

Abstract
Genome-wide association studies in Crohn's disease (CD) have identified 140 genome-wide significant loci. However, identification of genes driving association signals remains challenging. Furthermore, genome-wide significant thresholds limit false positives at the expense of decreased sensitivity. In this study, we explored gene features contributing to CD pathogenicity, including gene-based association data from CD and autoimmune (AI) diseases, as well as gene expression features (eQTLs, epigenetic markers of expression and intestinal gene expression data). We developed an integrative model based on a CD reference gene set. This integrative approach outperformed gene-based association signals alone in identifying CD-related genes based on statistical validation, gene ontology enrichment, differential expression between M1 and M2 macrophages and a validation using genes causing monogenic forms of inflammatory bowel disease as a reference. Besides gene-level CD association P-values, association with AI diseases was the strongest predictor, highlighting generalized mechanisms of inflammation, and the interferon-γ pathway particularly. Within the 140 high-confidence CD regions, 598 of 1328 genes had low prioritization scores, highlighting genes unlikely to contribute to CD pathogenesis. For select regions, comparably high integrative model scores were observed for multiple genes. This is particularly evident for regions having extensive linkage disequilibrium such as the IBD5 locus. Our analyses provide a standardized reference for prioritizing potential CD-related genes, in regions with both highly significant and nominally significant gene-level association P-values. Our integrative model may be particularly valuable in prioritizing rare, potentially private, missense variants for which genome-wide evidence for association may be unattainable.
Introduction
Genome-wide association studies (GWAS) in Crohn's disease (CD), a subtype of inflammatory bowel disease (IBD), have identified 140 high-confidence regions that demonstrate genome-wide significant evidence for association (1). Despite this large number, CD-associated variants account for only 13.6% of the variance for CD, indicating that additional regions, as well as additional variants within these 140 high-confidence regions, have yet to be identified. These 140 regions contain 1328 genes and a present challenge is to more precisely identify the genes contributing to CD pathogenesis underlying the association signals. Furthermore, while the application of strict significance thresholds reduces false positives, it comes at the expense of decreased sensitivity for disease gene identification.
The large majority of common associations identified by GWAS do not result in protein-coding changes, suggesting that many associated polymorphisms confer their pathogenic effects by modulating gene expression. Support for this concept is provided by the observation that GWAS signals are enhanced for expression quantitative trait loci (eQTL) (2,3). Furthermore, it is likely that presently identified eQTL represent only a fraction of biologically significant variants; as yet unidentified eQTL may only be identifiable under select activation and context-specific conditions (4–6). For these reasons, other gene expression features, such as epigenetic patterns and differential gene expression may be useful in defining DNA polymorphisms which modulate disease-relevant gene expression patterns.
An early observation from GWAS in immune-mediated diseases was the striking overlap of loci across distinct immune-mediated diseases, such as the association of IL23R (interleukin 23 receptor) to IBD (7), psoriasis (8), ankylosing spondylitis (9) and thyroiditis (10). These associations highlight the key contribution of the IL-23 pathway and select immune cell subsets [e.g. Th17 cells (11), innate lymphoid cells (12)] in driving numerous immune-mediated and autoimmune (AI) diseases. Therefore, association of genes with AI diseases is likely to be informative in models assessing disease contributions, based on the finding of pervasive sharing of genetic effects between AI diseases (13).
In this study, we explored gene features contributing to CD pathogenesis, including gene-based association data from CD and AI diseases, as well as gene expression features including eQTL, epigenetic and intestinal gene expression data. We developed an integrative model based on a reference set of genes with strong genetic and functional evidence for being pathogenic in CD. The integrative model performed well compared with an association-only based model. The significant contribution of genes implicated in other AI diseases highlights the pervasive contribution of the interferon (IFN)-γ pro-inflammatory pathway, common to a variety of immune-mediated diseases; in CD, IFN-γ plays a central role in differentiation of macrophage subsets. Finally, among the 1328 genes within the 140 high-confidence CD regions, we identify 598 genes with very low integrative scores, suggesting that they are unlikely to contribute to CD. This study provides a framework for integrating features identifying genes likely and unlikely to contribute to CD pathogenesis. This integrative framework can continually be refined and improved as new data and information accrues.
Results
We first sought to define gene features that are enriched among genes located within the 140 high-confidence, genome-wide significant CD-associated gene regions (1) compared with the rest of the genome. We explored gene-based CD association features, association with AI diseases and expression features including intestinal expression levels, eQTL and epigenetic data. These gene features formed the basis for development of an integrative framework of genes contributing to CD pathogenesis.
Enrichment of CD and AI GWAS signals in 140 high-confidence CD regions
We calculated gene-level CD-association P-values genome-wide using VEGAS (14) and applied a threshold for genome-wide significance of 3E−6. This threshold was based on a Bonferroni correction for 17 214 genes with gene-level CD-association P-values available. Using this threshold, 162 genes (12.2%) within the 140 high-confidence CD-associated gene regions (1) were genome-wide significant (Table 1, Fig. 1A). The Q–Q plot of gene-based P-values demonstrated an inflection at an approximate P-value of 0.05 (Fig. 1B), corresponding to 1566 genes demonstrating nominally significant gene-based association (P-values between 0.05 and 3E−6). As expected, compared with the rest of the genome, genes within high-confidence CD-associated regions demonstrated significant enrichment for gene-based association levels compared with rest of the genome at both genome-wide significant [log odds ratio (OR) 4.78, P-value 9.7E−155] and nominally significant (log OR 2.38, P-value = 3.14E−268) thresholds (Table 1).
