

Abstract
Aims
Premature ventricular complexes (PVCs) due to spontaneous calcium (Ca) release (SCR) events at the cell level can precipitate ventricular arrhythmias. However, the mechanistic link between SCRs and PVC formation remains incompletely understood. The aim of this study was to investigate the conditions under which delayed afterdepolarizations resulting from stochastic subcellular SCR events can overcome electrotonic source–sink mismatch, leading to PVC initiation.
Methods and results
A stochastic subcellular-scale mathematical model of SCR was incorporated in a realistic model of the rabbit ventricles and Purkinje system (PS). Elevated levels of diastolic sarcoplasmic reticulum Ca2+ (CaSR) were imposed until triggered activity was observed, allowing us to compile statistics on probability, timing, and location of PVCs. At CaSR≥ 1500 µmol/L PVCs originated in the PS. When SCR was incapacitated in the PS, PVCs also emerged in the ventricles, but at a higher CaSR (≥1550 µmol/L) and with longer waiting times. For each model configuration tested, the probability of PVC occurrence increased from 0 to 100% within a well-defined critical CaSR range; this transition was much more abrupt in organ-scale models (∼50 µmol/L CaSR range) than in the tissue strand (∼100 µmol/L) or single-cell (∼450 µmol/L) models. Among PVCs originating in the PS, ∼68% were located near Purkinje-ventricular junctions (<1 mm).
Conclusion
SCR events overcome source–sink mismatch to trigger PVCs at a critical CaSR threshold. Above this threshold, PVCs emerge due to increased probability and reduced variability in timing of SCR events, leading to significant diastolic depolarization. Sites of lower electronic load, such as the PS, are preferential locations for triggering.
Keywords: Delayed afterdepolarization, Triggered activity, Computer-based model, Purkinje system, Arrhythmia
1. Introduction
Arrhythmias are the leading cause of sudden death in patients with cardiac diseases such as myocardial ischaemia or heart failure (HF).1,2 Ectopic excitations are believed to be implicated in triggering a variety of these arrhythmias by causing premature ventricular complexes (PVCs). These are usually benign in healthy individuals3; however, under diseased conditions they can disrupt the normal sinus rhythm and precipitate life-threatening tachycardias.4,5
Ectopic excitations have been the topic of many experimental studies ranging from cellular to whole heart preparations.6–11 At the cellular level, they can occur when action potentials (APs) are elicited by suprathreshold afterdepolarizations caused by abnormalities at the level of ion channels. Afterdepolarizations occur either early during the plateau phase of a long-duration AP or ‘delayed’, when the AP is nearly or fully repolarized. With delayed afterdepolarizations (DADs), it is well established that abnormal calcium (Ca2+) cycling plays an essential role in their genesis.6,7,9,11,12 Ca2+ release from the sarcoplasmic reticulum (SR) due to random openings of Ryanodine receptors can generate spontaneous Ca2+ sparks in absence of any local trigger current provided through L-type Ca2+ channel activation.9 Such spontaneous Ca2+ release (SCR) events are favoured by conditions producing Ca2+ overload such as ischaemia, increased sympathetic nerve activity, hypertrophy, or HF.13–15 Spontaneous Ca2+ sparks can spread throughout the cell as waves, stimulating Ca2+-sensitive inward currents which can summate to produce a DAD-induced AP.
While such DADs may induce APs in isolated myocytes, this is not necessarily the case in tissue where myocytes are coupled via gap junctions to quiescent neighbouring myocytes. DADs induce electrotonic current flow, which greatly reduces its magnitude compared with isolated myocytes. For triggered APs to propagate, it is necessary that DADs occur within a sufficiently small window in time over a larger ensemble of adjacent myocytes to achieve suprathreshold depolarization. However, the mechanism by which spatiotemporal synchronization of stochastic SCR events is achieved in tissue remains poorly understood.
The objective of this study is to test the hypotheses that (i) a critical diastolic threshold in SR Ca2+ load (CaSR) exists above which PVCs emerge without the need for a mechanism to synchronize stochastic SCRs across cells, and (ii) electrotonic loading conditions in structurally healthy ventricles favour the origin of the first DAD-induced PVC in the Purkinje system (PS). These hypotheses are tested using a phenomenological model of SCR16 and AP17 which is incorporated into an anatomically accurate computer model of rabbit ventricles, equipped with a topologically realistic His-Purkinje system.18 Heterogeneities in cellular electrophysiology in ventricular as well as Purkinje cells are ignored to precisely assess the role of electrotonic load in PVC formation.
2. Methods
2.1. Model of spontaneous Ca2+ release and ventricular action potential
In ventricular myocytes, efflux of Ca2+ from the SR can occur spontaneously due to Ryanodine receptor (RyR) openings during diastole, independently of Ca2+ influx via L-type Ca2+ channels. Random RyR openings can induce spontaneous Ca2+ sparks, which can propagate to form Ca2+ waves, and depolarize the membrane to generate DADs. While computer models have been developed which explicitly account for spatio-temporal features of subcellular Ca2+ and stochastic transitions in individual RyRs, these are computationally prohibitive for investigating organ scale DAD formation.19–21 Thus, in this study, an experimentally based phenomenological model of SCR16 has been employed and coupled to the Mahajan-Shiferaw (MSH)17 rabbit ventricular AP model. Stochastic SCR events are represented as Ca2+ waves that are nucleated in the cell and then propagate as fire-diffuse-fire waves. Details of Ca2+ waves formation as well as their functional dependence on CaSR are given in the Supplementary material online. To generate DADs, key parameters of the MSH model were modified22 based on experimental data collected under HF conditions,23 which are known to increase the propensity for DADs (see Supplementary material online).
