned to receive hydrochlorothiazide (10–55 mg/kg/day) and reserpine (0.6–4.5 mg/kg/day) (HCTZ/Res) dissolved in drinking water until they were 12 weeks old, as described and published previously.22
2.2.2. Inhibitory oligonucleotide treatment
Male WKY and SHR were randomly assigned to receive four intraperitoneal (i.p.) injections with either a TLR9 antagonist [suppressive oligonucleotide (ODN2088); 60 µg/i.p.] (Invivogen, San Diego, CA, USA) or saline control (Vehicle; Veh), on 4 consecutive days. Blood pressure was measured pre-treatment (Day 0), throughout the treatment period (Days 1–4), and post-treatment (Days 5 and 12).
2.2.3. Stimulatory CpG oligonucleotide treatment
Male SD rats were randomly assigned to receive three i.p. injections with either a TLR9 agonist [type C synthetic CpG oligonucleotide (ODN2395); 0.1 mg/i.p.] (Invivogen), nuclear DNA (Salmon Sperm DNA; 0.1 mg/i.p.) (Invivogen), or Veh, across 4 days. On Days 0 and 5, blood pressure was measured, and on Day 6 the rats were euthanized.
To confirm the specificity of ODN2395 treatment to activate TLR9 signalling in the vasculature, mesenteric arteries (MRA) from naïve (untreated) SD rats were cleaned of perivascular adipose tissue and divided into four parts. One part was flash frozen to determine basal expression. The other three parts were incubated in 37°C PSS plus 2 µmol/L ODN2395 (this concentration is equivalent to the dose the treated SD received via i.p.), for 15, 30, or 30 min in conjunction with ODN2088 (1:10 ratio of agonist: antagonist, as per the instructions provided by the manufacturer). At the conclusion of the incubation period, MRA were flash frozen, and western blots (as described below) were used to examine the expression of canonical-inflammatory TLR9 signalling and non-canonical stress tolerance TLR9 signalling (see Supplementary material online, Table S1).
2.3. DNA isolation and purification
Arterial blood was collected from the abdominal aorta of male and female WKY and SHR in tubes containing heparin. Blood was immediately centrifuged at 1500 g for 15 min at 4°C. Plasma was then stored in multiple aliquots at −80°C until analysis. Circulating DNA was extracted and purified using the QIAamp DNA Blood Mini Kit (Qiagen, Germantown, MD, USA).
2.4. Mitochondrial DNA quantification
Isolated DNA from male and female WKY and SHR was amplified and quantified using real-time (RT)-PCR (MyiQ Single-Color Real-Time PCR Detection System). The primers (Invitrogen, Grand Island, NY, USA) that were used to amplify mtDNA were cytochrome B (Cyt B) (forward 5′-TCCACTTCATCCTCCCATTC-3′ and reverse 5′-CTGCGTCGGAGTTTAATCCT-3′) and NADH dehydrogenase subunit 6 (ND6) (forward 5′-ATTAGACCCTCAAGTCTCCG-3′ and reverse 5′-TTGACTGGTTGTCTAGGGTT-3′). Bacterial 16S rRNA (forward 5′-CGTCAGCTCGTGTTGTGAAA-3′ and reverse 5′-GGCAGTCTCCTTGAGTTCC-3′) was used to confirm no bacterial contamination of the samples. Using the products obtained from RT–PCR, endpoint PCR was also performed on a 2% agarose gel to confirm our findings. Primer sequences have no significant homology with DNA found in any bacterial species published on BLAST. RT–PCR results are presented as the inverse of cycle threshold (CT) for gene amplification.
2.5. Assessment of mitochondrial membrane potential (ψm)
Flow cytometry application of the JC-1 assay technique, as described previously,23 was used as an index of mitochondrial membrane potential (ψm). JC-1 is also a surrogate marker of mitochondrial permeability transition (MPT) pore opening, which is a critical event in early apoptosis. JC-1 is a cationic dye that accumulates in mitochondria. Monomers of JC-1 dye fluoresce in the green range while JC-1 aggregates fluoresce in the red range; therefore, an increase in green fluorescence intensity represents mitochondrial swelling. Since accumulation of JC-1 in the mitochondria is dependent on the ψm, loss of ψm indicates loss of JC-1 aggregates and is manifested by decreases in red fluorescence.
Representative tissues analysed included those whose function significantly influence blood pressure (kidney, heart, and brain), as well as bone marrow and whole heparinized blood. Bone marrow was isolated from the left femur, and blood was collected in microhematocrit capillary tubes (Thermo Fisher Scientific, Waltham, MA, USA). All other tissues were homogenized through a 100 µm nylon cell strainer (Thermo Fisher Scientific), lysed of red blood cells, and resuspended in binding buffer. Samples were then incubated for 15 min in the presence of 2 µmol/L JC-1 (37°C and 5% CO2) and then washed and resuspended in cold PBS. Subsequently, labelled cells were analysed and quantified by flow cytometry with excitation at 488 nm and emission at 530 nm (green) or 590 nm (red).
2.6. Deoxyribonuclease activity
Deoxyribonuclease (DNase) I and II activity in male WKY and SHR were measured using the single radial enzyme diffusion (SRED) assay, as previously described.24,25 For DNase I, 2 μL of serum was loaded into a 2 mm thick 2% agarose gel (Thermo Fisher Scientific) on an agarose-coated polyester Gelbond film (GE Healthcare, Uppsala, Sweden). The gel contained reaction buffer (0.1 mol/L sodium calcodylate, 0.02 mol/L MgCl2, and 0.002 mol/L CaCl2; pH 6.5), 10 mg/mL Type III salmon sperm DNA (all Sigma-Aldrich), and 5 mg/mL ethidium bromide (Thermo Fisher Scientific).
For DNase II, MRA and left ventricle (LV) were homogenized in ice-cold 0.1 mol/L sodium acetate buffer, containing 20 mmol/L EDTA and 0.3 mg/mL phenylmethanesulfonylfluoride (PMSF) (pH 4.7). Homogenates were kept on ice for at least 8 h and then centrifuged at 13 000 g for 20 min. Equal amounts of protein (2–15 µg) were loaded into a 2 mm thick 2% agarose gel (Thermo Fisher Scientific) on an agarose-coated polyester Gelbond film (GE Healthcare). The gel contained reaction buffer (0.1 mol/L sodium acetate buffer and 0.02 mol/L EDTA; pH 4.7), 0.1 mg/mL Type III salmon sperm DNA (all Sigma-Aldrich), and 0.01 mg/mL ethidium bromide (Thermo Fisher Scientific).
