

Abstract
Objective:
To further characterize mitochondrial dysfunction in LRRK2G2019S mutant Parkinson disease (PD) patient tissue (M-LRRK2G2019S), determine whether ursodeoxycholic acid (UDCA) also exerts a beneficial effect on mitochondrial dysfunction in nonmanifesting LRRK2G2019S mutation carriers (NM-LRRK2G2019S), and assess UDCA for its beneficial effect on neuronal dysfunction in vivo.
Methods:
Intracellular adenosine 5′-triphosphate (ATP) levels, oxygen consumption, and activity of the individual complexes of the mitochondrial respiratory chain as well as mitochondrial morphology were measured in M-LRRK2G2019S, NM-LRRK2G2019S, and controls. UDCA was assessed for its rescue effect on intracellular ATP levels in NM-LRRK2G2019S and in a LRRK2 transgenic fly model with dopaminergic expression of LRRK2G2019S.
Results:
Crucial parameters of mitochondrial function were similarly reduced in both M-LRRK2G2019S and NM-LRRK2G2019S with a specific decrease in respiratory chain complex IV activity. Mitochondrial dysfunction precedes changes in mitochondrial morphology but is normalized after siRNA-mediated knockdown of LRRK2. UDCA improved mitochondrial function in NM-LRRK2G2019 and rescued the loss of visual function in LRRK2G2019S flies.
Conclusion:
There is clear preclinical impairment of mitochondrial function in NM-LRRK2G2019S that is distinct from the mitochondrial impairment observed in parkin-related PD. The beneficial effect of UDCA on mitochondrial function in both NM-LRRK2G2019S and M-LRRK2G2019S as well as on the function of dopaminergic neurons expressing LRRK2G2019S suggests that UDCA is a promising drug for future neuroprotective trials.
The LRRK2G2019S mutation is the single most common monogenically inherited cause of Parkinson disease (PD).1 LRRK2G2019S leads to increased LRRK2 kinase activity with resulting cellular dysfunction and neuronal cell loss but the precise intracellular mechanisms leading to PD remain to be elucidated.2–4 There is age-dependent, reduced penetrance of LRRK2G2019S, which suggests the presence of biological rescue mechanisms in some individuals.5 This in turn raises hope that identification of suitable biological targets may prevent or at least reduce the risk of currently asymptomatic or nonmanifesting LRRK2G2019S carriers (NM-LRRK2G2019S) to undergo phenotypic conversion to manifesting carriers (M-LRRK2G2019S) by developing clinically manifest PD.
We and others have previously reported mitochondrial dysfunction in fibroblasts from patients with PD with the LRRK2G2019S mutation.6,7 We subsequently described complete rescue of mitochondrial dysfunction in PD tissue after treatment with ursodeoxycholic acid (UDCA).8 We now expand our studies to demonstrate a pattern of mitochondrial dysfunction in both M-LRRK2G2019S and NM-LRRK2G2019S that is distinct from the predominant complex I impairment observed in early-onset PD (EOPD) due to parkin or PINK1 mutations.9,10 Drosophila with specific expression of LRRK2G2019S in dopaminergic neurons display progressive loss of photoreceptor function. The photoreceptor mitochondria are dilated and have disorganized, aberrant cristae.11,12 We further demonstrate that UDCA exerts a beneficial effect on mitochondrial dysfunction in NM-LRRK2G2019S and report a beneficial effect of UDCA in vivo.
The results of our studies suggest that a clinical trial investigating the beneficial effect of UDCA in both NM-LRRK2G2019S and M-LRRK2G2019S should be considered.
METHODS
Research participants.
Punch skin biopsies were taken from 5 LRRK2G2019S mutation carriers with clinically manifest PD (M-LRRK2G2019S), 5 LRRK2G2019S mutation carriers without clinically manifest PD (NM-LRRK2G2019S), and 5 controls. There was no significant difference in age between the control and patient group (controls 62.8 ± 6.6 years, NM-LRRK2G2019S 49.8 ± 16.5 years, M-LRRK2G2019S 64.6 ± 9.3 years). Four of five M-LRRK2G2019S had been included in a previous study.6
Standard protocol approval, registrations, and patient consents.