2.2. Biventricular model
This DAD-prone myocyte model was incorporated into an anatomically accurate 3D computer model of the rabbit ventricles18 including a topologically realistic model of the PS. Electrical activity was solved with the CARP simulator24 using the monodomain approach. The same MSH myocyte model was used in both ventricles and PS to rule out any effects due to electrophysiological heterogeneities. This renders the formation of PVCs a sole function of dimensionality (3D ventricles and quasi-1D PS), biventricular anatomy, anisotropy, and CaSR.
2.3. Simulation protocol
Similar to experiments,15,25 myocyte models were paced at 2.5 Hz for 100 cycles to stabilize under varying extracellular Ca2+ concentrations, followed by a 1000-ms pause within which DAD formation was recorded. Extracellular Ca2+ concentration was increased to induce different diastolic levels of SR Ca2+ overload in the range 1000–1600 µmol/L (see Supplementary material online, Figure S1 and Table S1). Due to the stochastic nature of SCR, N = 1110 independent single myocyte experiments with different random seeds were performed for each CaSR. The exact same protocol was applied in the biventricular model, which was paced in space-clamped mode. Each myocyte in the model was assigned a unique random seed to ensure heterogeneous stochastic SCR evolution and preclude any dependency of location and timing of PVC formation on the preceding activation sequence. In the biventricular model, N = 100 experiments were performed for each CaSR.
2.4. Data analysis
For each CaSR, the number of experiments in which a SCR event with a recruitment rate >5 sparks/ms was observed within the pacing pause, n, was recorded to compute the probability pSCR = n/N. This recruitment rate criterion was sufficiently high to be only met if Ca2+ waves propagated throughout the whole cell. Given this definition, pSCR is equivalent to the probability of a propagating Ca2+ wave to occur in a given time interval within a single-cell. Likewise, pAP is the probability of a cell to trigger an AP during the pacing pause. The number of SCR events as well as the time until the onset of a SCR event, TSCR, and a triggered AP, TAP, were determined (Figure 1). Experiments were repeated with the fast sodium current, INa, and the L-type Ca2+ current, ICaL, inhibited, to compute the intrinsic spontaneous Ca2+ elevation in the submembrane space, ΔCas, and DAD amplitude, ΔVm, as a sole function of SCR. In tissue experiments, the location of the first PVC was also recorded.
Figure 1.

Ca-mediated triggered activity in an isolated myocyte. Shown are Cas and Vm traces of the last three out of 100 pacing cycles before a 1000-ms pause. (A) Ca2+ traces illustrate two different outcomes of the same stochastic model (blue: DAD, red: triggered AP). SCR (inset) led to elevations in the subsarcolemal Ca, ΔCas. (B) Vm traces taken from the same two experiments in (A). The elevations in ΔCas induced membrane depolarizations ΔVm which either turned into a subthreshold DAD (blue trace) or a full-blown AP (red trace).
3. Results
3.1. Ca2+-mediated triggered activity in isolated cells
Ca2+ and voltage traces during the last three of the 100 pacing cycles are shown in Figure 1 to illustrate different outcomes of the same stochastic model. In both experiments, SCR events elevated Cas, by ΔCas, which, in turn, led to a subthreshold (blue trace) depolarization, ΔVm, or an AP (red trace). Figure 2 summarizes results obtained for all prescribed CaSR. Both pSCR and pAP increased in a sigmoid fashion with CaSR (Figure 2A). SCR events occurred over the entire range of CaSR probed while triggered APs appeared only at CaSR ≥ 1100 µmol/L. SCRs or APs always occurred for CaSR ≥ 1350 and ≥1450 µmol/L, respectively. This was due to the gradual increase in the number of SCR events, a shortening of TSCR, and a reduced timing variability with increasing CaSR (Figure 2B and C). On average, the number of SCR events increased from 715 to 14 340, and average TSCR shortened from 732 to 142 ms, when increasing CaSR from 1000 to 1600 µmol/L.
Figure 2.
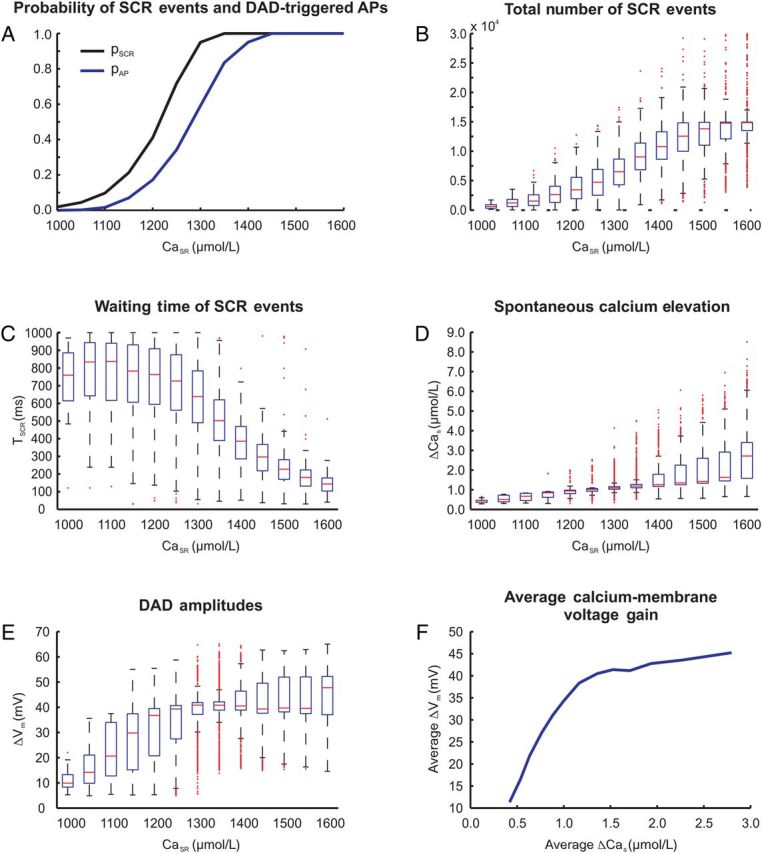
Statistics of triggered activity in N = 1110 single-cell experiments at various CaSR. (A) Probability of SCR events pSCR (black trace) and DAD-triggered APs pAP (blue trace). (B) Total number of SCR events. (C) Waiting time of SCR events, TSCR. (D) Spontaneous Ca2+ elevation ΔCas and (E) DAD amplitude ΔVm due to SCR events. (F) Average calcium-membrane voltage gain based on ΔVm and ΔCas averaged over all single-cell experiments.