Table 1.
Enrichment of gene features among CD region genes and among CD reference genes
| Gene feature | Category (n) | CD loci (#gene = 1328) | Non-CD loci (#gene = 23 134) | Log OR (95% CI) | P-value | CD reference (#gene = 54) | Non-CD reference (#gene = 24 408) | Log OR (95% CI) | P-value |
|---|---|---|---|---|---|---|---|---|---|
| Gene level CD association P-value | >0.05 (22 694) | 745 (56.1%) | 21 949 (94.9%) | 13 (24.1%) | 22 681 (92.9%) | ||||
| (3E−6, 0.05) (1566) | 421 (31.7%) | 1145 (4.9%) | 2.38 (2.25, 2.52) | 3.14E−268 | 22 (40.7%) | 1544 (6.3%) | 3.21 (2.53, 3.9) | 5.22E−20 | |
| ≤3E−6 (202) | 162 (12.2%) | 40 (0.2%) | 4.78 (4.43, 5.14) | 9.70E−155 | 19 (35.2%) | 183 (0.7%) | 5.2 (4.48, 5.92) | 1.94E−45 | |
| Association with other Al diseases | None (23 975) | 1196 (90.1%) | 22 779 (98.5%) | 24 (44.4%) | 23 951 (98.1%) | ||||
| In 1 study (337) | 70 (5.3%) | 267 (1.2%) | 1.61 (1.34, 1.88) | 1.37E−31 | 8 (14.8%) | 329 (1.3%) | 3.19 (2.38, 4) | 9.89E−15 | |
| In ≥2 studies (150) | 62 (4.7%) | 88 (0.4%) | 2.6 (2.27, 2.93) | 1.28E−53 | 22 (40.7%) | 128 (0.5%) | 5.14 (4.54, 5.75) | 1.44E−62 | |
| In eQTL with significant CD SNP | No (21 498) | 1003 (75.5%) | 20 495 (88.6%) | 29 (53.7%) | 21 469 (88%) | ||||
| Yes (2964) | 325 (24.5%) | 2639 (11.4%) | 0.92 (0.79, 1.05) | 4.76E−43 | 25 (46.3%) | 2939 (12%) | 1.84 (1.3, 2.38) | 1.76E−11 | |
| High expression level in intestine | No (10 858) | 431 (32.5%) | 10 427 (45.1%) | 6 (11.1%) | 10 852 (44.5%) | ||||
| Yes (13 604) | 897 (67.5%) | 12 707 (54.9%) | 0.54 (0.42, 0.65) | 5.20E−19 | 48 (88.9%) | 13 556 (55.5%) | 1.86 (1.01, 2.71) | 1.81E−05 | |
| Open Chromatin Thl7 | No (10 195) | 383 (28.8%) | 9812 (42.4%) | 6 (11.1%) | 10 189 (41.7%) | ||||
| Yes (14 267) | 945 (71.2%) | 13 322 (57.6%) | 0.6 (0.48, 0.72) | 5.87E−22 | 48 (88.9%) | 14 219 (58.3%) | 1.75 (0.9, 2.6) | 5.56E−05 | |
| Differential expression (CD versus control) | No (22 220) | 1182 (89%) | 21 038 (90.9%) | 43 (79.6%) | 22 177 (90.9%) | ||||
| Yes (2242) | 146 (11%) | 2096 (9.1%) | 0.21 (0.04, 0.39) | 0.018 | 11 (20.4%) | 2231 (9.1%) | 0.93 (0.27, 1.6) | 0.006 |
P-values and log odds ratios (log OR) are from a logistic regression model using single gene feature as the predictor.
Figure 1.
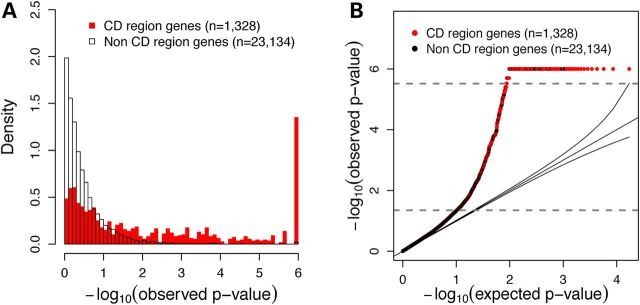
Distribution of gene-level P-values. (A) Density plot of gene level P-values within CD-associated regions (n = 1328) as defined by Jostins et al. (1), compared with all other genes (n = 23 134). (B) Q–Q plot of observed gene-level P-values compared with expected gene-level P-values. Observed P-values of 0.05 and 3E−6 are indicated by gray dashed lines.