We received approval from the regional ethical standards committee on human experimentation for this scientific project and written informed consent was obtained from all research participants in the study.
Mitochondrial function and UDCA treatment.
All biochemical measurements were performed on 3 separate cell preparations from different cell passages. Morphologic assessments were carried out on 30 fields of view in 3 wells per cell line per day and then on 3 separate occasions. All experiments were performed on cells from M-LRRK2G2019S, NM-LRRK2G2019S, and controls matched to within 1 passage of each other. Fibroblast culture conditions, measurements of total intracellular adenosine 5′-triphosphate (ATP) levels, and spectrophotometric assessment of respiratory chain complexes were undertaken as previously described.10 Fibroblasts from 5 NM-LRRK2G2019S and 5 matched controls were treated with 10 nM UDCA 24 hours after plating. Assays were performed after 24 hours of drug treatment. Mitochondrial oxygen consumption rate measurements were undertaken using the Seahorse Bioscience (North Billerica, MA) XF24 analyzer. All oxygen consumption rates are expressed normalized to cell number per well. See appendix e-1 on the Neurology® Web site at Neurology.org for further details of oxygen consumption measurement.
siRNA knockdown of LRRK2.
siRNA oligonucleotides were targeted to the sequence UUACCGAGAUGCCGUAUUA of the LRRK2 gene. Ten-nanometer siRNAs (against LRRK2 targeted or scramble negative or GAPDH positive) were transfected into control primary fibroblasts using 0.5 mM lipofectamine 2000 according to the manufacturers' instructions (Dharmacon, Loughborough, UK). Transfection efficiency was assessed by flow cytometry of scramble (fluorescently labeled) siRNA transfected cells. LRRK2 protein levels after siRNA knockdown was assessed by Western blotting at 72 hours post transfection as previously described.6 Seventy-two hours after transfection, cells were assayed for their cellular ATP levels as described above. Cellular toxicity was assessed after LRRK2 siRNA knockdown by trypan blue staining; there was no evidence of the LRRK2 siRNA being any more toxic than the scramble siRNA or transfection reagent alone, all of which elicited a small percentage of cellular toxicity of approximately 8%.
Effect of UDCA on neuronal function in vivo using dopaminergic expression of LRRK2G2019S in Drosophila.
Dopaminergic expression of the human transgenes hLRRK2 (wild-type) or LRRK2G2019S was achieved as previously described.11,12 Newly emerged female flies were transferred to vials of instant food (Carolina Biological Supply, Burlington, NC) containing no drug or 2.5 µM of UDCA in a pulsating light incubator at 29°C. Flies were transferred to fresh food every 2–3 days. Visual responses were recorded at 7 days using the steady-state visual evoked potentials (SSVEP) technique. Responses of photoreceptors, lamina neurons, and medulla neurons were separated using fast-Fourier transform (Matlab; MathWorks, Inc., Natick, MA).
Statistical analysis.
Cell culture experiments.
Values from multiple experiments were expressed as means ± SD. Statistical significance was assessed using a 2-way analysis of variance (ANOVA) with Bonferroni post correction.
Drosophila experiments.
The peak response for each genotype/drug combination was exported to SPSS (v22) (SPSS Inc., Chicago, IL) and tested by 2-way ANOVA. To achieve statistical independence, the contrast response functions were fitted by hyperbolic ratio functions and bootstrapped (Matlab, source code at https://github.com/wadelab/flyCode). The ANOVA of the bootstrapped parameters was performed in SPSS.
RESULTS
Mitochondrial dysfunction is independent of clinical manifestations in LRRK2G2019S mutation carriers.
Intracellular ATP levels were similarly decreased in M-LRRK2G2019S and NM-LRRK2G2019S with a decrease by 38% in M-LRRK2G2019S and a decrease by 35% in NM-LRRK2G2019S compared to controls (p < 0.05, see figure 1A). Similarly, basal mitochondrial oxygen consumption was reduced by 25% in both M-LRRK2G2019S and NM-LRRK2G2019S (p < 0.05, see figure 1B). At maximal respiration, there was more marked reduction still of oxygen consumption by 42% in both M-LRRK2G2019S and NM-LRRK2G2019S compared to controls (p < 0.05, see figure 1C). Coupled respiration was also reduced in both M-LRRK2G2019S and NM-LRRK2G2019S but more markedly so in M-LRRK2G2019S (80% reduction, p < 0.01) than in NM-LRRK2G2019S (60% reduction, p < 0.05, see figure 1D).
