

Abstract
Aims
To define the molecular mechanisms of cardiotoxicity induced by Sunitinib and to identify the role of biological sex in modulating toxicity.
Methods and results
Exposure of isolated cardiomyocytes to plasma-relevant concentrations of Sunitinib and other tyrosine kinase inhibitors produces a broad spectrum of abnormalities and cell death via apoptosis downstream of sexually dimorphic kinase inhibition. Phosphorylation of protein kinase C and phospholipase γ abrogates these effects for most tyrosine kinase inhibitors tested. Female sex and estradiol cause increased cardiotoxicity, which is mediated by reduced expression of a drug efflux transporter and a metabolic enzyme. Female but not male mice exposed to a 28-day course of oral Sunitinib exhibit similar abnormalities as well as functional deficits and their hearts exhibit differential expression of genes responsible for transport and metabolism of Sunitinib.
Conclusion
We identify the specific pathways affected by tyrosine kinase inhibitors in mammalian cardiomyocytes, interactions with biological sex, and a role for oestrogen in modulating drug efflux and metabolism. These findings represent a critical step toward reducing the incidence of cardiotoxicity with tyrosine kinase inhibitor chemotherapeutics.
Keywords: Sunitinib, Cardiotoxicity, Oestrogen, Multidrug-resistance-1, Cytochrome P450
1. Introduction
Small molecule inhibitors that occupy the ATP binding sites of receptor tyrosine kinases (RTKs) have been successful in inhibiting the abnormally high kinase activity and uncontrolled cell growth in many forms of cancer.1 Sunitinib, for example, was designed to inhibit multiple tyrosine kinases (TKs), including platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), and c-kit, each of which plays important roles in the growth of cancer cells and angiogenesis.2 Optimization of the pharmacological properties of tyrosine kinase inhibitors (TKIs) has resulted in more than 20 FDA-approved agents that target the kinases implicated in certain types of cancer. An emerging concern about TKIs, however, is detrimental effects on signalling pathways in healthy tissues, resulting in pathologies unrelated to the cancer. As with other TKIs, a significant number of patients developed cardiotoxicity during administration of Sunitinib,2 with evidence of congestive heart failure and electrocardiographic abnormalities.3,4 In one such study, 75% of patients receiving Sunitinib experienced at least a 15% reduction in left-ventricular ejection fraction, similar to the negative cardiac effects observed with Imatinib, another TKI.5 Despite these data, as recently as 2011, a Phase III trial testing Sunitinib in pancreatic cancer did not screen patients for cardiac dysfunction; heart failure was the second most common side effect of Sunitinib resulting in withdrawal from the trial.6
Because of the highly conserved nature of the TK active sites targeted by TKIs, these small molecules commonly inhibit more kinases than originally intended. Several mechanisms have been implicated in cardiotoxicity including loss of cardiomyocytes via mitochondrial dysfunction2 that may be induced by promiscuity of TKIs. Indeed, in an in vitro screen of 317 protein kinases implicated in cancer, 77 kinases interacted with Sunitinib with variable affinities.7 In vivo effects of TKIs on the heart have not been fully described, however, endomyocardial biopsies from patients receiving Sunitinib revealed abnormal swollen mitochondria and effaced cristae indicative of cellular stress.2 Considering the anatomical evidence for mitochondrial dysfunction, a mechanism involving AMP-activated protein kinase (AMPK), a novel target of Sunitinib, was proposed.8 Inhibition of AMPK by Sunitinib was confirmed in a kinase activity assay; however, no change in ATP levels was observed. Additionally, pretreatment with Metformin, a potent activator of AMPK did not rescue NRVMs from the cellular damage induced by Sunitinib.9
A confounding aspect of identifying the mechanisms of TKI-induced cardiotoxicity is that not all patients receiving these agents develop cardiotoxicity. The lack of uniform effect suggests possible genetic interactions that modulate the cardiotoxic effects of Sunitinib. In fact, a retrospective study revealed that female patients who receive Sunitinib exhibit more toxicities in multiple organ systems compared to men.10 A mechanism that has not been considered in the cardiotoxicity induced by Sunitinib is regulation of its metabolism and efflux from cardiomyocytes, which are processes known to be regulated by the sex hormone, estradiol (E2).
Although the basic inhibitory effects of Sunitinib on kinases have been described in an in vitro assay, to date, there has been no examination of the inhibitory effects on kinases in the adult heart and isolated cardiomyocytes. Additionally, no pre-clinical study has measured phosphorylation of these kinases in both males and females to determine the distinct sexually dimorphic effects of Sunitinib on TKIs. Here, we identify sexually dimorphic cardiotoxicity in mice and isolated cardiomyocytes. Our data demonstrate a role for the E2 in the modulation of drug handling within the cardiomyocyte.
2. Methods
2.1. Animals
Animal use was in accordance with a protocol approved by the Institutional Animal Care and Use Committee at the University of Colorado at Boulder and conformed with guidelines published by the US National Institutes of Health. Sunitinib (40 mg/kg/day2) or vehicle [dimethyl sulfoxide (DMSO)] was administered daily via oral gavage for 28 days. Mice were sacrificed via cervical dislocation after deep anesthetization with inhaled isoflurane on Day 29.
2.2. Neonatal rat ventricular myocytes isolation
Neonatal rat ventricular myocytes (NRVMs) were isolated from 1-day-old Sprague–Dawley rat cardiac ventricles, as previously described.11
2.3. Adult rat ventricular myocytes isolation
Adult rat ventricular myocytes (ARVMs) were isolated from the left ventricle of male and female rats using published protocols.12
2.4. RNA isolation and quantitative PCR
Total RNA was purified from cells or left-ventricular tissue using a TRIzol®-based reagent (Molecular Research Center, Inc., Cincinnati, OH, USA) and cDNA was synthesized, as previously described.12 Gene expression was quantified by measuring SYBR® Green (Invitrogen, Carlsbad, CA, USA) fluorescence using a Bio-Rad CFX 9600 Real-Time PCR System (Hercules, CA, USA).
2.5. Cell viability measurements
NRVMs were isolated and treated for 12–38 h. Adherent cells were stained with crystal violet dye (4-[(4-dimethylaminophenyl)-phenyl-methyl]-N,N-dimethyl-aniline, Sigma-Aldrich, St Louis, MO, USA). Intensity of dye, which is proportional to the number of viable cardiomyocytes,13 was measured and normalized to vehicle-treated NRVMs.