For both enzymes, cylindrical wells were punched into the agarose gel using a capillary tube (radius 1.5 mm). The gel was incubated for 23 h at 37°C in a moist chamber. The area of the dark circle produced by the degradation of salmon sperm DNA caused by enzyme diffusion was measured under an ultraviolet transluminator (312 nm). The area of diffusion, correlating to enzyme activity, was measured by Image J (NIH).
2.7. Blood pressure
Systolic blood pressure (SBP) was measured in non-anaesthetized rats via tail cuff (RTBP1001; Kent Scientific Corporation, Torrington, CT, USA). In SHR treated with HCTZ/Res, SBP was measured weekly until 12 weeks of age.22 For the WKY and SHR treated with ODN2088 or Veh, SBP was measured pre-treatment (Day 0), throughout the treatment period (Days 1–4), and post-treatment (Days 5 and 12). Finally, for the SD treated with Veh or ODN2395, SBP was measured pre-treatment (Day 0) and post-treatment (Day 5). For all blood pressure recordings, an average of the SBP from 10 cycles was taken from each rat and then averaged within group.
2.8. Vascular function
Mesenteric resistance arteries and thoracic aortas from Veh- or ODN2395-treated rats were excised and placed in 4°C physiological salt solution (PSS) containing (mmol/L): NaCl (130), NaHCO3 (14.9), KCl (4.7), KH2PO4 (1.18), MgSO4·7H2O (1.18), CaCl2·2H2O (1.56), EDTA (0.026), and glucose (5.5) (all Sigma-Aldrich). Excised third-order MRA and aortas were cleaned of perivascular adipose tissue and cut into 2 mm segments. Mesenteric resistance arteries and aortic segments were mounted on DMT wire and pin myographs (Danish MyoTech, Aarhus, Denmark), respectively. Aortic segments were set to a basal force of 30 mN. Mesenteric resistance arteries were normalized to their optimal lumen diameter for active tension development, as described previously.26 Briefly, normalization was determined based on the internal circumference/wall tension ratio of the arteries by setting the internal circumference (L0) to 90% of what the vessels would have if they were exposed to a passive tension equivalent of 100 mmHg (L100) transmural pressure.27 The diameter (I1) was then determined according to the equation I1 = L1/π, using specific software for normalization of resistance arteries (DMT Normalization Module; LabChart v.5.5.6, ADInstruments). Arteries were then bathed in 37°C PSS with continuous bubbling with 5% CO2 and 95% O2 for 30 min. All arteries were initially contracted with 120 mmol/L potassium chloride (KCl). Endothelium integrity was then tested with phenylephrine (PE)-induced contraction (3 × 10−6 mol/L) followed by endothelium-dependent relaxation with acetylcholine (ACh; 3 × 10−6 mol/L).
Cumulative concentration-response curves were performed to ACh (10−9–10−5 mol/L), sodium nitroprusside (SNP; 10−9–10−4 mol/L), and norepinephrine (NE; 10−11–10−4 mol/L) (all Sigma-Aldrich). Relaxation curves were performed after an initial contraction with 3 × 10−6 mol/L PE. Thirty minutes prior to NE concentration-response curves, some arteries were incubated with a nitric oxide synthase (NOS) inhibitor (L-NNA; 10−4 mol/L) (Sigma-Aldrich), reactive oxygen species (ROS) scavenger/superoxide dismutase (SOD) mimetic (tempol; 10−3 mol/L) (Tocris, Ellisville, MO, USA), cyclooxygenase (COX) inhibitor (indomethacin; 10−5 mol/L) (Sigma-Aldrich), or p38 MAPK inhibitor (SB203580; 10−5 mol/L) (EMD Millipore, Billerica, MA, USA). Results are presented as either %KCl for contraction or %PE contraction for relaxation. Some concentration-response curves were expressed as ‘differences in area under the concentration-response curves’ (ΔAUC), as described previously.28 Briefly, the change in AUC was calculated from the individual concentration-response curves, with or without inhibitors, using the GraphPad Prism 5.0 (La Jolla, CA, USA) statistical software. The ΔAUC was expressed as the difference in AUC in the presence of an inhibitor, compared with the corresponding control situation. Differences are denoted as ΔAUC (%KCl). Finally, the sample size indicated per vascular function experiment (see figure legends) is the number of independent rats from which isolated arterial segments were obtained, respective of treatment group.
2.9. Immunoblotting
Arteries were cleaned of perivascular adipose tissue, snapped frozen in liquid nitrogen, and then homogenized in ice-cold tissue protein extraction reagent (T-PER) (Thermo Fisher Scientific), with protease inhibitors (sodium orthovanadate, PMSF, protease inhibitor cocktail) and phosphatase inhibitors (sodium fluoride and sodium pyrophosphate) (all Sigma-Aldrich). Equal amounts of protein (30–40 µg) were loaded into polyacrylamide gels (8–12%) for western blots. Polyvinylidene difluoride (PVDF) membranes were probed for the expression of representative autophagy proteins, canonical-inflammatory proteins of TLR9 signalling, non-canonical stress tolerance proteins of TLR9 signalling, and proteins associated with vascular function (see Supplementary material online, Table S1). Phosphorylated protein expression was normalized to total protein expression; all other proteins were normalized to β-actin. Densitometric analysis was performed by Un-Scan-It software (Version 6.1) (Silk Scientific, Orem, UT, USA).
2.10. ROS measurement
ROS were measured in arteries from treated SD rats via dihydroethidium (DHE) fluorescence, as previously described.26 Arteries were embedded in tissue freezing medium (Triangle Biomedical Sciences, Durham, NC, USA) and frozen in liquid nitrogen. Transverse cross-sections (10 µm) of frozen arteries were equilibrated for 10 min in phosphate buffer in a light-protected humidified chamber at 37°C. Fresh buffer containing hydroethidine (1 µmol/L) (Sigma-Aldrich) was topically applied to each artery section. The slides were then incubated in a light-protected humidified chamber at 37°C for 30 min. Negative control sections received the same volume of phosphate buffer but in the absence of hydroethidine. Some arteries were incubated with NOS inhibitor (L-NNA; 10−4 mol/L) (Sigma-Aldrich) for 30 min prior to hydroethidine application, to assess the contribution of NOS uncoupling on ROS generation.29,30 Slides were viewed on an optical microscope equipped with a filter for rhodamine, using a ×20 fluorescent objective. Three different sections of each artery were analysed for the presence of ROS by examining the regions marked with red fluorescence from the oxidation products of DHE. The images were analysed using Image J.