Figure 1. Mitochondrial dysfunction in LRRK2G2019S.

(A) Adenosine 5′-triphosphate levels are reduced to similar levels in fibroblasts from both manifesting LRRK2G2019S carriers (M-LRRK2G2019S) and nonmanifesting LRRK2G2019S carriers (NM-LRRK2G2019S) compared to controls (*p < 0.05). (B) Oxygen consumption for control (circles, dotted line), LRRK2G2019S nonmanifesting (squares, gray line), and LRRK2G2019S manifesting (triangles, black line) patient fibroblasts under basal conditions and after the addition of oligomycin (•), FCCP (••), and rotenone (•••). Basal oxygen consumption is reduced in both M-LRRK2G2019S and NM-LRRK2G2019S compared to controls. (C) Maximal respiration (after the addition of FCCP) is reduced in both LRRK2G2019S nonmanifesting and LRRK2G2019S manifesting patient fibroblasts as compared to controls (*p < 0.05). (D) Coupled respiration (calculated using the basal rate and the rate after addition of oligomycin) is also reduced in both M-LRRK2G2019S and NM-LRRK2G2019S compared to controls (*p < 0.05, **p < 0.01).
siRNA knockdown of LRRK2 rescues cellular ATP levels.
LRRK2 protein levels were reduced by 80% in siRNA treated fibroblasts compared to mock transfected controls at 3 days post transfection (figure 2A). Cellular ATP levels were significantly higher in siRNA-mediated LRRK2 knockdown in NM-LRRK2G2019S and M-LRRK2G2019S fibroblasts at 3 days post transfection than scramble or GAPDH transfected fibroblasts (figure 2B, p < 0.01). Thus, reduction in LRRK2G2019S protein levels is sufficient to restore mitochondrial function in both NM-LRRK2G2019S and M-LRRK2G2019S fibroblasts.
Figure 2. siRNA knockdown of LRRK2 recovers intracellular adenosine 5′-triphosphate levels.
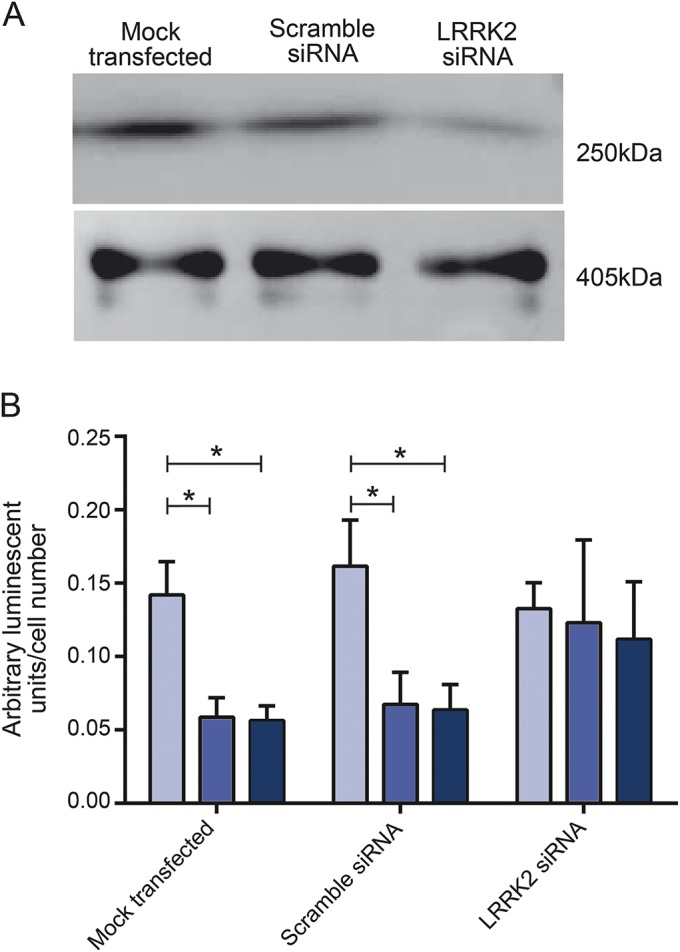
(A) Western blot shows LRRK2 protein levels (top panel) and actin protein levels (bottom panel) after mock transfection, scramble siRNA, or LRRK2 siRNA in manifesting LRRK2G2019S carrier (M-LRRK2G2019S) fibroblasts, showing knockdown of LRRK2 protein. (B) Adenosine 5′-triphosphate levels are reduced to similar levels in fibroblasts from both M-LRRK2G2019S (black bars) and nonmanifesting LRRK2G2019S carriers (NM-LRRK2G2019S) (gray bars) compared to controls (white bars) in both mock transfected and scramble transfected fibroblasts (*p < 0.05). Both M-LRRK2G2019S and NM-LRRK2G2019S fibroblasts transfected with LRRK2 siRNA have comparable levels to controls and significantly increased from scramble transfected levels (*p < 0.05).