2.6. Caspase activity assays
Caspase-3 activity in NRVMs was determined by measuring the cleavage of a fluorogenic caspase-3-specific substrate (Calbiochem, Darmstadt, Germany), as previously described.14
2.7. Immunohistochemistry
Sarcomeric organization was visualized in NRVMs using antibodies raised against sarcomeric myosins. Categorization of sarcomeric organization was performed by a blinded evaluator, as previously described.15
2.8. Echocardiography
Mice were anaesthetized using 3% isoflurane and subjected to echocardiography after 28 days of Sunitinib administration, as previously reported.12
2.9. Analysis of fibrosis
Quantification of collagen in left-ventricular tissue was performed using Picrosirius Red Stain Kit (Polysciences, Inc., Warren, PA, USA), polarized light microscopy, and ImageJ software, as previously described.12
2.10. RTK, intracellular kinase, and apoptosis arrays
Mouse Phospho-receptor Tyrosine Kinase Arrays, Human Phospho-kinase Arrays, or Human Apoptosis Array (R&D Systems, Minneapolis, MN) were used according to the manufacturer's protocol.12
2.11. Determination of cell volume and area in NRVMs
NRVM diameters were measured in triplicate in a Multisizer 3™ Coulter Counter® (Beckman Coulter, Brea, CA, USA), as previously described.16
2.12. Statistical analyses
Data are reported as mean ± standard error from the mean (SEM). Differences between groups were evaluated for statistical significance using Student's t-test or one- or two-way analysis of variance (ANOVA) followed by Tukey's post-hoc test, as indicated in legends. P-values < 0.05 were considered statistically significant.
3. Results
3.1. Sunitinib induces cardiotoxicity in cardiomyocytes in a concentration- and time-dependent manner
To examine the specific molecular effects of Sunitinib in cardiomyocytes, NRVMs were treated with 150 ng/mL Sunitinib. Although the target plasma concentration is 50–100 ng/mL, mean trough plasma concentrations can reach 1010 ng/mL.17 For the studies presented here, we used average plasma concentration in patients receiving Sunitinib2,18 or vehicle (1 μM DMSO). Expression of fetal genes in cardiomyocytes, a classical hallmark of cardiomyocyte toxicity,19 was measured using quantitative PCR; mRNAs encoding atrial natriuretic factor (Anf), brain natriuretic peptide (Bnp), and β-myosin heavy chain (β-MyHC) were significantly increased (4.1-, 8.2-, and 2.8-fold, respectively), whereas α-skeletal actin (αSkActin), which is up-regulated during cardiomyocyte dedifferentiation and hypertrophy,20 remained unchanged (Figure 1A).
Figure 1.
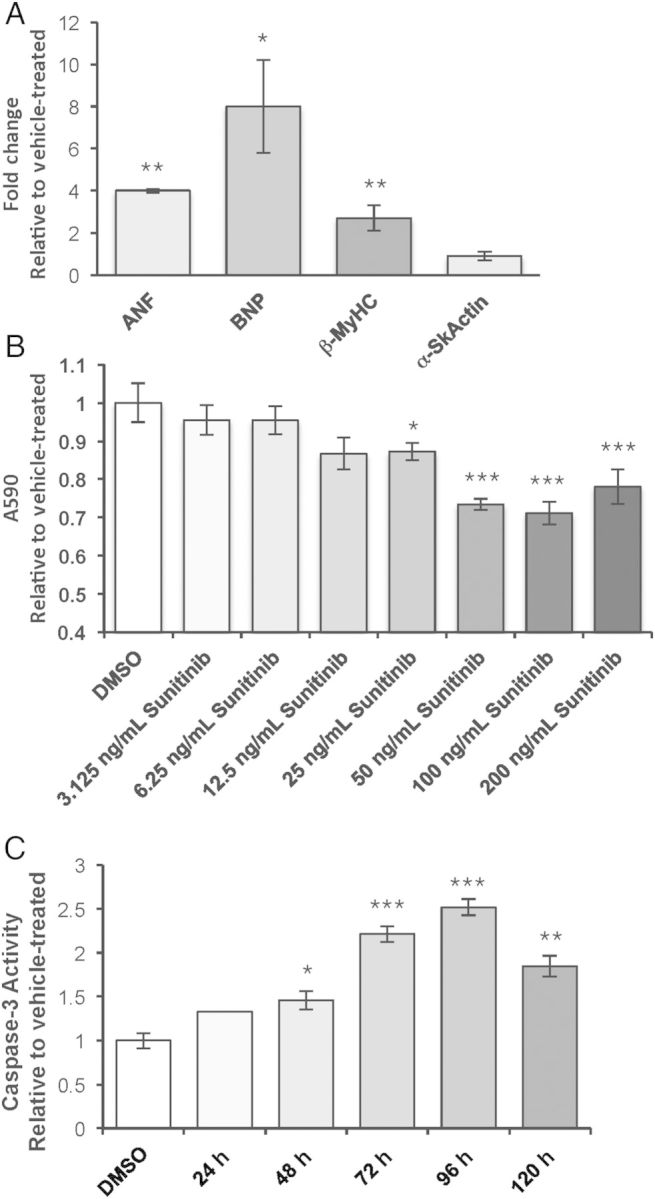
TKIs induce cardiotoxicity and cell death in NRVMs. (A) Fetal gene expression was measured in NRVMs treated with 150 ng/mL Sunitinib for 36 h using quantitative PCR. Fold change is relative to vehicle-treated. ANF, atrial natriuretic factor; BNP, brain natriuretic peptide; β-MyHC, β-myosin heavy chain; α-SkActin, α-Skeletal actin. n = 4 NRVM preparations analysed separately. Bars indicate the average fold change of the four preparations. Student's t-test, comparing Sunitinib- to vehicle-treated cells. (B) Absorbance at 590 nm (A590) of crystal violet-stained NRVMs treated with vehicle (DMSO), 75 ng/mL, 150 ng/mL, or 300 ng/mL Sunitinib (Sun) for 36 h. n = 6 NRVM preparations analysed separately. Bars indicate the average of the six preparations. One-way ANOVA followed by Tukey's post-hoc test. (C) Fold change in caspase-3 activity measured in lysates from NRVMs treated with 150 ng/mL Sunitinib for 24, 48, 72, 96, or 120 h. n = 3 NRVM preparations of 80–100 neonatal ventricles each. One-way ANOVA followed by Tukey's post-hoc test. For each panel, error bars represent standard error from the mean (SEM). *P < 0.05, **P < 0.01.
To examine viability with exposure to Sunitinib, NRVMs were treated with escalating concentrations of Sunitinib and were stained with crystal violet to visualize protein and DNA in live, adherent cells. A concentration-dependent loss of cells occurred after 24 h of exposure to Sunitinib (Figure 1B). A caspase-3 activity assay yielded inversely proportional results (Figure 1C). Therefore, Sunitinib induced cardiomyocyte death via apoptosis in NRVMs, a loss that occurred in a time-dependent manner.