Increased incidence of SCR events translated into larger Ca2+ elevations and DAD amplitudes. However, at ΔVm close to or above threshold, the intrinsic contribution of SCR events was masked by the onset of INa and IcaL. To measure the intrinsic driving force, i.e. the dependency of ΔCas and ΔVm on CaSR, all experiments were repeated with INa and ICaL currents inhibited. The average of ΔCas and ΔVm increased with CaSR, but to different extents (Figure 2D and E). With CaSR between 1200 and 1400 µmol/L, corresponding to the rising phase of pAP in Figure 2A, statistical variability was large, as reflected in many outliers. These were due to the differences between cells that elicited a triggered AP from those that did not (see differences in pSCR and pAP in Figure 2A). The relationship between ΔCas and ΔVm, referred to as ‘calcium-membrane voltage coupling gain’, was non-linear, where the average ΔCas per CaSR is plotted against the average ΔVm (Figure 2F).
3.2. Ectopic excitations in a 1D strand of coupled cells
To assess the effect of electrotonic load, all 1110 cells used in single-cell experiments were coupled to form a 1D strand. In this experiment, the same stochastic properties were assigned as previously to the individual cells. This experiment showed that intrinsic properties directly related to SCR events, such as their number, TSCR, and ΔCas (data not shown) remained virtually unaffected by coupling; however, noticeable differences were observed in ΔVm. Figure 3A compares ΔVm of uncoupled and coupled cells for CaSR ranging from 1400 to 1500 µmol/L. While the average ΔVm at 1400 µmol/L was much lower in the strand (20 vs. 41 mV), the difference decreased with increasing CaSR. At CaSR = 1500 µmol/L, ΔVm was 32 vs. 43 mV. Further, electrotonic load in the strand smoothed out variability in ΔVm and decreased calcium-membrane voltage coupling gain (Figure 3B). On average, SCR-induced ΔCas led to less depolarization; however, the difference attenuated with increasing CaSR.
Figure 3.
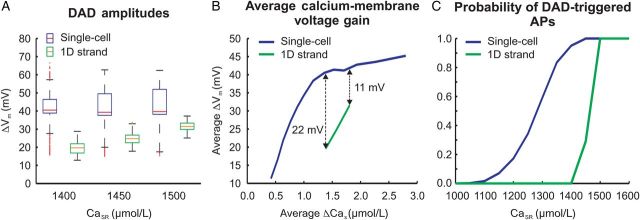
Differences between 1D strand and isolated myocytes. (A) ΔVm due to SCR events. (B) Ca-membrane voltage gain: relationship between averaged ΔVm and averaged ΔCas of all single-cell and 1D cable experiments. (C) pAP in 1D strand (green trace) relative to isolated myocyte data (blue trace).
The same simulation protocol was used to perform 100 strand experiments (Figure 3C). Overall, electrotonic coupling in the strand led to a significant shift in pAP towards higher CaSR along with a much steeper sigmoid rise around the threshold CaSR. Ectopic excitations only appeared at a CaSR of 1450 µmol/L with pAP of 29% and reached 100% at CaSR ≥ 1500 µmol/L.
3.3. PVCs in the biventricular model
Compared with the strand experiments, an even steeper transition in pAP from 0 to 100% was observed (Figure 4A, orange trace). For CaSR≤ 1450 µmol/L, no PVCs emerged at all. For CaSR ≥ 1500 µmol/L, PVCs emerged in all experiments, indicating a distinct narrow banded threshold zone in CaSR. Measured TAP were 349 ± 14 ms (1500 µmol/L) and 176 ± 18 ms (1600 µmol/L).
Figure 4.

Location and incidence of PVCs in the ventricles and PS. (A) pAP under different electrotonic loading conditions. (B) Location and incidence of PVCs at a CaSR of 1500 µmol/L, (C) 1600 µmol/L, and (D) 1600 µmol/L with SCR inhibited in the PS. Colour coding indicates number of ectopic foci.
Figure 4B and C show the distribution of foci at CaSR of 1500 and 1600 µmol/L. Despite the much larger number of cells in the ventricles, all foci arose exclusively, without exception, in the PS (see Supplementary material online, Video S1 for an example). The majority of PVCs (∼68%) originated in the distal branches of the PS in the immediate vicinity (<1 mm) of a Purkinje-ventricular junction (PVJ). The remaining foci were distributed throughout the PS without any obvious preference.