Distinct pattern of impaired mitochondrial function in LRRK2G2019S mutation carriers.
We previously reported specific reduction in complex I activity of the mitochondrial respiratory chain in fibroblasts of patients with EOPD due to homozygous or compound heterozygous parkin mutations.10 Analysis of the individual complexes of the mitochondrial respiratory chain was now also undertaken in M-LRRK2G2019S and NM-LRRK2G2019S to determine whether the pattern of mitochondrial dysfunction may be similar or distinct in PARK2-related EOPD and PARK8-related late-onset PD (figure 3). In contrast to our observations in parkin-mutant patient tissue, complex I and II activity was normal in both manifesting and nonmanifesting LRRK2G2019S mutation carriers (complex I, controls: 0.36 ± 0.12, NM-LRRK2G2019S: 0.52 ± 0.19, M-LRRK2G2019S: 0.35 ± 0.09; complex II, controls: 0.27 ± 0.08, NM-LRRK2G2019S: 0.32 ± 0.15, M-LRRK2G2019S: 0.35 ± 0.09; mean ± SD). Complex IV activity, however, was markedly decreased in both M-LRRK2G2019S and NM-LRRK2G2019S (controls: 100.6 ± 21.8, NM-LRRK2G2019S: 41.5 ± 8.8, M-LRRK2G2019S: 18.2 ± 5.1). There was also a decrease in complex III activity in M-LRRK2G2019S (p < 0.05) with an intermediate decrease in NM-LRRK2G2019S that did not reach significance (figure 3; controls: 48.5 ± 8.4, NM-LRRK2G2019S: 24.9 ± 6.2, M-LRRK2G2019S: 21.4 ± 4.2).
Figure 3. Activity of individual mitochondrial respiratory chain complexes in M-LRRK2G2019S and NM-LRRK2G2019S.
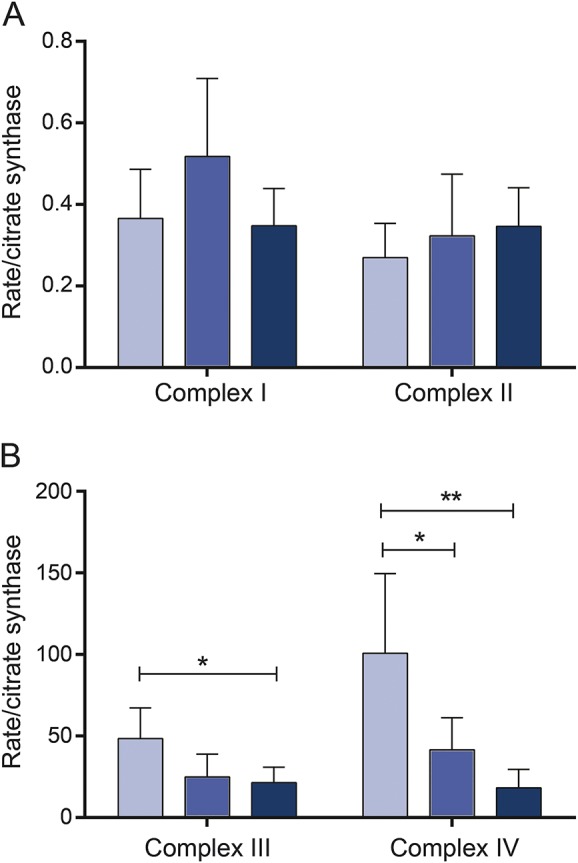
(A) Both complex I and II activity are unaltered in manifesting LRRK2G2019S carriers (M-LRRK2G2019S) (black bars) and nonmanifesting LRRK2G2019S carriers (NM-LRRK2G2019S) (gray bars) compared to controls (white bars). (B) Complex III function is reduced in M-LRRK2G2019S (*p < 0.05) and intermediate in NM-LRRK2G2019S (p > 0.05). Complex IV function is reduced in both M-LRRK2G2019S (**p < 0.01) and NM-LRRK2G2019S (*p < 0.05).