3.2. Sunitinib inhibits off-target kinases in cardiomyocytes
Although in vitro interactions between TKIs and a panel of 317 kinases implicated in oncogenesis have been previously measured,7 the RTKs targeted by each drug have not been examined in isolated cardiomyocytes. The patterns of off-target RTK inhibition that may be responsible for cardiotoxicity of several different TKIs in NRVMs or patients were therefore examined. The levels of phosphorylation of 39 RTKs in NRVMs treated with each of six US FDA-approved TKIs were measured, including 14 RTKs that are implicated in cardiac function (Figure 2A). Inhibition of the targeted RTKs was observed for each drug in addition to many off-target RTKs. For example, Sunitinib inhibited PDGFR and VEGFR but also 18 other off-target RTKs. Interestingly, Sunitinib treatment also increased phosphorylation of several RTKs including substantial increases in ErbB3 and neurotrophic tyrosine kinase receptor type 3 (TrkC). Although no individual or combination of inhibited TKs was strictly associated with cardiotoxicity in these analyses, the quantification of RTK inhibition largely agreed with graded toxicity in patients reported by other investigators9 such that more cell death was observed with drugs that induce more severe cytotoxicity in patients (Supplementary material online, Table S1 and Figure S2C). Combinatorial effects were not observed, such that exposure to two different TKIs did not increase cardiotoxicity (Supplementary material online, Figure S1).
Figure 2.

Off-target kinase inhibition induced by TKIs (150 ng/mL Sunitinib, 1 μg/mL Erlotinib, 3 μg/mL Imatinib, 100 ng/mL Dasatinib, 1.4 μg/mL Lapatinib, or 500 ng/mL Sorafenib) in NRVMs. (A) Levels of phosphorylation of RTKs in NRVMs treated with plasma-relevant doses of TKIs for 24 h were measured using an antibody array. Data were obtained from a single antibody array using three pooled NRVM preparations (80–100 neonatal ventricles each, 270 ventricle total) with each RTK tested in duplicate. Green indicates decreased phosphorylation and red indicates increased phosphorylation, relative to vehicle-treated NRVMs. (B) Absorbance of NRVMs treated with TKIs for 24 h and stained with crystal violet (A590). n = 4 NRVM preparations (80–100 neonatal ventricles per preparation) analysed separately and averaged. *P < 0.05, **P < 0.01, one-way ANOVA followed by Tukey's post-hoc test.
3.3. Cardiotoxicity induced by Sunitinib is abrogated by phosphorylation of phospholipase C
Because RTK signalling converges on activation of phosphorylation of phospholipase C (PLCγ) as well as protein kinase C (PKC),21 phenylephrine (PE), an α1-adrenergic receptor agonist,22 was used to activate these kinases in NRVMs treated with Sunitinib. Our hypothesis was that intervention downstream of RTK inhibition would abrogate the cardiomyocyte toxicity of Sunitinib. Concentrations of PE that do not induce cardiomyocyte hypertrophy23 or hypertension in vivo24 were used to activate PLCγ and PKC. NRVMs were treated for 36 h with PE at concentrations ranging from 2.8 pM to 100 μM. Toxicity was observed with PE concentrations over 25 μM, as indicated by reduced crystal violet staining compared with peak hypertrophy (Supplementary material online, Figure S2A). 375 pM up to 25 μM PE induced statistically significant hypertrophy of NRVMs and increased crystal violet staining (Supplementary material online, Figure S2A). Concentrations below 375 pM PE did not induce hypertrophy of NRVMs (Supplementary material online, Figure S2B and C).
To determine the effect of PE on cell death in NRVMs treated with Sunitinib, NRVMs were treated with Sunitinib alone or combined with dilutions of PE ranging from 2.80 pM to 47.6 nM for 48 h. Increased cell loss was observed in NRVMs co-treated with Sunitinib and hypertrophic concentrations of PE (>375 pM); however, significant cardioprotection was observed at lower concentrations (2.8–375 pM) (Figure 3A). These concentrations have also been shown to be subpressor in mice by specifically activating α-1 adrenergic receptors while avoiding activation of α-2 and β-adrenergic receptors.24 The role of PLCγ-PKC signalling in cardioprotection conferred by PE was validated using phorbol 12-myristate 13-acetate (PMA), a specific activator of PKC that also activates PLCγ.25 PMA alone did not alter crystal violet staining, however, co-treatment with Sunitinib reduced cell death induced by Sunitinib in NRVMs (Figure 3B). Indeed, the PMA-mediated abrogation of cell death induced by Sunitinib by PMA provided independent validation of the importance of this pathway intersection in cardioprotection from Sunitinib.
Figure 3.

Phosphorylation of PKC and PLCγ rescues cell loss in NRVMs treated with TKIs. A590 (absorbance of samples stained with crystal violet) was used to measure cell death in NRVMs treated with Sunitinib alone (light grey bar) or co-treated with decreasing concentrations of phenylephrine (PE, dark grey bars) for 24 h (A) with Sunitinib alone (white bar) or combined with the specific protein kinase C activator, phorbol 12-myristate 13-acetate (PMA) (hatched bar), for 24 h. Hatched bars indicate co-treatment with 50 nM PMA (B) or with TKIs alone (white bar) or combined with 2.8 pM PE for 24 h (hatched bars). (C and D) Caspase-3 activity measured in lysates obtained from NRVMs treated with TKIs alone or combined with 2.8 nM PE for 24 h. (A–C) n = 6 NRVM preparations; (D) n = 3 NRVM preparations. Error bars represent SEM. *P < 0.05, **P < 0.01, relative to vehicle using Student's t-test (A and C) or among all groups using ANOVA followed by Tukey's post-hoc test (B and D).
To determine whether the cardioprotective effect of phosphorylation of PLCγ is specific to Sunitinib or occurs with other TKIs, four additional TKIs associated with variable degrees of cardiotoxicity in patients (1.4 μg/mL Lapatinib,26 1 μg/mL Erlotinib,27 100 ng/mL Dasatinib,28 500 ng/mL Sorafenib29) were used at plasma-relevant concentrations in NRVMs; cells were stained with crystal violet to measure cell loss. All five TKIs induced cell loss compared with vehicle-treated NRVMs in a concentration-dependent manner (Figure 3C, open bars). Co-treatment with 2.8 pM PE, the lowest concentration that caused cardioprotection with Sunitinib, prevented cell loss in all TKIs tested except Lapatinib (Figure 3C, hatched bars). Additionally, reduced caspase-3 activity was observed when these TKIs were co-treated with 2.8 pM PE. For example, Sunitinib induced a 1.85-fold increase in caspase-3 activity. Combined Sunitinib and 2.8 pM PE treatment significantly reduced caspase-3 activity to just 1.28-fold higher than in vehicle-treated cardiomyocytes (Figure 3D).