To establish whether PVCs in the ventricles occurred, but were masked by foci emerging earlier in the PS, experiments were performed where SCR was inhibited in the PS. At a CaSR of 1500 µmol/L, where pAP was 100% with SCR in the PS enabled, no PVCs occurred (Figure 4A, red trace). However, after a minor increase in CaSR to 1550 µmol/L, PVCs always occurred. Foci were located at either the epicardial or endocardial surface, but none within the depth of the ventricular wall. Figure 4D shows the uneven clustering of focal sources along the edges of the base where the source–sink mismatch was less severe.
No major qualitative differences in pAP were found over all experiments from single-cell to ventricles, but quantitatively increased electrotonic load promoted a shift of pAP curves towards higher CaSR and a steepening of the transition from 0 to 100% probability (Figure 4A). The critical threshold in CaSR necessary for foci to occur with 100% probability was found to be 1450 µmol/L in the single-cell, 1500 µmol/L in the 1D strand and the 3D ventricles with PS, and 1550 µmol/L in the ventricles with SCRs inhibited in the PS.
3.4. PVC mechanism in 3D
In the absence of SCR events in the PS, PVCs in the ventricles appeared for CaSR ≥ 1550 µmol/L. The mechanism by which stochastic SCR events achieve sufficient synchronization to trigger a propagated AP is illustrated in Figures 5 and 6 (see also Supplementary material online, Video S2). The stochastic nature of SCR and associated Cas is shown in Figure 5A. At the focal site, Cas started to rise around 90 ms due to a SCR event (Figure 5B). Numerous other cells underwent SCRs earlier than this (Figure 5A2). At t3 = 120 ms, the instant of maximum rate of rise of spontaneous Cas, a Cas elevation of 2.2 µmol/L had built up at the focal site which was noticeably larger than the average Cas elevation of 1.26 µmol/L in the ventricles (Figure 5A3 and C). At t4 = 156.5 ms, when the upstroke velocity of the AP peaked, the average Cas elevation over the entire ventricles had arrived at 1.93 µmol/L, but numerous sites had higher elevation levels (Figure 5A4 and C). The fast increase in Cas at t4 was not solely due to a SCR event, but caused by Ca2+ flux via ICaL and Ca2+-induced Ca2+ release from the SR.
Figure 5.

Spontaneous Ca2+ elevation and genesis of a PVC in the absence of SCRs in the PS. (A) Spatial distribution of Cas at time instants: t1 = 0 ms, the start of pacing pause; t2 = 25 ms; t3 = 120 ms, the time of maximum rate of rise of Cas at focal site, and t4 = 156 ms, time of maximum rate of rise of the triggered AP. (B) Cas transient recorded at the ectopic focus. (C) Boxplots illustrating distribution of Cas over the ventricles at time instants t1–t4.
Figure 6.

Genesis of a PVC in absence of SCRs in the PS. (A) Spatial distribution of Vm at time instants: t1, t3–4 chosen as in Figure 5; t2 = 90.75 ms when averaged ΔVm = 5 mV. (B) Vm trace at focal site. (C) Boxplots illustrating distribution of Vm over the ventricles at the time instants t1 –t4.
Membrane depolarization in response to the elevation in Cas in the ventricles and at the focal site is shown in Figure 6A and B, respectively. Note that the PVC did not emerge from a cluster of synchronously firing cells; rather an overall depolarization of the ventricles occurred (Supplemental material online, Video S2). At time t4, when upstroke velocity peaked at the focal site, most cells had undergone significant depolarization (Figure 6A4 and C). This was driven by INCX currents in response to the rise in Cas caused by the large number of SCR events, occurring with an average TSCR of 102 ± 58 ms.
4. Discussion
This study used a multiscale computational model of the rabbit ventricles equipped with a topologically realistic model of the PS network to investigate probability, timing, and locations of SCR-mediated PVCs. Results show that the sigmoid rise of pAP is shifted towards higher CaSR and is markedly steeper in tissue relative to isolated myocytes. This gives rise to a narrow banded critical CaSR threshold where the regime in which no PVCs occur is separated from the high-probability regime by a minor difference in CaSR (50 µmol/L). This critical threshold depends on tissue dimensionality. In a 1D structure, such as a strand, pAP is higher and TAP is shorter than in higher dimensional tissue (Supplementary material online, Figure S2). As a consequence, the critical CaSR threshold is lower in the 1D PS network (1500 µmol/L) than in the 3D ventricular myocardium (1550 µmol/L) and therefore PVCs emerge, with overwhelming likelihood in the PS, with ∼68% occurring within a distance of <1 mm from a PVJ. At slightly higher CaSR above the critical threshold and with SCR inhibited in the PS, PVCs also originated in the ventricles mostly at sites of lower electrotonic load. These PVCs may emerge in the presence of the PS as well; however, they are masked by foci arising earlier in the PS due to the shorter average TAP there. Overall, the high probability and reduced timing variability of SCR events at CaSR beyond the critical threshold lead to a global diastolic depolarization, which drives the ventricles and the PS gradually to the firing threshold. Under such depolarized conditions, no explicit synchronization mechanism is required for a cluster of cells to overcome source–sink mismatches.