Effect of UDCA in asymptomatic LRRK2G2019S mutation carriers.
We have previously undertaken a drug screen of 2,000 compounds in PD mutant patient tissue, which led to the identification of UDCA as a potent mitochondrial rescue drug in parkin- and M-LRRK2G2019S patient fibroblasts.8 We now assessed the effect of UDCA in NM-LRRK2G2019S fibroblasts to further determine whether UDCA may also exert a beneficial effect in currently asymptomatic LRRK2G2019S mutation carriers. Treatment with UDCA recovered intracellular ATP levels in NM-LRRK2G2019S (control: DMSO treated 0.16 ± 0.06; UDCA treated 0.17 ± 0.4; NM-LRRK2G2019S DMSO treated 0.08 ± 0.02; UDCA treated 0.151 ± 0.04; p < 0.05; figure 4).
Figure 4. Rescue effect of ursodeoxycholic acid.
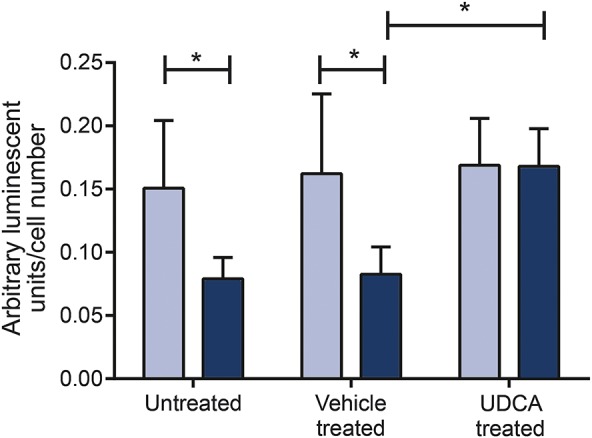
Adenosine 5′-triphosphate levels are reduced in in nonmanifesting LRRK2G2019S carriers (gray bars) compared to controls (white bars; *p < 0.05) but normalized after treatment with 10 nM ursodeoxycholic acid (UDCA).
Rescue effect of UDCA in LRRK2G2019S transgenic flies.
To determine the in vivo effects of UDCA on neuronal function, it was fed to flies with dopaminergic expression of LRRK2G2019S. As controls, we also fed these drugs to flies with dopaminergic expression of hLRRK2 or control flies that did not express any transgene. The visual contrast response function (CRF) was measured by SSVEP at 7 days. The CRF was split into components corresponding to the photoreceptors, lamina neurons, and medulla neurons. Transgenic flies with dopaminergic expression of LRRK2G2019S have markedly reduced CRFs, which are considerably smaller than those expressing hLRRK2 or nontransgenic controls. The photoreceptor response of the LRRK2G2019S flies was reduced to 32% of hLRRK2 transgenic flies (p < 0.001) and 27% of nontransgenic flies (p < 0.001). A more severe reduction still was seen in the neuronal layers (lamina of the LRRK2G2019S flies: 16% compared to hLRRK2 transgenic flies and 11% compared to nontransgenic flies; medulla of the LRRK2G2019S flies: 15% compared to hLRRK2 transgenic flies and 10% compared to nontransgenic flies, all p < 0.001, figure 5).