It has long been observed that select gene regions are more likely to be associated with multiple immune-mediated diseases, including AI diseases (1,13). We next explored the enrichment of CD-associated region genes among genes that have been reported to be associated with AI diseases. Among the 1328 CD region genes, 132 have been associated with at least one other AI disease, with 62 of these 132 genes being associated with two or more AI diseases (Table 1). We observed greater enrichment of CD-region genes compared with the rest of the genome for those genes associated with both one other AI disease (log OR 1.61, P-value = 1.37E−31) and with two or more AI disease (log OR 2.6, P-value = 1.28E−53). This observed enrichment indicates that association with other AI diseases is a logical feature to include in an integrative model of genes involved in CD pathogenesis.
Enrichment of gene expression features among genes in 140 high-confidence CD regions
We used RNA-Seq to obtain gene expression levels from 6 lamina propria mononuclear cell (LPMC) samples, 58 terminal ileal samples and 10 peripheral blood (PB) samples (including equal numbers of healthy control and CD cases, see the Materials and Methods section), and 16 other body tissue samples. We defined a gene as highly expressed in a sample if its expression level ranking was greater than the median of all genes within that sample type (see the Materials and Methods section). We found that the median number of genes highly expressed in PB was lower than that in LPMC and terminal ileum (one-sided Wilcoxon's test P-value = 7E−4 for LPMC versus PB; P-value = 2E−6 for TI versus PB). The median number of genes highly expressed in PB was higher than that in body tissues (one-sided Wilcoxon's test P-value = 4E−4). These analyses highlight the particular value of analyzing intestinal materials (Fig. 2A). We therefore used high expression in either LPMC or terminal ileum in our integrative model.
Figure 2.
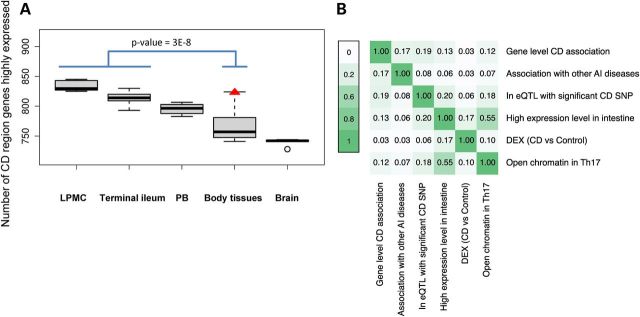
Characterization of gene features used in the integrative regression model. (A) Number of CD-associated region genes that are highly expressed in LPMC (n = 6), terminal ileal biopsies (n = 58), body tissues (one sample from each of 16 tissues) and brain (n = 5). The number of CD region genes highly expressed is significantly higher in the intestine tissues (LPMC and terminal ileum) than in other body tissues (Wilcoxon's test P-value = 3E−8). Among the body tissues, the number of CD region genes which are highly expressed is highest in white blood cells (red triangle) and lowest in the brain. (B) Pairwise correlation among gene features in the model. AI, autoimmune diseases; DEX, differentially expressed; PB, peripheral blood.
We sought to test whether various gene expression features are enriched for in genes within high-confidence CD regions, including (i) whether a significant expression quantitative trait locus (eQTL) was correlated with a nominally CD-associated SNP (see the Materials and Methods section, n = 2964 genes), (ii) whether the gene demonstrated high expression levels within intestinal tissues (n = 13 604 genes), (iii) whether the gene overlapped open chromatin sites (see the Materials and Methods section) in immune-related cells (for Th17 cells, n = 14 267 genes), or intestinal tissues and (iv) whether the gene demonstrated differential expression defined by a false discovery rate (FDR) <0.05 (see the Materials and Methods section) between terminal ileal tissues from CD patients and healthy controls (n = 2242 genes). For all four of these gene expression features, we observed significant enrichment of genes within high-confidence CD regions compared with those outside of CD regions (Table 1, Supplementary Material, Table S1).
A CD reference gene list shows stronger enrichment of gene features compared with all CD-region genes
To develop an integrative model including the aforementioned gene features to prioritize genes involved in CD pathogenesis, we selected a list of 54 CD reference genes based on previous literature (Supplementary Material, Table S2, see the Materials and Methods section). We then tested for enrichment of gene features for these genes compared with all other non-reference genes and found that all of the gene features examined were significantly enriched in CD reference genes. Furthermore, for all gene features, OR analyses showed greater enrichment of gene features for reference genes (n = 54) than for CD region genes (n = 1328) compared with the rest of the genome (Table 1). We compared genes near open chromatin sites from several immune cells (e.g. Th1, Th17, Th2, T regulatory cells and monocytes) and intestinal tissues and observed enrichment for all, with high levels of gene correlation between all of the immune cells tested (Supplementary Material, Table S1 and Supplementary Material, Fig. S1). Among the CD reference genes, the enrichment of genes near open chromatin sites was most significant in Th17 cells (Supplementary Material, Table S1) because of a high number of genes with Th17 signals. Our observation was consistent with that by Zhang et al. (15), who reported that within memory T cells, expression of immune disease-associated genes was typically increased in Th17-enriched rather than Th17-negative cells. Therefore, we chose to use open chromatin data from Th17 cells in subsequent analyses.