4.1. Triggered activity in tissue
Experimental studies indicate that most ventricular tachycardias in non-ischaemic HF are initiated by non-reentrant focal mechanisms9,10,26 such as SCR-triggered DADs. While underlying mechanisms are well understood in isolated myocytes, circumstances under which propagated APs are induced in the intact heart remain unclear. While APs are elicited in isolated myocytes once the membrane is depolarized above the threshold of INa activation, this is insufficient in tissue. According to the liminal length concept, the net ionic current generated by tissue around a focal site within the radius of electrotonic influence must become inward to initiate a propagated response.27,28 Therefore, a DAD-triggered event may propagate only if sufficiently well synchronized SCR events occur in a large cluster of adjacent cells.29 Moreover, due to electrotonic current flow via gap junctions, less current is available for local depolarization, requiring more INCX current for the same ΔVm as in isolated myocytes. Even at an elevated CaSR of 1450 µmol/L which was associated with a pAP of 100% in isolated myocytes, the average ΔVm was markedly reduced in the 1D strand and pAP was only 29%. No propagated APs were observed at CaSR < 1450 µmol/L in tissue, although intrinsic SCR metrics (total number of SCRs, TSCR and ΔCas) remained unaffected by coupling (data not shown). Both average TSCR and its variance were noticeably lowered at this CaSR (Figure 2C), but the temporal coincidence of SCR events was still insufficient to trigger a propagated AP. Thus, the combined effects of insufficient coincidence of SCR events and electrotonic loading led to a reduced ΔVm, preventing the initiation of PVCs.
The role of electrotonic currents in suppressing ectopic excitations has been studied experimentally and computationally.30–32 In a simulation study, Xie et al.31 aimed to establish quantitatively the size of a cluster of cells needed to undergo a DAD in synchrony to initiate AP propagation. As expected on geometric grounds, the source–sink mismatch was most severe in 3D under normal coupling conditions, necessitating a large cluster of cells (∼817 k cells) to fire. While this notion is compatible with our findings in that SCR events must occur in a large number of cells with a sufficiently close coincidence, our results suggest different conclusions regarding the underlying mechanism for spatial synchronization. This is mainly due to very different experimental conditions considered. Xie et al.31 tested a scenario where all cells within a given region synchronously underwent a DAD, whereas outside the region, no DADs occurred. Under these conditions, a sharp gradient builds up between depolarizing cells within the region and cells at rest in the outside. This represents a limiting case of a source–sink relationship, which maximizes the electrotonic load imposed on a focal site. In the scenario considered in this study, timing and location of DADs were determined entirely by the stochastic nature of SCR events and by electrotonic loading. Under such conditions, it is extremely unlikely that a sharp gradient between a focal cluster and adjacent tissue builds up. Rather, our findings suggest that ectopic foci form in tissue only at sufficiently high CaSR at which pAP is close to 100%. That is, essentially all cells undergo a SCR event and the variability in TSCR is narrowed down to achieve a sufficient degree of coincidence (Figure 2A and C). This gives rise to a global depolarization that drives the tissue gradually to the firing threshold (see Supplementary material online, Video S2). As all cells slowly depolarize, to slightly different extents and with some jitter in timing, electrotonic currents smooth out this ripple, but the electrotonic load upon individual cells is minor. Thus fairly small clusters of cells, which are electrotonically favoured or undergo by chance a larger SCR event, are able to trigger a propagated AP. However, under realistic conditions, we expect tissue scale heterogeneities in both Ca2+ cycling properties and ion channels regulating the AP. Thus, we expect that focal excitations will occur in regions where the number of SCR events is largest and coordinated. Indeed, in line scan images of Ca2+ waves in a whole rat heart, Wasserstrom et al.25 observed a PVC coinciding with the point when most of the cells within the experimental field undergo SCR or where ion channel conductances were such that an SCR event induced a larger ΔVm, i.e. in regions of lower (higher) IK1 (INCX) conductance. Moreover, as shown by Xie et al.,31 regional decreases in cellular coupling, as with reduced gap junction density or fibrosis, reduce conductivity, thereby lowering conduction velocity, and shorten space constant and liminal length. Under such conditions of reduced electrotonic load, the overall number of cells required to trigger a propagated response is much smaller, implicating a bias for PVCs to originate there. Indeed, Myles et al.30 demonstrated that gap junction uncoupling increases the occurrence of PVCs in the intact rabbit heart. This is also supported by our own simulations (data not shown) in which intracellular conductivities were significantly reduced by a factor of 10. This led to a reduction in conduction velocity by 74 and 78% in longitudinal and transverse direction, respectively. Under these conditions, the calcium-membrane voltage coupling gain increased, and the CaSR threshold for PVC formation was reduced by 50 µmol/L. However, location and spatial scale of such regions of altered coupling or electrophysiological properties is highly non-trivial and is likely dependent on the precise details of the electrophysiological gradients and structural heterogeneities involved.
4.2. Role of the His-Purkinje network in PVC formation
Several studies have documented the greater vulnerability of the PS to PVCs than the ventricular myocardium.10,26,33,34 Analysis of epicardial breakthrough patterns in ventricular tachycardia suggested ectopic focal sources in the His-Purkinje network.10 Higher susceptibility of the PS to PVCs is attributed either to differences in cellular electrophysiology34 or to tissue scale properties such as electrotonic load and liminal length.35,36 A recent modelling study of Li and Rudy34 provided evidence that in Purkinje cells, CaSR is higher and excitation threshold is lower due to the presence of the hyperpolarization-activated ‘funny current’ and a reduced IK1 current. This is corroborated by experimental findings showing that reduced IK1 is a key determinant for the greater calcium-membrane voltage coupling gain and genesis of PVCs in Purkinje fibres.37 On the other hand, Cerrone et al.10 used a simplified computer model of two thin tissue strips connected to a larger 2D sheet of tissue to mimic electrotonic loading conditions at PVJs. Their results support the notion that DADs are more likely to reach threshold within the thin fibres of the PS rather than in the 3D ventricular muscle due to the lower electrotonic load imposed on cells in the PS. This result is important as it demonstrates, for the first time, that tissue geometry may play an important role in predisposing locations of lowered electrotonic load to the formation of PVCs.