Figure 5. Rescue effect of ursodeoxycholic acid in vivo.

Contrast response functions (CRFs) for the visual response of flies with dopaminergic expression of LRRK2G2019S compared with those expressing (nonmutant) hLRRK2 or nontransgenic, wild-type controls with matched eye color (WT). CRFs are shown for photoreceptors, lamina neurons, and medulla neurons. At 7 days, all the LRRK2G2019S CRF responses are considerably lower than those observed in either of the other genotypes in untreated flies (red lines) but improve markedly after treatment with UDCA (green lines) in LRRK2G2019S flies at all 3 neuronal levels (p < 0.001 for photoreceptors, lamina neurons, and medulla neurons). In contrast, ursodeoxycholic acid does not increase any of the CRFs of hLRRK2 or control flies (light blue lines for untreated hLRRK2, dark blue lines for untreated WT, green lines for treated hLRRK2 and treated WT).
Feeding LRRK2G2019S flies with UDCA substantially increased all 3 components of the neuronal visual response, the photoreceptor response doubled, and the lamina and medulla neuronal responses increased by factors of 3 and 4, respectively (all p < 0.001). Thus it appears that UDCA has a powerful beneficial effect on neuronal signaling. However, the rescue was not complete: comparison of the LRRK2G2019S flies fed UDCA with control flies fed UDCA suggests that the rescue was ∼70% (photoreceptor response: 69% and 74% of hLRRK2 and nontransgenic controls; lamina: 66% and 57%; medulla: 75% and 66%; p < 0.001).
Our overall conclusion is that 2.5 μM UDCA provides a profound rescue of the LRRK2G2019S effect on dopaminergic signaling in vivo.
DISCUSSION
Parameters of mitochondrial function were similarly impaired in NM-LRRK2G2019S and M-LRRK2G2019S. Of note, our observation of a marked impairment of complex IV activity in LRRK2G2019S-related PD delineates this form of familial PD from EOPD due to parkin or PINK1 mutations with specific dysfunction of complex I, suggesting that different mechanisms lead to mitochondrial dysfunction in EOPD and LRRK2G2019S-associated PD. Our data are in keeping with a previous study investigating some basic aspects of mitochondrial function in a smaller cohort of NM-LRRK2G2019S.13 The observed normalization of mitochondrial function after siRNA-mediated LRRK2 knockdown supports the assumption that the observed mitochondrial dysfunction is a consequence of LRRK2G2019S rather than a nonspecific downstream effect or due to LRRK2 haploinsufficiency.
Taken together, our data strongly suggest that the observed changes in mitochondrial function are caused by LRRK2G2019S rather than mitochondrial dysfunction being secondary to mechanisms determining disease penetrance or a consequence of (clinically manifest) disease duration. Our data are in keeping with reports of mitochondrial dysfunction in LRRK2G2019S mutant induced pluripotent stem cell–derived neural cells but considerably more detailed.14 There is evidence to suggest that LRRK2 mutations lead to mitochondrial dysfunction via direct interaction of LRRK2 with the mitochondrial fission protein DLP1 (also known as DRP1).15 However, the observed upregulation of DLP1 in neuronal cell culture systems overexpressing wt or mutant LRRK2 could not be confirmed in LRRK2G2019S patient tissue.7 We only observed a mild increase of mitochondrial interconnectivity in M-LRRK2G2019S but not in NM-LRRK2G2019S (data not shown). The alterations in mitochondrial morphology did not correlate with cellular ATP levels or basal oxygen consumption. These data suggests that marked mitochondrial dysfunction may actually precede any changes in mitochondrial morphology in LRRK2G2019S mutant tissue rather than being a consequence of it.