2.11. Statistical analysis
The statistical procedures used included Student's t-tests, one-way and two-way analysis of variance (ANOVA), and non-linear regression analysis (LogEC50 and Emax). Tukey's post hoc testing was performed in the event of a significant omnibus ANOVA. All analyses were performed using data analysis software GraphPad Prism 5.0. Statistical significance was set at α = 0.05, and P-value <0.05 was considered significant for all statistical tests. The data are presented as mean ± SEM.
3. Results
Circulating mtDNA genes Cyt B and ND6 were elevated in plasma samples from male SHR, and expression of these genes were normalized with HCTZ/Res (Figure 1A). Bacterial 16S rRNA was not different among the groups. We confirmed these elevations in Cyt B and ND6 using endpoint PCR (Figure 1B). In female SHR, we did not observe any differences among WKY, SHR, and SHR + HCTZ/Res groups (see Supplementary material online, Figure S1). For this reason, all subsequent experiments employed only male rats.
Figure 1.

Circulating mitochondrial DNA (mtDNA) is elevated in male SHR, and this increased expression is pressure dependent. (A) Quantification of circulating mtDNA genes, cytochrome B (Cyt B), and NADH dehydrogenase subunit 6 (ND6), as measured by RT–PCR from plasma of WKY, SHR, and SHR treated with hydrochlorothiazide and reserpine (HCTZ/Res). 1/Ct denotes the reciprocal of the count where the sequence is detected; a direct function of gene concentration. Bacterial 16S rRNA showed no bacterial contamination of any specimen. (B) Representative blots from endpoint PCR. n = 3–8. One-way ANOVA: *P < 0.05 vs. WKY.
To ascertain where this mtDNA could be coming from in male SHR, we measured mitochondrial viability/early apoptosis marker (JC-1 monomer) in tissues whose function directly affects blood pressure (heart, kidney, and brain), as well as whole blood and bone marrow. We observed that SHR kidney and bone marrow cells have increased JC-1 monomer expression, representing MPT pore opening, decreased mitochondrial viability, and early apoptosis (Figure 2A and B).
Figure 2.

SHR has impaired mitochondrial viability and expression of early apoptosis marker JC-1 in kidney and bone marrow cells. Left, representative histograms for mitochondrial membrane potential (ψm) marker JC-1 monomer in (A) kidney and (B) bone marrow cells of WKY and SHR. Right, densitometric analysis. n = 5. Student's t-test: *P < 0.05 vs. WKY.
To support the finding that mtDNA is elevated in male SHR, we measured DNase I and II activity, as well as DNase II expression. DNase I had decreased activity in serum from SHR compared with WKY, and DNase II activity is decreased in SHR MRA and LV (Figure 3A). In aorta, we observed a decreased DNase II expression in SHR compared with WKY (Figure 3B). Additionally, representative autophagic proteins Beclin-1, ATG5, and LC3-I and -II were all decreased in SHR aorta compared with WKY (Figure 3C–E). Overall, these findings demonstrate an elevation of mtDNA in the circulation of hypertensive rats, as well as diminished DNase activity and decreased expression of representative autophagic proteins in cardiovascular tissues.
Figure 3.

SHR has impaired nucleic acid clearance capacity, and expression of autophagic machinery is diminished. Activities of (A) deoxyribonuclease (DNase) I in serum and DNase II in mesenteric resistance arteries and left ventricle from WKY and SHR. Left, representative images of agarose gels. Right, densitometric analysis. Aortic expression of representative autophagy proteins normalized to β-actin: (B) DNase II, (C) Beclin-1, (D) ATG5, and (E) LC3-I and -II. Above, representative images of immunoblots. Below, densitometric analysis. n = 3–6. Student's t-test: *P < 0.05 vs. WKY.
To test the hypothesis that circulating mtDNA and activation of TLR9 are involved in the pathogenesis of hypertension, we treated WKY and SHR with a TLR9 antagonist (ODN2088) and normotensive SD rats with a TLR9 agonist (ODN2395). To validate the specificity of ODN2395 to activate TLR9 signalling, and ODN2088 to inhibit it, we investigated protein expression changes of canonical-inflammatory TLR9 signalling (MyD88 and TRAF6) and also the newly established non-canonical stress tolerance TLR9 signalling (SERCA2, mitochondrial uniporter, and AMPKα) ex vivo and independently of the other physiological systems that integrate to control vascular function (i.e. autonomic nervous system, kidneys, and heart). Therefore, we incubated isolated MRA of naïve SD rats with ODN2395 for 15 and 30 min, as well as ODN2395 in conjunction with ODN2088 for 30 min. Upon normalization of expression relative to the unstimulated condition, we observed that ODN2395 changed protein expression of not only the traditional TLR9 inflammatory signalling (increased MyD88 and TRAF6 expression), but also the newly established non-canonical stress tolerance TLR9 signalling (increased SERCA2 and phosphorylated AMPKαThr172 expression) after 15 and 30 min compared with basal (Figure 4A–C). Moreover, incubation of ODN2395 with ODN2088 for 30 min inhibited these expression changes. Overall, these data support the specificity of the treatment with ODN2395 to activate TLR9 signalling in MRA, and ODN2088 to inhibit it. Interestingly, the in vivo treatment of three i.p. injections with ODN2395 did not change protein expression of the traditional TLR9 inflammatory signalling, nor the newly established non-canonical stress tolerance TLR9 signalling (see Supplementary material online, Figure S2A and B). Reasons for the differences in our two model systems with ODN2395 treatment (i.e. ex vivo vs. in vivo) remain to be clarified.
Figure 4.

Acute incubation of isolated MRA with TLR9 agonist ODN2395 activates TLR9 signalling. Incubation of naïve Sprague Dawley MRA with TLR9 agonist ODN2395 (2 µmol/L) for 15 or 30 min did not change protein expression of (A) TLR9. However, ODN2395 altered protein expressions of (B) canonical-inflammatory signalling (increased MyD88 and TRAF6) and (C) non-canonical stress tolerance signalling (increased SERCA2 and phosphorylated AMPKαThr172), relative to the unstimulated condition. Black bars: unstimulated, dark grey bars: 15 min stimulation, light grey bars: 30 min stimulation, and white bars: 30 min stimulation in the presence of TLR9 antagonist (ODN2088). Below, densitometric analysis. Above, representative images of immunoblots. n = 3–4. One-way ANOVA: *P < 0.05 vs. unstimulated.