In this study, a highly detailed computer model of rabbit ventricles and PS was employed to elucidate the influence of electrotonic load upon probability, location, and timing of PVCs. For this sake, any electrophysiological heterogeneity, which may be present in the rabbit heart in vivo, was ignored. Thus electrotonic load is the only tissue scale factor, which is subjected to spatial variation, any effects due to functional heterogeneities can be ruled out. PVCs of origin in the PS started to appear at a critical CaSR of 1500 µmol/L (Figure 4A and C). A further increase in CaSR did not entail a further increase in pAP, nor did it change the overall distribution of focal sites, rather a further reduction in TAP was observed. PVCs originated everywhere throughout the PS (Figure 4B and C and Supplementary material online, Video S1), but with a noticeable bias towards locations in the vicinity of PVJs, with about ∼68% of PVCs originating within a distance of <1 mm away from the closest PVJ. All sites, which fired more than once were in close proximity to a PVJ. This bias is explained by considering that the PVJ is modelled as a discrete resistive coupling between the terminal end of the PS and the ventricles. Coupling resistances, which were chosen to reproduce experimentally observed delays in anterograde and retrograde impulse transduction across the PVJ,18 were high enough to lower the electrotonic load in the vicinity of the PVJs. Indeed, increasing the resistance across the junctions to further reduce the electrotonic load imposed by the ventricles upon the PS (data not shown) reinforced this trend. The number of foci increased from 68% within an average distance of ∼0.7 mm to 84% within an average distance of 0.2 mm from a PVJ (CaSR of 1600 µmol/L).
4.3. Triggered activity in ventricles
At a CaSR of 1500 µmol/L, PVCs originated, without exception, in the PS (Figure 4A). When SCRs were inhibited in the PS, no PVCs were observed. However, at a marginally higher CaSR ≥ 1550 µmol/L, PVCs started to appear in the ventricles in all experiments (Figure 4D). PVC formation in this case is readily explained by the tissue-scale mechanisms elucidated earlier. SCR events occur virtually everywhere in the ventricles with sufficiently close coincidence such that the resulting inward currents summate to depolarize the resting potential gradually towards the firing threshold. PVCs tend to originate then in regions where the number of SCR events by chance is higher or where electrotonic load is lower. Indeed, almost all PVCs appeared along the tissue surface and tended to cluster along the edges of the ventricular base where electrotonic load is reduced.
While our results suggest that PVC formation at high CaSR is likely to occur in the ventricles, with an intact PS, PVCs always originated there first due to the shorter TAP (Figure 4C). Overall, our results strongly support the notion that the one-dimensional PS is a very likely source of PVCs in the intact ventricles. Results summarized in Figure 4 are compatible with findings reported by Cerrone et al.,10 where optical maps of the endocardium in an episode of non-sustained multifocal tachycardia revealed that focal discharges at multiple locations were present, all of which were shown to originate within the PS. Our findings are further corroborated by simulations in simpler geometric models (Supplementary material online, Figure S2) which emphasize the high sensitivity of PVCs to dimensionality. With increasing dimensionality, probability of PVCs decreases while TAP increases, thus indicating that PVCs appear not only with higher frequency, but also earlier in the 1D PS than in the 3D myocardium.
4.4. Steep sigmoid dependence of PVC probability upon CaSR
An important finding of this study is that the probability pAP in tissue has a very steep sigmoid dependence on CaSR (Figure 4A). This result is surprising since pAP in an isolated myocyte exhibits a much less steep dependence, suggesting that electrotonic coupling has a substantial effect on the shape of the pAP curve. This result can be explained by the stochasticity of SCR in a population of cells in tissue, along with the effect of electrotonic coupling. As shown in a theoretical study, the mean TAP is exponentially dependent on system parameters22 and, as a consequence, the fraction of ectopic excitations observed within a finite duration pause following pacing will go from 0 to 1 over a small change in CaSR (see Supplementary material online for details).