There is increasing evidence of an important role for impaired autophagy in the pathogenesis of LRRK2G2019S-related PD.16 Similarly, there is also strong evidence of impaired mitophagy in EOPD due to parkin or PINK1 mutations.17 Parkin interacts directly with LRRK2 protein in vitro and protects against LRRK2G2019S-induced dopaminergic neurodegeneration in Drosophila.18,19 However, the remarkably different pattern of abnormal mitochondrial function in LRRK2G2019S compared to parkin or PINK1 mutant patient tissue suggests that distinct (albeit possibly overlapping) mechanisms leading to mitochondrial dysfunction are present in the different forms of PD rather than providing further justification for a simplistic unifying mechanistic model of impaired mitochondrial recycling in all forms of PD.9,10 Recent work carried out in parkin and PINK1 mutant Drosophila models of PD supports the assumption of a specific impairment of mitophagy with selective alterations of respiratory chain turnover depending on the respective underlying PD gene defect.20 Future studies have to determine whether there is a possible link between the mitochondrial dysfunction in NM-LRRK2G2019S observed in this study and previously reported imaging or metabolomic profiling abnormalities in NM-LRRK2G2019S.21,22
In vivo as well as in vitro energy demand may contribute to the mechanisms by which LRRK2G2019S causes neurodegeneration. In our fly model, LRRK2G2019S is associated with mitochondrial deformity and the development of neuronal vacuoles, leading to loss of visual signalling.12 All physiologic visual defects resulting from the dopaminergic expression of LRRK2G2019S in older flies are reverted by UDCA, implying that it has a role in vivo in maintaining energy supply. However, further studies are necessary to firmly establish that the observed in vivo rescue effect of UDCA in LRRK2G2019S transgenic flies is indeed due to rescue of mitochondrial function rather than other, unrelated mechanisms. The structurally closely related (but not Food and Drug Administration [FDA]–licensed) compound ursocholic acid was equally effective (data not shown), similar to the group effect observed in our initial in vitro drug screen, which led to the identification of UDCA.8
The results of our study suggest that UDCA has a marked rescue effect on a biologically relevant pathomechanism for LRRK2G2019S-linked PD not only in NM-LRRK2G2019S and M-LRRK2G2019S patient or carrier tissue but also in a LRRK2G2019S transgenic animal model. Thus, mitochondrial rescue agents may be a promising novel strategy for disease-modifying therapy in LRRK2-related PD, either given alone or in combination with LRRK2 kinase inhibitors.23 UDCA is an FDA-licensed drug and has been in clinical use for several decades for the treatment of primary biliary cirrhosis.24 Drug repositioning is a powerful and comparatively cheap alternative to de novo drug development. CSF penetrance of UDCA has been established and the naturally occurring taurine conjugate (TUDCA) is already being tested for its neuroprotective effect in motor neuron disease (Clinical Trials registration: NCT00877604).25 TUDCA has displayed a partial neuroprotective effect in the MPTP-induced mouse model of PD.26 Taken together, our data and previously published studies suggest UDCA as a promising candidate for a clinical trial in PD to further investigate its neuroprotective effect.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the research participants for their help with the study and Alex Wade for his comments on the data.
GLOSSARY
- ANOVA
analysis of variance
- ATP
adenosine 5′-triphosphate
- CRF
contrast response function
- EOPD
early-onset Parkinson disease
- FDA
Food and Drug Administration
- M-LRRK2G2019S
manifesting LRRK2G2019S carriers
- NM-LRRK2G2019S
nonmanifesting LRRK2G2019S carriers
- PD
Parkinson disease
- SSVEP
steady-state visual evoked potentials
- TUDCA
taurine conjugate
- UDCA
ursodeoxycholic acid
Footnotes
Supplemental data at Neurology.org
Editorial, page 838
AUTHOR CONTRIBUTIONS
H.M.: acquisition of data, analysis and interpretation, drafting and critical revision of manuscript. R.F.: acquisition of data, analysis and interpretation, drafting and critical revision of manuscript. J.A.: patient recruitment, analysis and interpretation of data, critical revision of manuscript. G.B.: analysis and interpretation of data, critical requisition of manuscript. C.E.: overall study design and supervision, analysis and interpretation, drafting and critical revision of manuscript. O.B.: overall study design and supervision, analysis and interpretation, drafting and critical revision of manuscript.
STUDY FUNDING
Supported by Parkinson's UK (G-1007), The Wellcome Trust (097829), and the Norwegian Parkinson Foundation.