In SHR, four injections of ODN2088 significantly lowered SBP, and this decrease was sustained throughout the treatment period (Days 1–4) (Figure 5A). However, when ODN2088 treatment finished (days 5 and 12), SBP returned to pre-treatment (Day 0) values. On the other hand, three injections of ODN2395 to normotensive rats caused an increase in post-SBP (Figure 5B), but did not alter body mass, total heart mass, LV mass, right ventricle mass, or spleen mass (see Supplementary material online, Figure S3A–E). Treatment of normotensive rats with nuclear DNA (negative control) did not change SBP (Figure 5B).
Figure 5.

TLR9 antagonist ODN2088 lowers SBP in SHR, and TLR9 agonist ODN2395 increases SBP in previously normotensive rats. Systolic blood pressure responses (A) across ODN2088 treatment in WKY and SHR (arrows depict when ODN2088 or Veh were administered) and (B) post-ODN2395 treatment. n = 4–9. Repeated-measures ANOVA (with Bonferroni correction): *P < 0.05 vs. SHR-Veh; one-way ANOVA: *P < 0.05 vs. Veh.
Mesenteric resistance arteries from ODN2395-treated rats were less sensitive to the endothelium-dependent vasodilator ACh (LogEC50, Veh: −7.32 ± 0.03 vs. ODN2395: −6.98 ± 0.03, P < 0.05) (Figure 6A). In contrast, relaxation with the endothelium-independent vasodilator and nitric oxide (NO)-donor SNP was not different between groups (LogEC50, Veh: −6.48 ± 0.13 vs. ODN2395: −6.57 ± 0.08) (Figure 6B). Treatment with nuclear DNA did not alter MRA vascular reactivity to ACh or SNP (see Supplementary material online, Figure S4A and B). In the aorta, ODN2395 treatment impaired endothelium-dependent relaxation, as well as endothelium-independent relaxation (see Supplementary material online, Figure S5A and B). The reason for this difference in resistance and conduit artery function was not determined.
Figure 6.
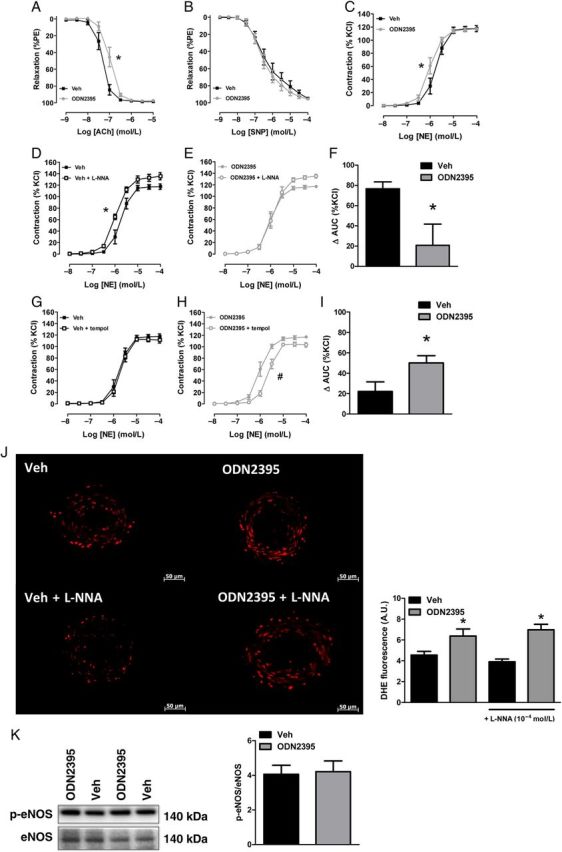
TLR9 agonist ODN2395 impairs endothelium-dependent relaxation in MRA and renders MRA more sensitive to NE via decreased nitric oxide and increased reactive oxygen species. Concentration-response curves for (A) acetylcholine (ACh), (B) sodium nitroprusside (SNP), and (C) NE in MRA from Veh- and ODN2395-treated rats. Concentration-response curves for NE, with or without L-NNA (10−4 mol/L), in MRA from (D) Veh- and (E) ODN2395-treated rats. (F) Area under the curve (AUC) analysis for NE concentration-response curves, with or without L-NNA. Concentration-response curves for NE, with or without tempol (10−3 mol/L), in MRA from (G) Veh- and (H) ODN2395-treated rats. (I) AUC analysis for NE concentration-response curves, with or without tempol. (J) Dihydroethidium (DHE) fluorescent staining (×20 magnification) in MRA from Veh- and ODN2395-treated rats, with or without L-NNA (10−4 mol/L). Left, representative images of fluorescent staining. Right, densitometric analysis. (K) MRA expression of phosphorylated eNOSSer1177 normalized for total eNOS. Left, representative images of immunoblot. Right, densitometric analysis. n = 6/6 (independent rats/MRA segments). LogEC50 or Student's t-test: *P < 0.05 vs. Veh; LogEC50: #P < 0.05 vs. ODN2395.
Mesenteric resistance arteries from ODN2395-treated rats were also more sensitive to the adrenergic agonist NE (LogEC50, Veh: −5.75 ± 0.04 vs. ODN2395: −6.01 ± 0.04, P < 0.05) (Figure 6C), whereas NE contraction in MRA from nuclear DNA-treated rats was unaffected (see Supplementary material online, Figure S4C). To evaluate the role of NO in this increased sensitivity of MRA after ODN2395 treatment, the NOS inhibitor L-NNA (10−4 mol/L) was used with some concentration-response curves. NOS inhibition with L-NNA increased the sensitivity to NE in MRA from Veh-treated rats (LogEC50, Veh: −5.75 ± 0.04 vs. Veh + L-NNA: −5.97 ± 0.03, P < 0.05) (Figure 6D), with no effect in MRA from ODN2395-treated rats (LogEC50, ODN2395: −6.01 ± 0.04 vs. ODN2395 + L-NNA: −5.90 ± 0.04) (Figure 6E). Therefore, arteries from ODN2395-treated rats had a decreased ΔAUC in the presence of L-NNA [ΔAUC (%KCl), Veh: 77 ± 7 vs. ODN2395: 21 ± 21, P < 0.05] (Figure 6F), indicating that a reduction in NO contributed to the increased sensitivity to NE.