Levels of phosphorylation of intracellular signalling molecules was also examined using protein lysates from NRVMs treated with vehicle, Sunitinib, 100 pM PE (the mean concentration that promoted cardioprotection) or Sunitinib co-treated with PE to identify the pathways required for cardiomyocyte survival. With PE treatment alone, kinases associated with α1-adrenergic stimulation were phosphorylated: MEK1/2, Erk1/2, and PLCγ, and increased phosphorylation reversed the inhibition induced by Sunitinib. Other signalling pathways including MSK1-p38-cAMP response element-binding protein (CREB) demonstrated a similar pattern in NRVMs co-treated with Sunitinib and PE. Notably, several kinases associated with apoptosis were altered with Sunitinib exposure but normalized with combined treatment with PE. For example, a pan-phospho-JNK antibody detected a 37.4% decrease in phosphorylation with Sunitinib treatment. Sunitinib treatment of NRVMs reduced phosphorylation of JNK by 42% relative to vehicle-treated cells; addition of PE abrogated this reduction to only 8.2% (Figure 4A).
Figure 4.

(A) Heat map demonstrating phosphorylation of intracellular signalling molecules in NRVMs treated with Sunitinib, PE, or both for 24 h. (B) Heat map demonstrating levels of protein involved in apoptosis in NRVMs treated for 24 h with increasing doses of Sunitinib (150, 300, 450 ng/mL), 100 pM PE, or 150 ng/mL Sunitinib plus 100 pM PE. (A and B) n = 3 pooled NRVM preparations, 80–100 neonatal ventricles per preparation.
Because Sunitinib-induced apoptosis was reduced by the addition of PE, an antibody array was used to identify the components of apoptosis involved in Sunitinib-induced cell death and the cardioprotective effects of PE. As expected, no change in expression of DNA damage and cell cycle proteins (p53, p21, p27) was observed when NRVMs were exposed to escalating plasma-relevant concentrations of Sunitinib for 24 h. Increased expression of pro-apoptotic proteins (Cytochrome c, Bax, Hsp70, and cleaved caspase-3) related to mitochondrial stress was observed in a concentration-dependent manner (data not shown), as suggested by other groups who demonstrated mitochondrial abnormalities in the hearts of mice treated with Sunitinib.2 Interestingly, proteins involved in the extrinsic pathway of apoptosis [tumour necrosis factor-related apoptosis-inducing ligand receptor-1 (TRAIL R1), Fas-associated death domain protein (FADD), Fas] were also increased in a concentration-dependent manner in NRVMs exposed to Sunitinib; expression of these proteins was also reduced with co-treatment with PE (Figure 4B). Statistical analyses are not presented because NRVM preparations were pooled and it would be inappropriate to form statistical conclusions based on these data.
3.4. Sunitinib induces sexually dimorphic cardiotoxicity in isolated adult cardiomyocytes
In light of increased toxicity of Sunitinib in women, the role of biological sex was examined in ARVMs isolated from both male and female adult rats and subsequently treated with Sunitinib.30 In agreement with our data in NRVMs exposed to Sunitinib, expression of the fetal gene program was increased with Sunitinib treatment. However, this effect was observed in female ARVMs only (Figure 5A). This sex difference was also observed in the effects of Sunitinib on apoptosis in ARVMs. Caspase-3 activity was significantly increased in a concentration-dependent manner in female ARVMs but not in male ARVMs with 36 h of Sunitinib treatment (Figure 5B); there was a non-significant trending increase in caspase-3 activity in male ARVMs to three-fold above vehicle-treated cells.
Figure 5.

(A) Expression of the fetal gene program (ANF, BNP, βMyHC, SERCA) in female (light grey bars) and male (dark grey bars) ARVMs treated with 150 ng/mL Sunitinib or vehicle (DMSO) for 36 h. n = ventricles from four male and four female rats evaluated individually and averaged, Student's t-test, comparing Sunitinib with vehicle-treated cells. (B) Caspase-3 activity measured using a colorimetric assay in female (light grey bars) and male (dark grey bars) ARVMs treated with Sunitinib or vehicle for 36 h. n = ventricles from four male and four female rats evaluated separately and averaged, Student's t-test, comparing Sunitinib to vehicle-treated. (C) Heat map demonstrating activation states of RTKs in female and male ARVMs treated with Sunitinib, relative to vehicle-treated. n = ventricles from four male and fur female rats. Two RTKs antibody arrays were performed using two pooled male and female ARVM preparations each. Student's t-test, comparing Sunitinib to vehicle-treated cells. For A and B, error bars represent SEM. *P < 0.05, **P < 0.01.
3.5. RTKs are differentially inhibited in male and female adult cardiomyocytes exposed to Sunitinib
Because of the sexual dimorphism observed in fetal gene expression and caspase-3 activation induced by Sunitinib, we quantitated the RTK inhibition by Sunitinib in male and female ARVMs. Different RTKs were inhibited in males and females with more RTKs being inhibited to a larger degree in female cells. In agreement with measurements of TKI targeting in vitro,7 there is a lack of specificity in the TK inhibition in cardiomyocytes by Sunitinib, which has been implicated in the cardiac pathology induced by Sunitinib.7 Thirty RTKs were inhibited in female ARVMs while only 19 were inhibited in males; there were 12 inhibited RTKs shared between the two groups. Notably, seven out of 13 RTKs required for normal cardiac function were inhibited more in female than in male ARVMs (Figure 5C).
3.6. Expression of genes responsible for drug transport and metabolism is differentially regulated in male and female cardiomyocytes
In light of sexual dimorphisms in expression of genes encoding drug transporters and metabolic enzymes in hepatocytes, we examined expression of Mdr1 (which encodes the multidrug resistance protein 1 that is responsible for efflux of Sunitinib) and Cyp1A1 (cytochrome P450, family 1, subfamily A; one of the two metabolic enzymes responsible for metabolizing Sunitinib) in male and female ARVMs. Male ARVMs exhibited increased expression of Mdr1, suggesting greater clearance of Sunitinib in male cells (Figure 6A). However, Mdr1 was reduced when female ARVMs were exposed to Sunitinib for 36 h (Figure 6A), suggesting that expression of these mRNAs may be influenced by the presence of sex hormones in vivo. Sex hormones including E2 modify metabolism and transport of several drugs and xenobiotics primarily though regulation of gene expression.31
Figure 6.

(A) Expression of MDR1 in female (light grey bars) and male ARVMs treated with Sunitinib. MDR1 (B) or CYP1A1 (C) expression in NRVMs treated with Sunitinib or plasma-relevant concentrations of oestrogen (E2) alone or combined with 100 nM E2 or 1 μM E2 (hatched bars) for 24 h. (D) ANF expression in NRVMs treated with Sunitinib alone (white bar) or with E2 or PE (white hatched bars), or with Ellipticine (black bar). Error bars indicate SEM. (A) Student's t-test, compared with Sunitinib treated. n = 4 rats per sex. (B–D) One-way ANOVA followed by Tukey's post-hoc test. n = 4 rats per sex. (A–D) *P < 0.05, **P < 0.01, ***P < 0.001.