DISCLOSURE
H. Mortiboys is the recipient of a Parkinson's UK Senior Research Fellowship. R. Furmston is the recipient of a White Rose, PhD, Studentship. G. Bronstad and J. Aasly report no disclosures relevant to the manuscript. C. Elliott is the recipient of a Wellcome Trust Institutional Strategic Support Fund Grant (097829). O. Bandmann is the recipient of a Parkinson's UK grant on mitochondrial dysfunction in LRRK2 (G-1007), a member of the Editorial Board of Neurology®, and vice-chair of the research advisory panel of Parkinson's UK. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lesage S, Durr A, Tazir M, et al. LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N Engl J Med 2006;354:422–423. [DOI] [PubMed] [Google Scholar]
- 2.Greggio E, Jain S, Kingsbury A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 2006;23:329–341. [DOI] [PubMed] [Google Scholar]
- 3.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 2006;9:1231–1233. [DOI] [PubMed] [Google Scholar]
- 4.Tsika E, Moore DJ. Mechanisms of LRRK2-mediated neurodegeneration. Curr Neurol Neurosci Rep 2012;12:251–260. [DOI] [PubMed] [Google Scholar]
- 5.Healy DG, Falchi M, O'Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol 2008;7:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010;75:2017–2020. [DOI] [PubMed] [Google Scholar]
- 7.Papkovskaia TD, Chau KY, Inesta-Vaquera F, et al. G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum Mol Genet 2012;21:4201–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mortiboys H, Aasly J, Bandmann O. Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson's disease. Brain 2013;136:3038–3050. [DOI] [PubMed] [Google Scholar]
- 9.Hoepken HH, Gispert S, Morales B, et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol Dis 2007;25:401–411. [DOI] [PubMed] [Google Scholar]
- 10.Mortiboys H, Thomas KJ, Koopman WJ, et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol 2008;64:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Afsari F, Christensen KV, Smith GP, et al. Abnormal visual gain control in a Parkinson's disease model. Hum Mol Genet 2014;23:4465–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hindle S, Afsari F, Stark M, et al. Dopaminergic expression of the Parkinsonian gene LRRK2-G2019S leads to non-autonomous visual neurodegeneration, accelerated by increased neural demands for energy. Hum Mol Genet 2013;22:2129–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grunewald AK, Arns B, Meier B, Brockmann K, Tadic V, Klein C. Does uncoupling protein 2 expression qualify as marker of disease status in LRRK2-associated PD? Antioxid Redox Signal 2014;20:1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper O, Seo H, Andrabi S, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson's disease. Sci Transl Med 2012;4:141ra90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Yan MH, Fujioka H, et al. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet 2012;21:1931–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orenstein SJ, Kuo SH, Tasset I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 2013;16:394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vives-Bauza C, Przedborski S. Mitophagy: the latest problem for Parkinson's disease. Trends Mol Med 2011;17:158–165. [DOI] [PubMed] [Google Scholar]
- 18.Ng CH, Mok SZ, Koh C, et al. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci 2009;29:11257–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith WW, Pei Z, Jiang H, et al. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci USA 2005;102:18676–18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vincow ES, Merrihew G, Thomas RE, et al. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci USA 2013;110:6400–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johansen KK, Wang L, Aasly JO, et al. Metabolomic profiling in LRRK2-related Parkinson's disease. PLoS One 2009;4:e7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sierra M, Sanchez-Juan P, Martinez-Rodriguez MI, et al. Olfaction and imaging biomarkers in premotor LRRK2 G2019S-associated Parkinson disease. Neurology 2013;80:621–626. [DOI] [PubMed] [Google Scholar]
- 23.Lee BD, Shin JH, VanKampen J, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat Med 2010;9:998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hohenester S, Oude-Elferink RP, Beuers U. Primary biliary cirrhosis. Semin Immunopathol 2009;31:283–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parry GJ, Rodrigues CM, Aranha MM, et al. Safety, tolerability, and cerebrospinal fluid penetration of ursodeoxycholic acid in patients with amyotrophic lateral sclerosis. Clin Neuropharmacol 2010;33:17–21. [DOI] [PubMed] [Google Scholar]
- 26.Castro-Caldas M, Carvalho AN, Rodrigues E, et al. Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson's disease. Mol Neurobiol 2012;46:475–486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.