We hypothesized that increased ROS generation after ODN2395 treatment may quench NO bioavailability. To test this, we performed concentration-response curves to NE in the presence of tempol (10−3 mol/L). Tempol had no effect on the contraction to NE in MRA from Veh-treated rats (LogEC50, Veh: −5.75 ± 0.04 vs. Veh + tempol: −5.70 ± 0.03) (Figure 6G). However, in MRA from ODN2395-treated rats, tempol decreased the sensitivity to NE (LogEC50, ODN2395: −6.01 ± 0.04 vs. ODN2395 + tempol: −5.65 ± 0.03, P < 0.05) (Figure 6H). Therefore, arteries from ODN2395-treated rats had an increased ΔAUC in the presence of tempol [ΔAUC (%KCl), Veh: 22 ± 9 vs. ODN2395: 50 ± 7, P < 0.05] (Figure 6I), indicating that ROS generation contributed to the increased sensitivity to NE. In the aorta, ODN2395 treatment increased sensitivity to NE similarly—in a NO- and ROS-dependent manner (see Supplementary material online, Figure S5C–I).
Consistent with the above data, DHE fluorescence, indicating ROS generation, was significantly greater in MRA (Figure 6J) and aorta (see Supplementary material online, Figure S5J) of ODN2395-treated rats, compared with Veh-treated rats. To evaluate the role of uncoupled NOS on ROS generation, the NOS inhibitor L-NNA (10−4 mol/L) was incubated with some MRA sections prior to hydroethidine application. The presence of L-NNA did not affect DHE fluorescence, indicating that uncoupled NOS does not contribute to the increased ROS generation (Figure 6J). Finally, there were no differences in protein expression of phosphorylated eNOSSer1177 in MRA (Figure 6K). However, we did observe an increase in phosphorylated eNOSSer1177 in the aorta of ODN2395-treated rats (see Supplementary material online, Figure S5K). This increase could be explained as a compensatory mechanism to combat increases in ROS31,32 and decreases in NO bioavailability.33
We hypothesized that COX enzymes may contribute to this increased ROS generation and increased sensitivity to NE after ODN2395 treatment due to COX 2 being a downstream effector of NF-κB,34 as well as COX enzymes being a prominent source of ROS in the vasculature.35 Additionally, given that p38 MAPK stimulates ROS generation36 and MAPKs are downstream of TLR signalling,2 we hypothesized that p38 MAPK may also play a role. Therefore, we performed concentration-response curves to NE in the presence of the COX inhibitor indomethacin (10−5 mol/L) or the p38 MAPK inhibitor SB203580 (10−5 mol/L). In MRA from Veh-treated rats, COX inhibition had no effect on the contraction to NE (LogEC50, Veh: −5.75 ± 0.04 vs. Veh + indomethacin: −5.68 ± 0.03) (Figure 7A). However, in MRA from ODN2395-treated rats, COX inhibition decreased the sensitivity to NE (LogEC50, ODN2395: −6.01 ± 0.04 vs. ODN2395 + indomethacin: −5.62 ± 0.05, P < 0.05) (Figure 7B). Therefore, arteries from ODN2395-treated rats had an increased ΔAUC in the presence of indomethacin [ΔAUC (%KCl), Veh: 9 ± 7 vs. ODN2395: 63 ± 20, P < 0.05] (Figure 7C), indicating that COX activation contributed to the increased sensitivity to NE. Supporting these concentration-response curves with indomethacin, ODN2395 treatment significantly increased MRA protein expression of COX 2 (Figure 6D), and COX 1 was trending to increase, although this did not reach statistical significance (Figure 7E). In MRA from Veh-treated rats, p38 MAPK inhibition had no effect on the contraction to NE (LogEC50, Veh: −5.75 ± 0.04 vs. Veh + SB203580: −5.76 ± 0.03) (Figure 7F). However, in MRA from ODN2395-treated rats, p38 MAPK inhibition decreased the sensitivity to NE (LogEC50, ODN2395: −6.01 ± 0.04 vs. ODN2395 + SB203580: −5.68 ± 0.05, P < 0.05) (Figure 7G). Supporting these data, ODN2395 treatment significantly increased MRA protein expression of phosphorylated p38Thr180/Tyr182 MAPK (Figure 7H). In the aorta, ODN2395 treatment increased sensitivity to NE similarly—in a COX- and p38 MAPK-dependent manner (see Supplementary material online, Figure S6A–D).
Figure 7.

Cyclooxygenase (COX) and p38 MAPK inhibition normalizes ODN2395-induced sensitivity to NE in MRA. Concentration-response curves for NE, with or without indomethacin (10−5 mol/L), in MRA from (A) Veh- and (B) ODN2395-treated rats. (C) Area under the curve (AUC) analysis for NE concentration-response curves, with or without indomethacin. MRA expression of (D) COX 2 and (E) COX 1 normalized to β-actin. Above, representative images of immunoblots. Below, densitometric analysis. Concentration-response curves for NE in MRA from (F) Veh- and (G) ODN2395-treated rats with or without SB203580 (10−5 mol/L). (H) Phosphorylated p38Thr180/Tyr182 MAPK normalized for total p38 MAPK. Above, representative images of immunoblot. Below, densitometric analysis. n = 6/6 (independent rats/MRA segments). Student's t-test: *P < 0.05 vs. Veh; LogEC50: #P < 0.05 vs. ODN2395.
4. Discussion
The first significant observation of the present investigation was that male SHR have elevated circulating mtDNA, increased early apoptosis, impaired mitochondrial viability, and diminished nucleic acid clearance machinery, and their blood pressure is sensitive to TLR9 inhibition. The second major finding was that a CpG oligonucleotide, specific for TLR9, induced an increase in blood pressure and caused endothelial dysfunction.
Recently, it was observed that hypertensive patients have elevated levels of cell-free CpG-DNA and antibodies targeted to CpG-DNA injected into infant SHR, delayed the age-dependent increase in blood pressure.20 However, whether this CpG-DNA was mitochondria derived, and the contribution of TLR9 to hypertension pathogenesis, was not determined. Some investigators have suggested that the elevated CpG-DNA in hypertensive patients is a result of chronic (periodontal) infections.37 Our results in male SHR suggest that this circulating CpG-DNA in hypertensive patients could be mtDNA, as our RT–PCR results with bacterial 16S ribosomal subunit indicated that there was no difference in the presence of bacteria between WKY and SHR. Circulating mtDNA as a biomarker of injury was first established in trauma patients11 and therefore could also serve as a novel biomarker and/or therapeutic target in the treatment or management of hypertension.