3.7. Oestrogen negatively regulates expression of Mdr1 and Cyp1a1
To examine the effect of the sex hormone E2 on transport and metabolism of Sunitinib in cardiomyocytes, NRVMs were exposed to Sunitinib for 36 h in the presence or absence of physiological concentrations of E2. Expression of Mdr1 was increased in NRVMs treated with Sunitinib, and this increase was blocked by co-treatment with 100 nM or 1 μM E2 (Figure 6B). As with other cell types,32 upon treatment with Sunitinib, expression of Cyp1A1 in NRVMs was increased significantly. However, when co-treated with 100 nM, a physiological concentration for rat, or 1 μM E2, a supraphysiological concentration, this increase was reduced (Figure 6C). Expression of Anf mRNA was increased in the presence of E2 alone or combined with Sunitinib relative to Sunitinib alone. Co-treatment of Sunitinib with 100 pM PE abrogated the increased in Anf induced by Sunitinib. Combined treatment of 10 μM Ellipticine, a CYP1A1 inhibitor, increased Anf expression to above the level induced by Sunitinib alone (Figure 6D). Therefore, the presence of E2 reduces expression of proteins required for metabolism and efflux of Sunitinib, thereby implicating E2 in the increased cardiotoxicity induced by Sunitinib.
3.8. Functional and molecular cardiotoxicity is observed in Sunitinib-treated mice
In light of modulation of cardiotoxicity by biological sex and E2, male and female mice were administered Sunitinib to determine whether the cardiotoxicity and proposed mechanisms apply in vivo. Our analyses revealed cardiac dysfunction, increased left-ventricular fibrosis indicative of cell death, and increased fetal gene expression in female mice after 28 days of Sunitinib administration (6–7 animals per group). Relative to vehicle-treated sex-matched mice, percent fractional shortening (%FS) significantly decreased in females (−15.0%, p < 0.05) and was unchanged in male mice treated with Sunitinib. There was evidence for ventricular dilation only in females treated with Sunitinib, including a 15% (P < 0.01) decrease in anterior ventricular wall thicknesses, 40.5% (P < 0.01) increase in left-ventricular volume during systole (LV Vol;s), and 28.5% (P < 0.05) decrease in LV mass. Unlike in females, the hearts of males treated with Sunitinib exhibited no significant functional or morphological alterations (Figure 7A–C).
Figure 7.

Mice receiving Sunitinib exhibit sexually dimorphic functional and molecular cardiotoxicity. (A) Percent fractional shortening (%FS), (B) Left-ventricular anterior wall thickness during systole (LVAW;s) in female (light grey bars) and male (dark grey bars) mice receiving vehicle (open bars) or Sunitinib (hatched bars) for 28 days. (C) Left-ventricular volume during systole (LV Vol;s), (D–F) Expression of fetal genes (D) MDR1 (E) and CYP1A1 (F) in the left ventricles of female and male mice. Female mice, light grey bars; male mice, dark grey bars. (A–C, E and F) two-way ANOVA followed by Tukey's post-hoc test, (D) Student's t-test comparing expression of individual genes between males and females. (A–F) Error bars represent SEM. *P < 0.05, **P < 0.01,***P < 0.001, n = 6–7 mice per sex.
To determine whether the fetal program of gene expression is induced as in isolated cardiomyocytes, expression of fetal genes was measured in the left ventricles of 13 females [seven receiving Sunitinib and six receiving vehicle (DMSO)] and 14 males (seven receiving Sunitinib and seven receiving vehicle). Levels of Anf, Bnp, β-MyHC, and αSkActin mRNAs were significantly elevated in the hearts of female mice receiving Sunitinib, whereas levels were unchanged in the hearts of male mice (Figure 7D). For example, mRNA levels for Anf and Bnp were increased to 5.9-fold (P < 0.001) and 1.8-fold (P < 0.01), respectively, in females and did not change significantly in males. Our data reveal a sexual dimorphism in the cardiac response to just one course of Sunitinib exposure, in agreement with a recent clinical study that identified female sex as a risk factor for severe general toxicity requiring discontinuation of treatment in females receiving Sunitinib therapy.10
Sex differences in basal expression of genes encoding efflux transporters and enzymes involved in xenobiotic metabolism have been observed in other tissues.33 To examine the expression of Mdr1 in the hearts of male and female mice, mRNA was isolated from ventricles from 6 to 8 mice of each sex that received one 28-day course of vehicle or Sunitinib. Although there were no differences in baseline expression between males and females, the induction of Mdr1 expression was 22% greater in males than females when treated with Sunitinib for 28 days (Figure 7E), in agreement with our experiments in ARVMs. In mice, expression of Cyp1A1 was significantly higher in males than females (Figure 7F). As expected, with Sunitinib, expression of Cyp1A1 was increased, however, this was observed only in males but not females (Figure 7F).
4. Discussion
In the studies presented here, we used both in vitro and in vivo methods to: (i) define the specific kinases inhibited by Sunitinib in cardiomyocytes and in cardiac tissue and how it compares with that of other TKIs; (ii) examine the effects of biological sex on Sunitinib-induced cardiotoxicity, and (iii) identify the molecular mechanism by which Sunitinib induces cardiomyocyte death. Importantly, using antibody arrays, we measured phosphorylation states of 39 RTKs and 46 signalling molecules to characterize the effect of Sunitinib on molecular signalling in cardiomyocytes. We identified sexually dimorphic cardiotoxicity that negatively affected females but not males, and by targeting adrenergic receptors, demonstrated that activation of the PKC pathway could abrogate this toxicity. Our results implicate E2 in a mechanism involving drug efflux as contributing to the cardiotoxicity observed in females.
4.1. Mechanisms of cardiotoxicity induced by Sunitinib
The data presented here suggest that a lack of targeting specificity contributes to cardiotoxicity in mammalian cardiomyocytes treated with Sunitinib as well as with other TKIs. Although AMPK was previously implicated as mediating cardiotoxicity in patients receiving Sunitinib,5 a complex network of inhibited intracellular signalling molecules includes p38-MSK1-CREB was also observed; activation of this pathway is required for cardiac hypertrophy and is induced by α1-adrenergic stimulation by PE.22,34 However, a novel mechanism involving inhibition by Sunitinib that converges with the PKC and PLCγ pathways as well as activation of extrinsic pathways of apoptosis is supported by our data.