We next measured the overlap between each pair of aforementioned gene features using the Pearson correlation. The highest gene feature correlation was between open chromatin in Th17 cells and high expression levels in the intestine (0.55), followed by high expression levels in the intestine and eQTL (0.20). Correlations between other feature pairs were lower (Fig. 2B), suggesting that each feature provides complementary information useful in developing an integrative model for prioritizing CD-related genes.
Building and validating an integrative logistic regression model to prioritize CD genes
We next developed an integrative model, which combines GWAS results with gene expression features to better identify CD-related genes. We built a logistic regression model in which CD reference genes were used to label the dependent variable and all of the gene features listed in Table 1 used as predictors. As expected, gene-level CD association P-values were the most significant predictors, with both high and nominal CD GWAS association being strong predictors (Supplementary Material, Table S2). Association with AI diseases was the next most significant predictor, with genes reported to be associated with other AI diseases in more than one study being more predictive than genes reported in only one study. Despite the fact that gene expression features showed significant enrichment in CD-region genes and CD reference genes (Table 1), these features were not significant in the model; gene-level P-values may capture a significant fraction of expression feature contributions. For example, 41 of the 54 reference genes had highly or nominally significant gene-level P-values, among which 25 were in eQTL with nominally associated CD SNP(s); in such cases, the contribution of eQTL would be captured by the association signal itself.
The median rank of CD reference genes was 142 based on rankings produced by the integrative model, compared with 446 based solely on gene-level P-values, indicating that our model more highly prioritized the reference genes (Fig. 3A). The receiver operating characteristic (ROC) curve suggested that our model fit the data well, since the area under the curve (AUC) was 0.94 (Fig. 3B), compared with 0.90 when the log-transformed gene level CD-association P-value was used as the only predictor. We compared the area under the two ROC curves using the DeLong test in the R package pROC (16), and found that our integrative model performed significantly better than the CD GWAS P-value model (one-sided test P = 0.005); a bootstrap test with 500 replicates drawn from the data also showed that our integrative model consistently outperforms the GWAS-only model (one-sided test P = 0.005).
Figure 3.

Performance of the integrative model. (A) Boxplot ranking of 54 CD reference genes comparing the integrative model and GWAS-only model. (B) ROC curves of four regression models. Blue curve: full integrative model. The red cross at the inflection of the ROC curve, corresponds to the cut-off of the top 1685 genes. Black curve: GWAS-only model. Green curve: model with all predictors except for GWAS P-value. Gray curve: model with randomization of all predictors except GWAS P-value (box plots along the gray line reflect the variation observed in the randomization analyses). Distribution of integrative model prioritization scores for all human genes (C), and for the top 1685 ranked genes (D). Red carets indicate scores of genes with nominally significant GWAS P-values and three or more expression features.
In order to confirm that overfitting did not occur, we carried out randomization analyses. In each randomization step, CD GWAS P-values remained attached to the genes and all other predictors were randomized. Randomization and model fitting were performed 100 times. Models with randomized non-CD GWAS P-value predictors had a median AUC of 0.87, below the integrative model (AUC of 0.94) and below the GWAS-only model (AUC of 0.90). Including the randomized non-GWAS P-value predictors actually impeded the performance of the model, suggesting that overfitting did not occur in the integrative model (Fig. 3B). We also performed cross-validation on both the integrative and GWAS-only models and compared the performance of these two models. We randomly split the CD reference genes and non-CD genes into half, then used half of the data to train the model and the other half to test the model and calculate the AUC. We repeated this 500 times, and found that the median AUC of the integrative model was 0.93, while the median AUC of the GWAS-only model was 0.90 (one-sided paired t-test P < 2E−16). We further investigated the performance of a model with all predictors except for CD GWAS P-value. It had an AUC of 0.90 (Fig. 3B). This model performed well because the other AI disease and eQTL with significant CD SNP predictors carry information that overlapped CD GWAS P-values.
All genes having moderately significant gene-level P-values and at least three expression features are ranked among the top 1685 genes; this gene-level threshold also marked the inflection point of ROC curve (Fig. 3B). The distribution of prioritization scores of all genes (Fig. 3C) and of the top 1685 genes (Fig. 3D) are shown. The prioritization scores for all genes are listed in Supplementary Material, Table S3.
The integrative model demonstrates enrichment of GO pathways and differential expression between M1 and M2 macrophage subsets compared with GWAS-only rankings
We compared genes prioritized by our integrative model with those prioritized by GWAS gene-level P-values alone using Gene Ontology (GO) analyses. Among the top 213 prioritized genes (corresponding to a gene level P-value threshold of ∼3E−6), 118 genes were prioritized by both methods, with 95 genes unique to either one or the other method. GO analyses with GOrilla (17) indicated that the top 213 genes specifically prioritized by the integrative model were enriched for a variety of CD-related biological processes such as regulation of T cell activation, cellular response to IFN-γ, antigen processing and presentation, and enrichment for immune system processes generally was far more significant with the integrative model (FDR = 2.98E−29) compared with the GWAS-only model (FDR = 7.35E−15) (Supplementary Material, Tables S4 and S5). Regarding specific cytokine pathways implicated by the integrative model, the most significant was for IFN-γ signaling (FDR = 5.4E−22).