4.5. SCRs and PVC formation in HF
In this study, key parameters of the MSH myocyte model were modified, in the context of HF remodelling, to increase its propensity for DADs. However, the failing heart undergoes a more complex set of electrophysiological changes, where altered Ca2+ cycling, AP prolongation, and gap junction remodelling are hallmark features favouring triggered activity arising from afterdepolarizations.2,12,23,30 Abnormal RyR function is a key pathophysiological mechanism in HF. Experimental studies indicate that the SR content is reduced in HF.12,38 Thus, it seems contradictory that SCR events due to Ca2+ overload play a significant role in PVC formation. However, unlike in this study where SCR events have a functional dependence on CaSR, in HF they have been attributed to an increased Ca2+ leak from the SR due to RyR phosphorylation.38 In this scenario, the threshold level for SCRs is lowered and the amount of CaSR required to induce SCRs is reduced. Thus pharmacological strategies aiming to raise the threshold for SCRs in HF by stabilizing the RyRs may have potential benefits in PVC prevention. Indeed, Dantrolene has been demonstrated to preserve inotropy while inhibiting diastolic SCRs but not systolic releases in HF cells.39
4.6. Study limitations
While the computer model used in this study is state-of-the-art with regard to the representation of biventricular anatomy, topology of the PS network, modelling of PVJs and multiscale representation of stochastic aspects of Ca2+ cycling, it is simplistic in other aspects. First, although a Ca-binding site on the luminal side of the RyR channel is believed to play a role in SCR, only a functional dependence on CaSR is considered in our phenomenological Ca2+ model. However, a luminal sensor would only pose additional sensitivity to the gradient of Ca2+ across the SR membrane making SCRs even more sensitive to CaSR. Secondly, the cell model was modified to represent some key electrophysiological changes observed under HF conditions, namely up- and down-regulation of INCX and IK1, respectively. The rationale behind this choice is that DAD-triggered activity, the focus of this study, is extremely sensitive to the ratio of inward to outward currents near the threshold for an AP with INCX and IK1 being the two main factors. Investigating how PVC formation is affected by other remodelling process, such as AP prolongation, enhanced heterogeneity or conduction slowing, would require numerous adjustments, which would be a study in its own right. Finally, all known spatial heterogeneities in cellular electrophysiology have been fully ignored. This deliberate choice in the design of this in-silico model allowed us to dissect out the role of the main tissue-scale parameter of interest of this study—the influence of electrotonic load. By using the exact same cellular makeup for both ventricles and PS, any influence of functional heterogeneities can be ruled out.
5. Conclusion
Using an anatomically accurate computer model of rabbit ventricles and His-Purkinje system this study found that the probability of SCR-mediated ectopic excitations in tissue increases dramatically due to small changes in CaSR. This high sensitivity is observed close to a critical CaSR above which pAP increases rapidly, from 0 to 100%. The 0 and 100% probability regime is separated by a very narrow corridor of CaSR of only ∼3.3%. This critical threshold is due to the influence of electrotonic load on the probabilistic nature of SCR. As a consequence, in the ventricles there is a range of CaSR where PVCs occur exclusively in the PS, mostly in the immediate vicinity of a PVJ. While PVCs may also occur in the 3D myocardium at higher CaSR, due to longer TAP, these are masked by ectopic excitations occurring earlier in the PS, suggesting the PS as the lone source of PVCs. The strong sensitivity of ectopic excitations on CaSR suggests that therapeutic approaches that target this relationship can be highly effective to prevent PVCs.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the National Institutes of Health (grant number 1RO1 HL 10119601) and by the Austrian Science Fund (grant number F3210-N18).
Acknowledgements
The authors gratefully acknowledge the valuable suggestions of Dr Simon Sedej, Dr Brigitte Pelzmann, and Dr Klaus Zorn-Pauly in reading the manuscript.
Conflict of interest: none declared.
References
- 1.Mehta D, Curwin J, Gomes JA, Fuster V. Sudden death in coronary artery disease: acute ischemia versus myocardial substrate. Circulation 1997;96:3215–3223. [DOI] [PubMed] [Google Scholar]
- 2.Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, Kass D, Feldman AM, Marban E. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation 1994;90:2534–2539. [DOI] [PubMed] [Google Scholar]
- 3.Zimetbaum P, Josephson ME. Evaluation of patients with palpitations. N Engl J Med 1998;338:1369–1373. [DOI] [PubMed] [Google Scholar]
- 4.Zhang S, Skinner JL, Sims AL, Rollins DL, Walcott GP, Smith WM, Ideker RE. Three-dimensional mapping of spontaneous ventricular arrhythmias in a canine thrombotic coronary occlusion model. J Cardiovasc Electrophysiol 2000;11:762–772. [DOI] [PubMed] [Google Scholar]
- 5.Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, Smits JF, Flameng W, Clancy CE, Moons L, Vos MA, Dewerchin M, Benndorf K, Collen D, Carmeliet E, Carmeliet P. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med 2001;7:1021–1027. [DOI] [PubMed] [Google Scholar]
- 6.Capogrossi MC, Houser SR, Bahinski A, Lakatta EG. Synchronous occurrence of spontaneous localized calcium release from the sarcoplasmic reticulum generates action potentials in rat cardiac ventricular myocytes at normal resting membrane potential. Circ Res 1987;61:498–503. [DOI] [PubMed] [Google Scholar]
- 7.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization: underlying mechanism and threshold for triggered action potentials. Circ Res 2000;87:774–780. [DOI] [PubMed] [Google Scholar]