Proteins involved in extrinsic induction of apoptosis including Fas and FasL were up-regulated by Sunitinib in cardiomyocytes. Concomitant increases in FADD, a member of the Fas receptor complex, and TRAIL, which also forms a receptor complex with FADD were also observed. Interestingly, Fas signalling in cardiomyocytes can have functions unrelated to apoptosis including induction of hypertrophy35 as well as up-regulation in the failing human heart near areas of fibrosis.36 Up-regulation of Fas has also been observed in human heart failure. In agreement with this finding, using the same Sunitinib treatment paradigm in mice, skin biopsies revealed a significant up-regulation of Fas and Fas ligand (FasL) and increased cleaved caspase-3 in keratinocytes.37
4.2. Drug metabolism and transport may be altered by Sunitinib in a sexually dimorphic manner
Several drugs negatively modulate expression and activity of drug efflux transporters and metabolic enzymes and increase effective intracellular concentrations of drug.38,39 Indeed, polymorphisms in these genes have been identified as possible risk factors among women who experience toxicities with Sunitinib.40 For example, genetic polymorphisms in cancer cells that reduce the activity of CYP1A1 and CYP3A4, the enzymes primarily responsible for the metabolism of Sunitinib and other TKIs, and MDR1, are responsible for increased progression-free survival via increased intracellular drug concentrations.40 We therefore examined the role of these important proteins in the cardiotoxicity induced by Sunitinib. The relationship between Sunitinib and expression of these genes may be explained by the involvement of the aryl hydrocarbon receptor (AhR), a nuclear receptor that is activated by exposure to xenobiotics and toxic environmental agents and regulates transcription of metabolizing enzymes such as CYP1A1. Ligand-independent activation of AhR by Sunitinib has been shown to regulate expression of CYP1A1 and MDR1 in breast cancer cells.32 A limitation of these studies is that, despite mRNA expression being indicative of protein expression and activity,41–43 these measurements were not made so definitive conclusions require further study.
4.3. E2 negatively modulates expression of a drug metabolizing enzyme and efflux transporter in cardiomyocytes
There exists an ongoing debate about the role of E2 in protection from genetic and environmental cardiovascular insult.44 Models of cardiac disease such as myocardial infarction and atherosclerosis demonstrate cardioprotection in females that is attributable to the presence of E2.45 However, considering our data in mice and in ARVMs, it appears that E2 is detrimental to females receiving Sunitinib. Indeed, female sex is a risk factor for developing toxicities with several types of chemotherapeutic, including Sunitinib.40,46 Female sex may not have been considered because the incidence of gastrointestinal stromal tumour, renal cell carcinoma, and pancreatic cancer, diseases for which Sunitinib is approved, is significantly higher in males.47–49
Sunitinib is primarily metabolized by the hepatic cytochrome P450, CYP3A4. However, the extrahepatic enzyme, CYP1A1, has recently been identified as a participant in the metabolism of Sunitinib.32 CYP1A1 is expressed in cardiomyocytes, and expression is induced by Sunitinib in cancer cells32,50 as well as in the hearts of rats receiving Sunitinib via a mechanism involving ligand-independent activation of AhRs.51 Interestingly, CYP1A1 is also involved in metabolism of E2, and E2 itself can reduce the induction of CYP1A1 in endometrial cells.52 E2 is also capable of down-regulating expression of the MDR1 efflux transporter that eliminates Sunitinib from cells, thus increasing cellular sensitivity to drugs and other toxins.53 Sex differences in hepatic drug metabolism have been well-described and implicate cytochrome P450s like CYP1A1 and CYP3A4 that are differentially regulated by sex, thyroid, and growth hormones.30 Our studies are the first to our knowledge to examine this phenomenon in the heart. By reducing expression of the proteins primarily responsible for Sunitinib clearance, Cyp1A1 and MDR1, the concentration of Sunitinib in cardiomyocytes would be higher and increased inhibition of kinases would be observed.
We are currently examining the molecular interactions between E2 and TKIs and their relationship to biological sex. We hypothesize that inhibition mediated by Sunitinib is exacerbated in females by the presence of endogenous E2, which itself has inhibitory effects on kinase phosphorylation levels (data not shown). Low levels of E2 in males could prevent this effect in males receiving Sunitinib; however, we postulate that administration of exogenous E2 would have more detrimental effects in males likely due to lower basal expression/phosphorylation of RTKs and intracellular kinases in the hearts of males. Indeed, when male mice are treated with E2, Inhibition of kinases by E2 has been observed in other systems; administration of E2 inhibits activity of MAP kinase activity in smooth muscle cells in an E2 receptor-dependent manner.54
4.4. Subpressor concentrations of phenylephrine may confer cardioprotection in patients receiving Sunitinib
The intersection of pathways involved in α-adrenergic signalling and RTKs suggested that PE, an α1-adrenergic activator, could be used to examine the roles of PKC and PLCγ in the cardiotoxicity induced by Sunitinib and could represent a viable intervention in the development of cardiotoxicity with TKIs. The cardioprotective effects of PE and α1-adrenergic receptor stimulation have been explored in pathological stimuli in the heart including ischaemia.55 Concentrations used to achieve this effect cause hypertension,55 a known risk factor for developing cardiotoxicity with Sunitinib. Additionally, alteration of ion channel function and availability of ATP via activation of PKC have been implicated; however, detailed analyses of the signaling pathways involved in the cardioprotective effect have not been performed.56 Although beyond the scope of the current study, it would be interesting to test PE as a cardioprotective agent that could be co-administered with Sunitinib. We hypothesize that co-administration of subpressor PE and Sunitinib would not interfere with the effects of Sunitinib on cancer cells because expression of α-1 adrenergic receptors is largely restricted to cells in the cardiovascular system.57
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by National Institutes of Health (2R01HL050560 to L.A.L.) and the Marisco Chair of Excellence (L.A.L.), National Institutes of Health Postgraduate Training Grant in Cardiovascular Research (T32 HL-07822 to P.A.H.), and American Heart Association Postdoctoral Fellowship (11POST7780011 to P.A.H.).
Acknowledgements
We are grateful to the following colleagues who provided technical assistance and advice on these experiments: Kelly Ambler, Ann Robinson, Ben Barthel, and Kristen Barthel. We also thank Sarah Aguirre, who participated in the optimization of immunohistochemistry experiments.
Conflict of interest: none declared.