Given the enrichment of IFN-γ pathways by GO analyses, we sought to further explore differential gene expression in M1 (classically activated) compared with M2 (alternatively activated) macrophages. M1 macrophages are enriched in genes contributing to microbial killing, whereas M2 macrophages are involved in tissue repair. Macrophage differentiation is modulated by a variety of environmental and ontologic factors; in vitro models include treatment with IFN-γ and IL-4 to induce M1 and M2 differentiation, respectively (18). We compared genes prioritized by our integrative model and those prioritized solely based on CD GWAS P-values (GWAS-only model) to determine how many of each were differentially expressed between M1 and M2 macrophages.
Using macrophage microarray data, 1634 (8%) of human genes showed differential expression between M1 and M2 (M1–M2 DEX genes). We examined the proportion of M1–M2 DEX genes on different gene ranking cutoffs, according to both integrative model and GWAS-only model. Consistently throughout the gene rankings, we observed a higher proportion of differentially expressed genes with the integrative model compared with the association-only rankings (Fig. 4A). This difference was most marked among the highest ranked genes. For example, comparing the top 213 genes, the proportion of M1 compared M2 differentially expressed genes was 24% (n = 51 genes) compared with 19% (n = 40), for the integrative and GWAS-only rankings, respectively (one-sided binomial test P = 0.04). The enrichment of genes involved in M1–M2 polarization was significantly higher among these highly prioritized CD genes than among all human genes (P = 1E−13). A heat map presenting the expression pattern of these 51 genes is shown in Figure 4B after treatment with IFN-γ and IL-4 for M1 and M2 differentiation, respectively. Among genes in eQTL with nominally CD-associated SNPs, a modestly higher fraction of genes demonstrated higher expression in M1 versus M2 cells, reflecting a central role for M1 cells in CD pathogenesis. Interestingly, genes that were highly expressed in M1 were more likely to be down-regulated by CD risk alleles than all other genes after both 6 h (P = 0.01) and 24 h (P = 0.02) of polarization (Supplementary Material, Table S6).
Figure 4.
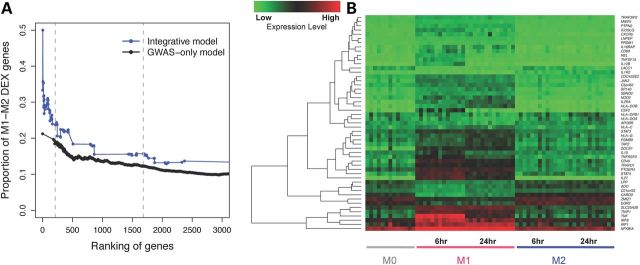
Genes highly ranked in the integrative model and differential expression (DEX) between macrophage subtypes M1 and M2. (A) Proportion of genes that show differential expression between M1 and M2 macrophages, corresponding to different cutoffs on gene rankings. Genes highly ranked by the integrative model (blue curve) are more likely to be M1–M2 DEX than genes highly ranked by gene-based GWAS-only (black curve). Dashed lines indicate the top 213 genes (corresponding to gene-based genome-wide significant association, P = 3E−6), and the top 1685 genes in the integrative model. (B) Heatmap showing expression levels of 51 genes both highly prioritized by our integrative model (top 213 genes) and differentially expressed between polarized macrophage subtypes M1 and M2 at either time point 6 or 24 h after treatment with IFN-γ or IL4. M0 denotes macrophages that have not yet been polarized to be M1-like or M2-like. Red colors correspond to higher expression level and green corresponds to lower expression level. Genes are hierarchically clustered according to their expression pattern.
The integrative model performs better than a GWAS-only ranking on a set of genes causing monogenic disorders associated with IBD-like immunopathology
To test the applicability of our model, we used this approach using a different set of reference genes taken from monogenic disorders associated with early onset of intestinal inflammation as summarized by Uhlig (19). We compared the performance of our integrative model and a GWAS-only model using the 32 autosomal monogenic disease genes listed. We directly applied the scores from the integrative regression model and from a GWAS-only model to these genes and performed ROC analyses. The AUC of our integrative model when using the new list of CD genes was 0.80, which was nominally better than the model using only GWAS P-values included as a predictor, which had an AUC of 0.72 (one-sided DeLong's test P = 0.056). The decrease in the absolute values of predictive power was likely the result of using many genes not specifically related to CD—many of these monogenic disorders present with primarily colonic disease; that the difference between the integrative and CD GWAS P-value models was only nominally significant is likely a result of including fewer genes in the reference list.