- 8.Boyden PA, Pu J, Pinto J, Keurs HE. Ca(2+) transients and Ca(2+) waves in purkinje cells: role in action potential initiation. Circ Res 2000;86:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pogwizd SM. Nonreentrant mechanism underlying spontaneous ventricular arrhythmias in a model of nonischemic heart failure in rabbits. Circulation 1995;92:1034–1048. [DOI] [PubMed] [Google Scholar]
- 10.Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O'Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res 2007;101:1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai Y, Jones PP, Guo J, Zhong X, Clark RB, Zhou Q, Wang R, Vallmitjana A, Benitez R, Hove-Madsen L, Semeniuk L, Guo A, Song LS, Duff HJ, Chen SR. Phospholamban knockout breaks arrhythmogenic Ca2+ waves and suppresses catecholaminergic polymorphic ventricular tachycardia in mice. Circ Res 2013;113:517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sipido KR. Calcium overload, spontaneous calcium release, and ventricular arrhythmias. Heart Rhythm 2006;3:977–979. [DOI] [PubMed] [Google Scholar]
- 13.Kass RS, Lederer WJ, Tsien RW, Weingart R. Role of calcium ions in transient inward currents and aftercontractions induced by strophanthidin in cardiac Purkinje fibers. J Physiol (Lond) 1978;281:187–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sipido KR, Volders PG, de Groot SH, Verdonck F, Van de Werf F, Wellens HJ, Vos MA. Enhanced Ca(2+) release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation 2000;102:2137–2144. [DOI] [PubMed] [Google Scholar]
- 15.Vassalle M, Lin CI. Calcium overload and cardiac function. J Biomed Sci 2004;11:542–565. [DOI] [PubMed] [Google Scholar]
- 16.Chen W, Aistrup G, Wasserstrom JA, Shiferaw Y. A mathematical model of spontaneous calcium release in cardiac myocytes. Am J Physiol Heart Circ Physiol 2011;300:H1794–H1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahajan A, Shiferaw Y, Sato D, Baher A, Olcese R, Xie LH, Yang MJ, Chen PS, Restrepo JG, Karma A, Garfinkel A, Qu Z, Weiss JN. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys J 2008;94:392–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyle PM, Deo M, Plank G, Vigmond EJ. Purkinje-mediated effects in the response of quiescent ventricles to defibrillation shocks. Ann Biomed Eng 2010;38:456–468. [DOI] [PubMed] [Google Scholar]
- 19.Restrepo JG, Karma A. Spatiotemporal intracellular calcium dynamics during cardiac alternans. Chaos 2009;19:037115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rovetti R, Cui X, Garfinkel A, Weiss JN, Qu Z. Spark-induced sparks as a mechanism of intracellular calcium alternans in cardiac myocytes. Circ Res 2010;106:1582–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaur N, Rudy Y. Multiscale modeling of calcium cycling in cardiac ventricular myocyte: macroscopic consequences of microscopic dyadic function. Biophys J 2011;100:2904–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen W, Asfaw M, Shiferaw Y. The statistics of calcium-mediated focal excitations on a one-dimensional cable. Biophys J 2012;102:461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res 2001;88:1159–1167. [DOI] [PubMed] [Google Scholar]
- 24.Vigmond EJ, Hughes M, Plank G, Leon LJ. Computational tools for modeling electrical activity in cardiac tissue. J Electrocardiol. 2003;36(suppl.):69–74. [DOI] [PubMed] [Google Scholar]
- 25.Wasserstrom JA, Shiferaw Y, Chen W, Ramakrishna S, Patel H, Kelly JE, O'Toole MJ, Pappas A, Chirayil N, Bassi N, Akintilo L, Wu M, Arora R, Aistrup GL. Variability in timing of spontaneous calcium release in the intact rat heart is determined by the time course of sarcoplasmic reticulum calcium load. Circ Res 2010;107:1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rushton WAH. Initiation of the propagated disturbance. Proc R Soc B 1937;124:210–243. [Google Scholar]
- 27.Joyner RW, Wang YG, Wilders R, Golod DA, Wagner MB, Kumar R, Goolsby WN. A spontaneously active focus drives a model atrial sheet more easily than a model ventricular sheet. Am J Physiol Heart Circ Physiol 2000;279:H752–H763. [DOI] [PubMed] [Google Scholar]
- 28.Houser SR. When does spontaneous sarcoplasmic reticulum Ca(2+) release cause a triggered arrhythmia? Cellular versus tissue requirements. Circ Res 2000; 87:725–727. [DOI] [PubMed] [Google Scholar]
- 29.Joyner RW, Sugiura H, Tan RC. Unidirectional block between isolated rabbit ventricular cells coupled by a variable resistance. Biophys J 1991;60:1038–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res 2012;110:1454–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J 2010;99:1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res 2006;99:292–298. [DOI] [PubMed] [Google Scholar]
- 33.Deo M, Boyle PM, Kim AM, Vigmond EJ. Arrhythmogenesis by single ectopic beats originating in the Purkinje system. Am J Physiol Heart Circ Physiol 2010;299:H1002–H1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li P, Rudy Y. A model of canine purkinje cell electrophysiology and Ca(2+) cycling: rate dependence, triggered activity, and comparison to ventricular myocytes. Circ Res 2011;109:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fozzard HA, Schoenberg M. Strength-duration curves in cardiac Purkinje fibres: effects of liminal length and charge distribution. J Physiol 1972;226:593–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindemans FW, Denier Van der Gon JJ. Current thresholds and liminal size in excitation of heart muscle. Cardiovasc Res 1978;12:477–485. [DOI] [PubMed] [Google Scholar]
- 37.Maruyama M, Joung B, Tang L, Shinohara T, On YK, Han S, Choi EK, Kim DH, Shen MJ, Weiss JN, Lin SF, Chen PS. Diastolic intracellular calcium-membrane voltage coupling gain and postshock arrhythmias: role of purkinje fibers and triggered activity. Circ Res 2010;106:399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belevych AE, Radwański PB, Carnes CA, Györke S. ‘Ryanopathy’: causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc Res 2013;98:240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maxwell JT, Domeier TL, Blatter LA. Dantrolene prevents arrhythmogenic Ca2+ release in heart failure. Am J Physiol Heart Circ Physiol 2012;302:H953–H963. [DOI] [PMC free article] [PubMed] [Google Scholar]