References
- 1.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009;9:28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, Woulfe K, Pravda E, Cassiola F, Desai J, George S, Morgan JA, Harris DM, Ismail NS, Chen JH, Schoen FJ, Van den Abbeele AD, Demetri GD, Force T, Chen MH. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 2007;370:2011–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall PH, Harshman LC, Srinivas S, Witteless RM. The frequency and severity of cardiovascular toxicity from targeted therapy in advanced renal cell carcinoma patients. JACC Heart Failure 2013;1:72–78. [DOI] [PubMed] [Google Scholar]
- 4.Telli ML, Witteles RM, Fisher GA, Srinivas S. Cardiotoxicity associated with the cancer therapeutic agent sunitinib malate. Ann Oncol 2008;19:1613–1618. [DOI] [PubMed] [Google Scholar]
- 5.Kerkela R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand JB, Force T. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med 2006;12:908–916. [DOI] [PubMed] [Google Scholar]
- 6.Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, Chen JS, Horsch D, Hammel P, Wiedenmann B, Van Cutsem E, Patyna S, Lu DR, Blanckmeister C, Chao R, Ruszniewski P. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 2011;364:501–513. [DOI] [PubMed] [Google Scholar]
- 7.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008;26:127–132. [DOI] [PubMed] [Google Scholar]
- 8.Kerkela R, Woulfe KC, Durand JB, Vagnozzi R, Kramer D, Chu TF, Beahm C, Chen MH, Force T. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of amp-activated protein kinase. Clin Transl Sci 2009;2:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasinoff BB, Patel D, O'Hara KA. Mechanisms of myocyte cytotoxicity induced by the multiple receptor tyrosine kinase inhibitor sunitinib. Mol Pharmacol 2008;74:1722–1728. [DOI] [PubMed] [Google Scholar]
- 10.van der Veldt AA, Boven E, Helgason HH, van Wouwe M, Berkhof J, de Gast G, Mallo H, Tillier CN, van den Eertwegh AJ, Haanen JB. Predictive factors for severe toxicity of sunitinib in unselected patients with advanced renal cell cancer. Br J Cancer 2008;99:259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maass AH, Buvoli M. Cardiomyocyte preparation, culture, and gene transfer. Methods Mol Biol 2007;366:321–330. [DOI] [PubMed] [Google Scholar]
- 12.Haines CD, Harvey PA, Luczak ED, Barthel KK, Konhilas JP, Watson PA, Stauffer BL, Leinwand LA. Estrogenic compounds are not always cardioprotective and can be lethal in males with genetic heart disease. Endocrinology 2012;153:4470–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi MS, Tong L, Cook AC, Schanbacher BL, Huang H, Han B, Ayers LW, Bauer JA. Increased myocardial prevalence of C-reactive protein in human coronary heart disease: Direct effects on microvessel density and endothelial cell survival. Cardiovasc Pathol 2012;21:428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stauffer BL, Konhilas JP, Luczak ED, Leinwand LA. Soy diet worsens heart disease in mice. J Clin Invest 2006;116:209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q, Moncman CL, Winkelmann DA. Mutations in the motor domain modulate myosin activity and myofibril organization. J Cell Sci 2003;116:4227–4238. [DOI] [PubMed] [Google Scholar]
- 16.Cosper PF, Harvey PA, Leinwand LA. Interferon-gamma causes cardiac myocyte atrophy via selective degradation of myosin heavy chain in a model of chronic myocarditis. Am J Pathol 2012;181:2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lankheet NA, Knapen LM, Schellens JH, Beijnen JH, Steeghs N, Huitema AD. Plasma concentrations of tyrosine kinase inhibitors imatinib, erlotinib, and sunitinib in routine clinical outpatient cancer care. Ther Drug Monit 2014;36:326–334. [DOI] [PubMed] [Google Scholar]
- 18.Lankheet NA, Blank CU, Mallo H, Adriaansz S, Rosing H, Schellens JH, Huitema AD, Beijnen JH. Determination of sunitinib and its active metabolite n-desethylsunitinib in sweat of a patient. J Anal Toxicol 2011;35:558–565. [DOI] [PubMed] [Google Scholar]
- 19.Harvey PA, Leinwand LA. The cell biology of disease: Cellular mechanisms of cardiomyopathy. J Cell Biol 2011;194:355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Driesen RB, Verheyen FK, Debie W, Blaauw E, Babiker FA, Cornelussen RN, Ausma J, Lenders MH, Borgers M, Chaponnier C, Ramaekers FC. Re-expression of alpha skeletal actin as a marker for dedifferentiation in cardiac pathologies. J Cell Mol Med 2009;13:896–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fantl WJ, Johnson DE, Williams LT. Signalling by receptor tyrosine kinases. Annu Rev Biochem 1993;62:453–481. [DOI] [PubMed] [Google Scholar]
- 22.Montagne O, Le Corvoisier P, Guenoun T, Laplace M, Crozatier B. Impaired alpha1-adrenergic responses in aged rat hearts. Fundam Clin Pharmacol 2005;19:331–339. [DOI] [PubMed] [Google Scholar]
- 23.Prasad AM, Ma H, Sumbilla C, Lee DI, Klein MG, Inesi G. Phenylephrine hypertrophy, Ca2+-ATPase (SERCA2), and Ca2+ signaling in neonatal rat cardiac myocytes. Am J Physiol Cell Physiol 2007;292:C2269–C2275. [DOI] [PubMed] [Google Scholar]
- 24.Vecchione C, Fratta L, Rizzoni D, Notte A, Poulet R, Porteri E, Frati G, Guelfi D, Trimarco V, Mulvany MJ, Agabiti-Rosei E, Trimarco B, Cotecchia S, Lembo G. Cardiovascular influences of alpha1b-adrenergic receptor defect in mice. Circulation 2002;105:1700–1707. [DOI] [PubMed] [Google Scholar]
- 25.Lee IH, You JO, Ha KS, Bae DS, Suh PG, Rhee SG, Bae YS. AHNAK-mediated activation of phospholipase C-gamma1 through protein kinase C. J Biol Chem 2004;279:26645–26653. [DOI] [PubMed] [Google Scholar]
- 26.Chatsiproios D, Haidinger R, Suter T. Practical management recommendations for anti-ErbB2 therapy with lapatinib. Breast Care (Basel) 2008;3:13–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen MH, Kerkela R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation 2008;118:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu X, Cang H, Li C, Zhao ZK, Li H. Quartz crystal microbalance sensor array for the detection of volatile organic compounds. Talanta 2009;78:711–716. [DOI] [PubMed] [Google Scholar]
- 29.Schmidinger M, Zielinski CC, Vogl UM, Bojic A, Bojic M, Schukro C, Ruhsam M, Hejna M, Schmidinger H. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 2008;26:5204–5212. [DOI] [PubMed] [Google Scholar]
- 30.Kato R, Yamazoe Y. Sex-specific cytochrome p450 as a cause of sex- and species-related differences in drug toxicity. Toxicol Lett 1992;64–65 Spec No:661–667. [DOI] [PubMed] [Google Scholar]