Summary of the integrative model performance within high-confidence CD regions
Finally, we asked how well the integrative model functioned in both prioritizing likely CD genes, as well as excluding genes highly unlikely to contribute to disease pathogenesis. Among the 140 high-confidence CD regions, 30 of the regions contained precisely one gene ranked within the top 213 genes of our integrative model, and for 69 of the regions, no genes in the locus ranked among the top 213 genes. Conversely, four of the CD regions had six or more genes ranked among the top 213 genes. Of note, these four regions include both the MHC (major histocompatibility complex) on chromosome 6p21, the IBD5 locus on chromosome 5q31 and a gene-rich locus on chromosome 3p21 encompassing 71 genes (Supplementary Material, Table S3). Inspection of the gene score distribution genome-wide (Fig. 3C) demonstrates major peaks at −7.35 (n = 7110 genes) and −8.61 (n = 7189 genes). The score of −8.61 corresponds to the lowest score possible, with no positive gene features listed in Table 1. The second major peak in Figure 3C with a score of −7.35 results from the dual presence of high expression within the intestine, together with open chromatin marks within Th17 cells, with the absence of any other positive gene features. The significant correlation (r = 0.55, Fig. 2B) between these two expression features accounts for the large number of genes (n = 7110) having this score. Consistent with the ROC estimates (Fig. 3B), four CD reference genes had integrative gene prioritization scores less than or equal to −7.35, namely CXCL5 (Chemokine C-X-C Motif, Ligand 5, IBD_25 locus, Supplementary Material, Table S3), IRGM (Immunity-Related GTPase Family, IBD_33 locus, Supplementary Material, Table S3), DOK3 (docking protein 3, IBD_35 locus, Supplementary Material, Table S3) and CD48 (CD48 molecule, IBD_8 locus, Supplementary Material, Table S3). The IBD_25 locus contains a cluster of chemokine genes, and none of the genes in this region ranked within the top 1685 genes. Interestingly, the IRGM gene at the IBD_33 locus has been implicated as a likely CD gene due in large part to its role in autophagy. However, at this locus, the TNIP (TNFAIP3 interacting protein 1) is the highest ranking gene, and given the critical role of the TNIP-TNFAIP3 complex in down-modulating TNF function, the contribution of IRGM to CD pathogenesis cannot be considered definitive. Furthermore, RNA-Seq analyses reveal that IRGM expression is extremely low in both CD and healthy control samples with the median FPKM values of 0.016 and 0.0 in RNA samples isolated from LPMC and in terminal ileal biopsies, respectively. Taken together, these data strongly support the concept that genes with integrative scores of −7.35 and lower are unlikely to contribute to CD pathogenesis. Across the 140 high-confidence CD regions containing 1328 genes, 598 genes had scores less than or equal to −7.35, thereby highlighting a significant fraction of genes that are much less likely to contribute to CD pathogenesis.
Discussion
GWAS have typically reported the most associated marker in a region, and defined boundaries containing possible disease-associated genes based on potentially long-range cis effects of polymorphisms modulating gene expression (1). In this study, we sought to systematically explore gene association and expression features to prioritize genes contributing to CD pathogenesis. Toward these ends, we performed gene-based association tests based on the most significant marker within each gene, accounting for linkage disequilibrium patterns and number of markers in specific genomic windows (14).
To develop an integrative model, we developed a reference list of CD pathogenicity genes, based on the presence of functional differences mapped to associated missense mutations [e.g. NOD2 (20), ATG16L1 (21)], Mendelian forms of IBD [e.g. IL10 (22), IL10RA, IL10RB (23,24), XIAP (25)], within a GWAS signal presence of a gene containing a preponderance of missense mutations [e.g. CARD9 (26)] and within a given locus, the presence of a single gene having a uniquely high number of implicating factors, including literature-based support [GRAIL (27)], protein–protein interactions [DAPPLE (28)], associated cSNP, associated eQTL and gene inclusion in a causative Bayesian network (1). For our study, we chose reference genes conservatively, including only genes which had very strong evidence of CD involvement. At this point, the number of genuine CD genes is unknown. By incorrectly assigning genes as ‘non-CD’ genes, this will reduce the power of our model, the extent of which would increase with increasing numbers of genuine CD genes; the power of our model will be significantly decreased if the number of genuine CD genes is quite large.
Our integrative approach proved to be more powerful than gene-based association signals alone in identifying CD-related genes based on statistical validation, GO annotation enrichment analyses and consideration of the number of prioritized genes which were differentially expressed between M1 and M2 macrophage subpopulations. Although we are using classes of immune cells both for prioritization (Th17 cells) and for validation (macrophages), Th17 cells are a type of CD4+ T cell, whereas M1/M2 macrophages are derived from PB monocytes, and represent fundamentally distinct hematopoietic cell lineages. The biology and genomics of adaptive (Th17 cells) and innate (M1/M2, monocytes) are fundamentally different; in support of this concept is the much greater enrichment of IBD GWAS signals in H3K27Ac marks in CD4+ T cell subsets was observed compared with PB monocytes (29). Because M1/M2 biology is so distinct from Th17 cells, the presence of differential M1 versus M2 gene expression provides experimental validation of our model.
Apart from gene-level CD association P-values, associations with AI diseases were the strongest predictor in our model. This finding highlights the key role of common mechanisms of inflammation, and the IFN-γ pathway in particular. To a significant extent, CD is a Th1-mediated disease, with high levels of IFN-γ mediating critical cell-mediated immune functions, such as killing of intracellular pathogens (30). M1 macrophage subsets, induced by IFN-γ differentiation, play a critical role in CD pathogenesis. Interestingly, however, among genes demonstrating both higher expression in M1 compared with M2 cells and nominal CD association, a higher fraction of eQTL were associated with decreased, as opposed to increased, gene expression. This observation highlights the complexity of IFN-γ signaling and the possible role of innate immune deficiencies (here, M1 function) in CD pathogenesis.