Generally, it has been thought that necrotic cell death was the primary source of pro-inflammatory DAMPs, due to disintegration of the plasma membrane and release of intracellular constituents.38 However, apoptosis can also be immunogenic as a result of the programmed release of immunostimulatory molecules.6,39 Given that mitochondria play a pivotal role in regulating apoptosis (including the increased permeability of the outer mitochondrial membrane, the participation of pro- and anti-apoptotic Bcl-2 family proteins, and the release of caspase activators), it is not surprising that we observed increased circulating mtDNA in conjunction with impaired mitochondrial viability and increased early apoptosis in SHR kidney and bone marrow. These tissues are consistent with the increased JC-1 monomer expression observed in the aorta of angiotensin II hypertensive rats.40 Finally, our data supports that apoptosis is a significant contributor to hypertension pathophysiology41; however, programmed release of DAMPs could be novel characteristic contributing to the detrimental effects of apoptosis in hypertension.
The impaired clearance capacity of serum and cardiovascular tissues in SHR increases the possibility that mtDNA is able to activate TLR9 and contribute to the pathogenesis of hypertension. In fact, pressure-overload released mtDNA, which escaped autophagic degradation, led to TLR9-mediated inflammatory responses in cardiomyocytes, and subsequently contributed to the development of myocarditis and dilated cardiomyopathy in mice.12 Moreover, decreased DNase activity has been a well-established phenotype of patients with SLE,42 which is associated with increased cardiovascular risk, partially attributable to hypertension.43 In the future, therapeutics targeting clearance mechanisms in hypertension may be a way of reducing circulating DAMPs, such as mtDNA and therefore pattern recognition receptor activation, without modulating or compromising the immune system.
We observed that treatment of SHR with TLR9 antagonist ODN2088 lowered SBP significantly as soon as 24 h after the onset of treatment. This decreased blood pressure was sustained until the conclusion of treatment, at which point SBP returned to pre-treatment values. On the other hand, SBP increased after only 4 days with treatment with TLR9 agonist ODN2395. Rapid changes in SBP like these suggest that TLR9 is modulating effector systems that control blood pressure (e.g. cardiac output and/or total peripheral resistance). These blood pressure data, taken in conjunction with our data observing an increased sensitivity to NE and decreased sensitivity to ACh in MRA from rats treated with ODN2395, suggest that TLR9 is able to cause increases in total peripheral resistance, and this increase in total peripheral resistance contributes to the rapid changes in blood pressure that we observed. Toll-like receptor 9 modulation of arterial blood pressure is consistent with recent literature observing TLR9 as a negative regulator of cardiac vagal tone and baroreflex function.44
Cyclooxygenase may provide a mechanism for vascular dysfunction, especially in SHR.45,46 In hypertension, COX 2-derived vasoconstrictor prostanoids exacerbate vasoconstrictor responses to α-adrenergic agonists47 and COX 2-derived ROS generation, which quenches NO bioavailability, impairs vasorelaxation to ACh.48 In addition, p38 MAPK activation is associated with endothelial dysfunction and hypertension, with its inhibition improving indices of oxidative stress, vascular relaxation to ACh, and blood pressure.36 Our findings that TLR9 agonist ODN2395 activated COX and p38 MAPK is consistent with the literature in macrophages that demonstrate TLR9 and MyD88-dependent activation of NF-κB and p38 MAPK are required for transcriptional regulation of COX 2 expression induced by CpG-DNA.34
Interestingly, ODN2395 treatment also increased non-receptor tyrosine kinase c-Src phosphorylation (see Supplementary material online, Figure S7). Despite the establishment of c-Src as an upstream signalling molecule of MAPKs in vascular dysfunction and hypertension,49,50 how tyrosine kinases regulate TLRs and TLR9 signalling is only emerging.51 A tyrosine motif in the cytoplasmic domain of TLR9 has been shown to be required for the intracellular trafficking of TLR9 to the endolysosomal compartment and activation of NF-κB signalling.52 Moreover, it has been observed that CpG oligonucleotides activate a TLR9-independent pathway initiated by two Src family kinases, Hck and Lyn in human monocytes.53 Sanjuanet al.53 observed that inhibition of Src family kinases blocked MyD88-dependent signalling and cytokine secretion. As expected, chloroquine, an inhibitor of lysosomal acidification, blocked TLR9 and MyD88-dependent cytokine secretion, but failed to inhibit CpG-induced Src family kinase activation. This suggests that tyrosine phosphorylation is both a CpG-induced event and an upstream requirement for the activation of TLR9.53 In light of this, our data support the specificity of the ODN2395 treatment to activate MyD88-dependent signalling, despite no change in TLR9 expression.
In conclusion, we observed that in a genetic model of essential hypertension, mitochondrial viability is impaired, early apoptosis is increased, circulating mtDNA is elevated, and the autophagic enzymes and proteins that would be responsible for its degradation are diminished. We went on to observe that inhibition of TLR9 in SHR lowers blood pressure, and short-term treatment of normotensive rats with a CpG oligonucleotide, specific for TLR9, was able to activate COX and p38 MAPK, increase ROS generation, and quench NO bioavailability. These factors contributed to dysfunctional vascular reactivity and an elevation in arterial blood pressure. With the ability of ODN2395 to cause a hypertensive phenotype, we infer that mtDNA released from injured cells could potentially initiate a feed-forward cascade of pressure- and ischaemic-induced events that would aggravate even more cell injury and death in SHR (see Supplementary material online, Figure S8). Collectively, we propose a novel mechanism by which the DAMP, mtDNA, and innate immune system receptor TLR9 contribute to the pathogenesis of hypertension.
Our investigation adds to the exponentially expanding field of immune system activation and inflammation in hypertension. Nonetheless, the exact mechanisms that initiate this activation are not well understood, and this is where the novelty of our investigation resides. Despite a number of original articles supporting the contribution of the adaptive immune system in hypertension,54–56 participation of the innate immune system is only emerging, and even fewer have described the role of DAMPs.2,57 Although this contribution of the adaptive immune system to the pathogenesis of hypertension is not in question, the participation of innate immune system in hypertension needs further investigation. Our data demonstrate the involvement of the DAMP mtDNA and the innate immune system pattern recognition receptor TLR9. This investigation supports Bomfim et al.21 who first observed a role for TLRs (with TLR4) in dysfunctional vasoreactivity and hypertension in SHR. Collectively, these investigations promote the exploration of novel therapies that target DAMPs and/or the innate immune system in hypertensive patients that are resistant to all existing treatment strategies.