- 31.Isoherranen N, Thummel KE. Drug metabolism and transport during pregnancy: How does drug disposition change during pregnancy and what are the mechanisms that cause such changes? Drug Metab Dispos 2013;41:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maayah ZH, El Gendy MA, El-Kadi AO, Korashy HM. Sunitinib, a tyrosine kinase inhibitor, induces cytochrome P450 1A1 gene in human breast cancer MCF7 cells through ligand-independent aryl hydrocarbon receptor activation. Arch Toxicol 2013;87:847–856. [DOI] [PubMed] [Google Scholar]
- 33.Navas JM, Segner H. Estrogen-mediated suppression of cytochrome P4501A (CYP1a) expression in rainbow trout hepatocytes: role of estrogen receptor. Chem Biol Interact 2001;138:285–298. [DOI] [PubMed] [Google Scholar]
- 34.Markou T, Hadzopoulou-Cladaras M, Lazou A. Phenylephrine induces activation of CREP in adult rat cardiac myocytes through MSK1 and PKA signaling pathways. J Mol Cell Cardiol 2004;37:1001–1011. [DOI] [PubMed] [Google Scholar]
- 35.Wollert KC, Heineke J, Westermann J, Ludde M, Fiedler B, Zierhut W, Laurent D, Bauer MK, Schulze-Osthoff K, Drexler H. The cardiac Fas (APO-1/CD95) receptor/Fas ligand system: relation to diastolic wall stress in volume-overload hypertrophy in vivo and activation of the transcription factor AP-1 in cardiac myocytes. Circulation 2000;101:1172–1178. [DOI] [PubMed] [Google Scholar]
- 36.Filippatos G, Leche C, Sunga R, Tsoukas A, Anthopoulos P, Joshi I, Bifero A, Pick R, Uhal BD. Expression of Fas adjacent to fibrotic foci in the failing human heart is not associated with increased apoptosis. Am J Physiol 1999;277:H445–H451. [DOI] [PubMed] [Google Scholar]
- 37.Yeh CN, Chung WH, Su SC, Chen YY, Cheng CT, Lin YL, Chang WC, Chung-Yee Hui R, Chiang KC, Chen TW, Jan YY, Chen CW, Chen TJ, Yang CH, Hung SI. Fas/Fas ligand mediates keratinocyte death in sunitinib-induced hand-foot skin reaction. J Invest Dermatol 2014;134:2768–2775. [DOI] [PubMed] [Google Scholar]
- 38.Harker WG, Bauer D, Etiz BB, Newman RA, Sikic BI. Verapamil-mediated sensitization of doxorubicin-selected pleiotropic resistance in human sarcoma cells: selectivity for drugs which produce DNA scission. Cancer Res 1986;46:2369–2373. [PubMed] [Google Scholar]
- 39.Hsiao P, Sasongko L, Link JM, Mankoff DA, Muzi M, Collier AC, Unadkat JD. Verapamil p-glycoprotein transport across the rat blood-brain barrier: cyclosporine, a concentration inhibition analysis, and comparison with human data. J Pharmacol Exp Ther 2006;317:704–710. [DOI] [PubMed] [Google Scholar]
- 40.van der Veldt AA, Eechoute K, Gelderblom H, Gietema J, Guchelaar HJ, van Erp NP, van den Eertwegh AJ, Haanen JB, Mathijssen RH, Wessels JA. Genetic polymorphisms associated with a prolonged progression-free survival in patients with metastatic renal cell cancer treated with sunitinib. Clin Cancer Res 2011;17:620–629. [DOI] [PubMed] [Google Scholar]
- 41.Krishnamurthy K, Vedam K, Kanagasabai R, Druhan LJ, Ilangovan G. Heat shock factor-1 knockout induces multidrug resistance gene, MDR1b, and enhances P-glycoprotein (ABCB1)-based drug extrusion in the heart. Proc Natl Acad Sci USA 2012;109:9023–9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laguens RP, Lazarowski AJ, Cuniberti LA, Vera Janavel GL, Cabeza Meckert PM, Yannarelli GG, del Valle HF, Lascano EC, Negroni JA, Crottogini AJ. Expression of the MDR-1 gene-encoded P-glycoprotein in cardiomyocytes of conscious sheep undergoing acute myocardial ischemia followed by reperfusion. J Histochem Cytochem 2007;55:191–197. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Nachtigal MW, Kardami E, Cattini PA. FGF-2 protects cardiomyocytes from doxorubicin damage via protein kinase c-dependent effects on efflux transporters. Cardiovasc Res 2013;98:56–63. [DOI] [PubMed] [Google Scholar]
- 44.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women's health initiative randomized controlled trial. JAMA 2002;288:321–333. [DOI] [PubMed] [Google Scholar]
- 45.Barta E, Strec V, Styk J, Okolicany J, Rajecova O. Protective effect of oestradiol on the heart of rats exposed to acute ischaemia. Physiol Bohemoslov 1989;38:193–200. [PubMed] [Google Scholar]
- 46.Stein BN, Petrelli NJ, Douglass HO, Driscoll DL, Arcangeli G, Meropol NJ. Age and sex are independent predictors of 5-fluorouracil toxicity. Analysis of a large scale phase iii trial. Cancer 1995;75:11–17. [DOI] [PubMed] [Google Scholar]
- 47.Chiang NJ, Chen LT, Tsai CR, Chang JS. The epidemiology of gastrointestinal stromal tumors in Taiwan, 1998–2008: a nation-wide cancer registry-based study. BMC Cancer 2014;14:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chow WH, Dong LM, Devesa SS. Epidemiology and risk factors for kidney cancer. Nat Rev Urol 2010;7:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013;144:1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alfieri RR, Galetti M, Tramonti S, Andreoli R, Mozzoni P, Cavazzoni A, Bonelli M, Fumarola C, La Monica S, Galvani E, De Palma G, Mutti A, Mor M, Tiseo M, Mari E, Ardizzoni A, Petronini PG. Metabolism of the EGFR tyrosin kinase inhibitor gefitinib by cytochrome P450 1A1 enzyme in EGFR-wild type non small cell lung cancer cell lines. Mol Cancer 2011;10:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maayah ZH, Ansari MA, El Gendy MA, Al-Arifi MN, Korashy HM. Development of cardiac hypertrophy by sunitinib in vivo and in vitro rat cardiomyocytes is influenced by the aryl hydrocarbon receptor signaling pathway. Arch Toxicol 2014;88:725–738. [DOI] [PubMed] [Google Scholar]
- 52.Ricci MS, Toscano DG, Mattingly CJ, Toscano WA., Jr Estrogen receptor reduces CYP1A1 induction in cultured human endometrial cells. J Biol Chem 1999;274:3430–3438. [DOI] [PubMed] [Google Scholar]
- 53.Mutoh K, Tsukahara S, Mitsuhashi J, Katayama K, Sugimoto Y. Estrogen-mediated post transcriptional down-regulation of P-glycoprotein in MDR1-transduced human breast cancer cells. Cancer Sci 2006;97:1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dubey RK, Jackson EK, Gillespie DG, Zacharia LC, Imthurn B, Keller PJ. Clinically used estrogens differentially inhibit human aortic smooth muscle cell growth and mitogen-activated protein kinase activity. Arterioscler Thromb Vasc Biol 2000;20:964–972. [DOI] [PubMed] [Google Scholar]
- 55.Cohen MV, Yang XM, Liu GS, Heusch G, Downey JM. F, bradykinin, opioids, and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial k(atp) channels. Circ Res 2001;89:273–278. [DOI] [PubMed] [Google Scholar]
- 56.Pyle WG, Smith TD, Hofmann PA. Cardioprotection with kappa-opioid receptor stimulation is associated with a slowing of cross-bridge cycling. Am J Physiol Heart Circ Physiol 2000;279:H1941–H1948. [DOI] [PubMed] [Google Scholar]
- 57.Graham RM, Perez DM, Hwa J, Piascik MT. Alpha 1-adrenergic receptor subtypes. Molecular structure, function, and signaling. Circ Res 1996;78:737–749. [DOI] [PubMed] [Google Scholar]