Within high-confidence CD regions, 598 of 1328 genes had low prioritization scores, thereby highlighting genes unlikely to contribute to CD pathogenesis. In contrast, comparably high integrative model scores were observed for multiple genes in select, high-confidence CD regions. Prime examples of these included regions which had very strong association signals (e.g. NOD2, IL23R) or extensive linkage disequilibrium (e.g. MHC region on chromosome 6p21). At select regions, such as the IBD5 locus (31) on chromosome 5q31, the presence of both extensive linkage disequilibrium and multiple genes with cis-eQTL in the region (specifically, PDLIM4, SLC22A4, SLC22A5, IRF1 within the IBD5 locus) highlights the impossibility in some cases of defining a single causal gene driving association signals. Rather, the disease associations in such regions regulate multiple genes; dissection of the particular contribution of any single gene in such genomic regions will be particularly difficult, especially given the modest, largely additive, contributions of any single locus to overall disease expression.
Our analyses provide a standardized reference for prioritizing potential CD-related genes, allowing for further investigation of genes with both highly significant and nominally significant gene-level association P-values. Our integrative model may be particularly valuable in prioritizing rare, potentially private, missense variants for which genome-wide evidence for association may be unattainable. As more genome-scale data become available, the identification of disease genes will be improved with these additional data, with continual refinement of CD reference genes.
Materials and Methods
Intestinal tissue collection and RNA extraction
Informed consent was obtained as approved by the Institutional Review Boards for intestinal tissue collections. Terminal ileal biopsy samples from CD cases were submerged in 1 ml of RNAlater Stabilization Reagent (Qiagen) and stored at −80°C until extraction. LPMC samples: 8–10 colonoscopic biopsy specimens were obtained from each individual. Biopsies were collected in RPMI 1640 media with 2% fetal bovine serum. After washing with Hank's buffered saline, the biopsies were incubated in the presence of 5 mm EDTA at 37°C for 30 min to remove the epithelial layer. Then, the biopsies were digested with 1 mg/ml collagenase D (Roche, Germany) for 2 h, and after which, the cells were collected in RPMI 1640 with 10% fetal bovine serum. After spinning, the cells were diluted in 40% Percoll, loaded with 100% Percoll and centrifuged at 400g for 20 min. The cells between the two layers were collected and used as LPMC. The biopsies were homogenized in QIAzollysis reagent (Qiagen) using a tissue homogenizer (Omni International, Kennesaw, GA, USA) for RNA extraction. Total RNAs were extracted using the QiagenmiRNAeasy mini kit (Qiagen) according to the manufacturer's protocol.
RNA-Seq data and expression microarray data
The following sources of RNA-Seq data were analyzed:
Full biopsies from the terminal ileum were collected by the Denson Lab (Cincinnati Children's Hospital) from 58 individuals, including 28 CD patients and 30 healthy controls. Samples were barcoded up to 12 per lane and sequenced by the Illumina HiSeq 2000 machine. Thirteen samples were paired-end sequenced and 45 underwent single-end sequencing.
LPMC samples were collected in our lab from the intestine of six individuals, including six CD patients and six healthy controls. Paired end mRNA sequencing was performed using an Illumina HiSeq 2000 machine with a single sample per lane.
RNA-Seq data from 16 human tissues were included from the Body Map 2.0 project (32). Tissues included adipose, adrenal, brain, breast, colon, heart, kidney, liver, lung, lymph node, ovary, prostate, skeletal muscle, testes, thyroid and white blood cells (Supplementary Material, Table S1).
Additional brain tissue data from four samples were collected by the Sestan Lab (Fig. 2A, Courtesy of Nenad Sestan Laboratory, Yale University).
We used PB microarray data of five control and five CD human subjects published by Sipos et al. (33).The microarray included 17 098 of the 24 462 autosomal genes which were in the RNA-Seq analyses (1062 out of 1328 CD-region genes).
Gene expression level analyses based on RNA-Seq data and expression microarray data
Sequencing data were analyzed with TopHat and Cufflinks (34,35) to obtain gene-level expression estimates using the UCSC human reference gene annotation list. Quality control metrics included (i) excluding samples with <10 million reads mapped and (ii) excluding terminal ileum and LPMC samples having the median Spearman's rank correlation <0.9 with other samples in their groups. In total, 6 LPMC samples and 58 terminal ileum samples were kept for final analyses. Sample quality control details are summarized in Supplementary Material, Table S7.
We defined a gene as highly expressed in the intestine if it had a median rank within any tissue type (i.e. CD terminal ileum, control terminal ileum, CD LPMC and control LPMC) >50% of the median ranks of all genes in that tissue type. We defined a gene as highly expressed in a PB sample if it had an expression level ranking greater than the median of all genes within that sample. The microarray included 17 098 of the 24 462 autosomal genes which were in the RNA-Seq analyses (1062 out of 1328 CD-region genes). Since the microarray typed fewer genes than RNA-Seq, we estimated the number of CD-region genes highly expressed in a PB sample to be (number of CD-region genes highly expressed identified in microarray/1062 × 1328). In that way, the estimated number of CD-region genes which were highly expressed based on microarray data was directly comparable with the number obtained from RNA-Seq data.