Authors’ contributions
C.G.M. was responsible for study design, data collection, statistical analyses, and manuscript preparation. C.F.W. assisted with study design, data collection, and manuscript preparation. S.G., T.M., and R.C.W. assisted with study design and manuscript preparation. S.O., B.B., and J.S. assisted with data collection. All authors read and approved the final manuscript.
Supplementary material
Supplementary material is available at Cardiovascular Research online
Funding
This study was supported in part by the American Heart Association (#13PRE14080019, #13SDG17050056), the National Institutes of Health (R01 HL071138, R01 DK083685), and CNPq, Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil.
Acknowledgements
We acknowledge the technical support provided by Dr Wagner Reis of the Stern laboratory (Department of Physiology, GRU) and Dr ShaLi Zhang of the Inscho laboratory (Division of Nephrology, University of Alabama and formerly Department of Physiology, GRU).
Conflict of interest: none declared.
References
- 1.Singh MV, Chapleau MW, Harwani SC, Abboud FM. The immune system and hypertension. Immunol Res 2014;59:243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol 2014;306:H184–H196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994;12:991–1045. [DOI] [PubMed] [Google Scholar]
- 4.Matzinger P. The danger model: a renewed sense of self. Science 2002;296:301–305. [DOI] [PubMed] [Google Scholar]
- 5.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 2008;8:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 2011;32:157–164. [DOI] [PubMed] [Google Scholar]
- 7.Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol 2004;5:190–198. [DOI] [PubMed] [Google Scholar]
- 8.Stacey KJ, Young GR, Clark F, Sester DP, Roberts TL, Naik S, Sweet MJ, Hume DA. The molecular basis for the lack of immunostimulatory activity of vertebrate DNA. J Immunol 2003;170:3614–3620. [DOI] [PubMed] [Google Scholar]
- 9.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol 2006;7:49–56. [DOI] [PubMed] [Google Scholar]
- 10.Sagan L. On the origin of mitosing cells. J Theor Biol 1967;14:255–274. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010;464:104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012;485:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells 2013;36:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klinman DM. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol 2004;4:249–258. [DOI] [PubMed] [Google Scholar]
- 15.Shintani Y, Drexler HC, Kioka H, Terracciano CM, Coppen SR, Imamura H, Akao M, Nakai J, Wheeler AP, Higo S, Nakayama H, Takashima S, Yashiro K, Suzuki K. Toll-like receptor 9 protects non-immune cells from stress by modulating mitochondrial ATP synthesis through the inhibition of SERCA2. EMBO Rep 2014;15:438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shintani Y, Kapoor A, Kaneko M, Smolenski RT, D'Acquisto F, Coppen SR, Harada-Shoji N, Lee HJ, Thiemermann C, Takashima S, Yashiro K, Suzuki K. TLR9 mediates cellular protection by modulating energy metabolism in cardiomyocytes and neurons. Proc Natl Acad Sci USA 2013;110:5109–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okoshi MP, Matsubara LS, Franco M, Cicogna AC, Matsubara BB. Myocyte necrosis is the basis for fibrosis in renovascular hypertensive rats. Braz J Med Biol Res 1997;30:1135–1144. [DOI] [PubMed] [Google Scholar]
- 18.Gardner DL. Arteriolar necrosis and the prenecrotic phase of experimental hypertension. Q J Exp Physiol Cogn Med Sci 1963;48:156–163. [DOI] [PubMed] [Google Scholar]
- 19.Tan LB, Jalil JE, Pick R, Janicki JS, Weber KT. Cardiac myocyte necrosis induced by angiotensin II. Circ Res 1991;69:1185–1195. [DOI] [PubMed] [Google Scholar]
- 20.Veiko NN, Konorova IL, Neverova ME, Fidelina OV, Mkrtumova NA, Ershova ES, Kon'kova MS, Postnov AI. Delayed appearance of hypertension in spontaneously hypertensive rat (SHR) injected with CpG-rich DNA early in ontogenesis. Biomed Khim 2010;56:686–699. [DOI] [PubMed] [Google Scholar]
- 21.Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, Fortes ZB, Webb RC, Carvalho MH. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond) 2012;122:535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tipton AJ, Baban B, Sullivan JC. Female spontaneously hypertensive rats have a compensatory increase in renal regulatory T cells in response to elevations in blood pressure. Hypertension 2014;64:557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baban B, Liu JY, Mozaffari MS. Endoplasmic reticulum stress response and inflammatory cytokines in type 2 diabetic nephropathy: role of indoleamine 2,3-dioxygenase and programmed death-1. Exp Mol Pathol 2013;94:343–351. [DOI] [PubMed] [Google Scholar]
- 24.Nadano D, Yasuda T, Kishi K. Measurement of deoxyribonuclease I activity in human tissues and body fluids by a single radial enzyme-diffusion method. Clin Chem 1993;39:448–452. [PubMed] [Google Scholar]
- 25.Koizumi T. Deoxyribonuclease II (DNase II) activity in mouse tissues and body fluids. Exp Anim 1995;44:169–171. [DOI] [PubMed] [Google Scholar]
- 26.Wenceslau CF, Rossoni LV. Rostafuroxin ameliorates endothelial dysfunction and oxidative stress in resistance arteries from deoxycorticosterone acetate-salt hypertensive rats: the role of Na+K+-ATPase/ cSRC pathway. J Hypertens 2014;32:542–554. [DOI] [PubMed] [Google Scholar]
- 27.Mulvany MJ, Halpern W. Mechanical properties of vascular smooth muscle cells in situ. Nature 1976;260:617–619. [DOI] [PubMed] [Google Scholar]
- 28.Rossoni LV, Salaices M, Marin J, Vassallo DV, Alonso MJ. Alterations in phenylephrine-induced contractions and the vascular expression of Na+,K+-ATPase in ouabain-induced hypertension. Br J Pharmacol 2002;135:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao L, Mann GE. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res 2009;82:9–20. [DOI] [PubMed] [Google Scholar]
- 30.d'Uscio LV. eNOS uncoupling in pulmonary hypertension. Cardiovasc Res 2011;92:359–360. [DOI] [PubMed] [Google Scholar]
- 31.Simao S, Gomes P, Pinto V, Silva E, Amaral JS, Igreja B, Afonso J, Serrao MP, Pinho MJ, Soares-da-Silva P. Age-related changes in renal expression of oxidant and antioxidant enzymes and oxidative stress markers in male SHR and WKY rats. Exp Gerontol 2011;46:468–474. [DOI] [PubMed] [Google Scholar]
- 32.Ulker S, McMaster D, McKeown PP, Bayraktutan U. Impaired activities of antioxidant enzymes elicit endothelial dysfunction in spontaneous hypertensive rats despite enhanced vascular nitric oxide generation. Cardiovasc Res 2003;59:488–500. [DOI] [PubMed] [Google Scholar]
- 33.Vaziri ND, Ni Z, Oveisi F. Upregulation of renal and vascular nitric oxide synthase in young spontaneously hypertensive rats. Hypertension 1998;31:1248–1254. [DOI] [PubMed] [Google Scholar]
- 34.Yeo SJ, Gravis D, Yoon JG, Yi AK. Myeloid differentiation factor 88-dependent transcriptional regulation of cyclooxygenase-2 expression by CpG DNA: role of NF-kappaB and p38. J Biol Chem 2003;278:22563–22573. [DOI] [PubMed] [Google Scholar]
- 35.Wong MS, Vanhoutte PM. COX-mediated endothelium-dependent contractions: from the past to recent discoveries. Acta Pharmacol Sin 2010;31:1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bao W, Behm DJ, Nerurkar SS, Ao Z, Bentley R, Mirabile RC, Johns DG, Woods TN, Doe CP, Coatney RW, Ohlstein JF, Douglas SA, Willette RN, Yue TL. Effects of p38 MAPK Inhibitor on angiotensin II-dependent hypertension, organ damage, and superoxide anion production. J Cardiovasc Pharmacol 2007;49:362–368. [DOI] [PubMed] [Google Scholar]
- 37.Desvarieux M, Demmer RT, Jacobs DR, Jr, Rundek T, Boden-Albala B, Sacco RL, Papapanou PN. Periodontal bacteria and hypertension: the oral infections and vascular disease epidemiology study (INVEST). J Hypertens 2010;28:1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyake Y, Yamasaki S. Sensing necrotic cells. Adv Exp Med Biol 2012;738:144–152. [DOI] [PubMed] [Google Scholar]
- 39.Davidovich P, Kearney CJ, Martin SJ. Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol Chem 2014;395:1163–1171. [DOI] [PubMed] [Google Scholar]
- 40.Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Abe Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension 2005;45:438–444. [DOI] [PubMed] [Google Scholar]
- 41.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension 2001;38:581–587. [DOI] [PubMed] [Google Scholar]
- 42.Chitrabamrung S, Rubin RL, Tan EM. Serum deoxyribonuclease I and clinical activity in systemic lupus erythematosus. Rheumatol Int 1981;1:55–60. [DOI] [PubMed] [Google Scholar]
- 43.Aranow C, Ginzler EM. Epidemiology of cardiovascular disease in systemic lupus erythematosus. Lupus 2000;9:166–169. [DOI] [PubMed] [Google Scholar]
- 44.Rodrigues FL, Silva LE, Hott SC, Bomfim GF, da Silva CA, Fazan R, Jr, Resstel LB, Tostes RC, Carneiro FS. Toll-like receptor 9 (TLR9) plays a key role in the autonomic cardiac and baroreflex control of arterial pressure. Am J Physiol Regul Integr Comp Physiol 2015;308:R714–R723. [DOI] [PubMed] [Google Scholar]
- 45.Luscher TF, Vanhoutte PM. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension 1986;8:344–348. [DOI] [PubMed] [Google Scholar]
- 46.Spitler KM, Matsumoto T, Webb RC. Suppression of endoplasmic reticulum stress improves endothelium-dependent contractile responses in aorta of the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 2013;305:H344–H353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alvarez Y, Briones AM, Balfagon G, Alonso MJ, Salaices M. Hypertension increases the participation of vasoconstrictor prostanoids from cyclooxygenase-2 in phenylephrine responses. J Hypertens 2005;23:767–777. [DOI] [PubMed] [Google Scholar]
- 48.Virdis A, Bacca A, Colucci R, Duranti E, Fornai M, Materazzi G, Ippolito C, Bernardini N, Blandizzi C, Bernini G, Taddei S. Endothelial dysfunction in small arteries of essential hypertensive patients: role of cyclooxygenase-2 in oxidative stress generation. Hypertension 2013;62:337–344. [DOI] [PubMed] [Google Scholar]
- 49.Ishida M, Ishida T, Thomas SM, Berk BC. Activation of extracellular signal-regulated kinases (ERK1/2) by angiotensin II is dependent on c-Src in vascular smooth muscle cells. Circ Res 1998;82:7–12. [DOI] [PubMed] [Google Scholar]
- 50.Touyz RM, He G, Wu XH, Park JB, Mabrouk ME, Schiffrin EL. Src is an important mediator of extracellular signal-regulated kinase 1/2-dependent growth signaling by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients. Hypertension 2001;38:56–64. [DOI] [PubMed] [Google Scholar]
- 51.Chattopadhyay S, Sen GC. Tyrosine phosphorylation in Toll-like receptor signaling. Cytokine Growth Fact Rev 2014;25:533–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chockalingam A, Rose WA, 2nd, Hasan M, Ju CH, Leifer CA. Cutting edge: a TLR9 cytoplasmic tyrosine motif is selectively required for proinflammatory cytokine production. J Immunol 2012;188:527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanjuan MA, Rao N, Lai KT, Gu Y, Sun S, Fuchs A, Fung-Leung WP, Colonna M, Karlsson L. CpG-induced tyrosine phosphorylation occurs via a TLR9-independent mechanism and is required for cytokine secretion. J Cell Biol 2006;172:1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ba D, Takeichi N, Kodama T, Kobayashi H. Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J Immunol 1982;128:1211–1216. [PubMed] [Google Scholar]
- 55.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 2007;204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res 2010;107:263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singh MV, Abboud FM. Toll-like receptors and hypertension. Am J Physiol Regul Integr Comp Physiol 2014;307:R501–R504. [DOI] [PMC free article] [PubMed] [Google Scholar]