We identify genes differentially expressed (DEX) between CD cases and controls by analyzing full biopsy terminal ileal RNA-Seq data with DESeq2 (36) at an FDR cut-off of 0.05.
Gene level CD association P-values
Gene-level CD association P-values were calculated based on a previous GWAS data set including 6333 CD and 15 056 control samples, where the HapMap3 imputed SNP-level CD association P-values were available (37). SNP-level P-values for 1 235 490 SNPs were first transformed, so that the genomic inflation factor was 1 (38). After that, gene-level P-values for 17 031 genes were calculated with VEGAS (14), taking into account the P-value of the most significant SNP and linkage disequilibrium structure within ±50 kb of the gene. Next, 1 000 000 simulations were run to obtain a P-value for each gene, allowing the most significant possible P-value to be 1E−6, which reached the significance threshold of the Bonferroni corrected P-values (i.e. 3E−6). Gene level P-values were further categorized into three groups, i.e. group 0 genes (not significant) had gene-level P > 0.05; group 1 genes (nominally significant) had gene-level P-values between 0.05 and 3E−6; group 2 genes had gene-level P ≤ 3E−6. The 3E−6 threshold was based on the Bonferroni correction (i.e. P = 0.05 for testing 17 214 genes which had SNP-level CD-association data). For the GWAS-only model, we used log (gene-level CD GWAS P-value). In the integrative model, we categorized the gene-level CD GWAS P-value along with all other predictors so that the regression result was easier to interpret. To be specific, we categorized gene level CD GWAS P-value into three groups, i.e. group 0 genes (not significant) had gene level P > 0.05; group 1 genes (nominally significant) had gene-level P-values between 0.05 and 3E−6; group 2 genes had gene-level P-value ≤ 3E−6. Genes without available SNP association data were also coded as group 0 in the analyses.
Gene eQTL information
We looked for genes in cis or trans eQTL with SNP(s) significantly associated with CD (SNP-level CD association P ≤ 0.01). We integrated four different sources of eQTL. In all cases, except for the intestinal data set, a P-value cut-off of 1E−5 was applied to the eQTL.
The University of Chicago eQTL database (http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl), containing eQTL collected from multiple studies (39).
The Dixon et al. eQTL data set (http://www.sph.umich.edu/csg/liang/asthma/), containing eQTL inferred from 400 lymphoblastoid cell lines of asthmatic children (40).
The Merck Research Laboratories eQTL data set, containing eQTL of four tissues from 1000 morbidly obese patients (41).
An intestinal eQTL data set, comprised 173 samples. For the intestinal eQTL data set, an FDR method was applied, using an α-level of 5% corrected for multiple testing (42).
Association of genes with other AI diseases
Information on genes implicated in AI diseases was obtained from the GWAS catalog (43). The GWAS catalog listed significant SNP-disease associations (P < 1E−5) from large-scale GWAS as well as the most likely disease-associated genes reported in those studies. In our analyses, we looked for genes reported in the GWAS catalog associated with the following 14 AI diseases: ankylosing spondylitis, asthma, atopic dermatitis, celiac disease, IgA level (nephropathy), multiple sclerosis, primary biliary cirrhosis, primary sclerosing cholangitis, psoriasis, rheumatoid arthritis, systemic lupus erythematosus, systemic sclerosis, type 1 diabetes and vitiligo. We categorized the association with other auto-immune diseases into three groups (group 0: no association with any AI disease; group 1: one reported association with other AI disease; group 2: more than one reported association with other AI disease).
Open chromatin data
Open chromatin data were downloaded from the ENCODE database (https://genome.ucsc.edu/ENCODE/) (44), where open chromatin regions were summarized in the ‘narrowPeak’ files for DNase-seq experiments. We defined that a gene had open chromatin if its gene region or 2 kb upstream region overlapped with any region identified to have open chromatin.
Selection of CD reference genes
We selected our 54 CD reference genes based on the following criteria. (i) Immunochip-based genes. From the 140 CD or IBD loci identified by Jostins et al. (1), we included those genes which were prioritized by two bioinformatic methods utilized previously (GRAIL, DAPPLE, cSNP, eQTL and Bayesian network analysis). For those regions in which more than one candidate gene was prioritized by two or more methods, we did not include either gene in our CD reference list. For those regions containing gene(s) prioritized by only one bioinformatic method, we included the gene that was the closest one to the SNP showing maximal association evidence in the region. (ii) Genes demonstrating evidence for Mendelian forms of IBD were obtained by literature and OMIM searches and were included in our CD reference list. (iii) Genes demonstrating an excess of rare variants in cases or controls on deep re-sequencing. The list of our CD reference genes and their literature sources are included in Supplementary Material, Table S8.
Logistic regression model
A logistic regression model was built in which the dependent variable was coded as 0 (non CD reference gene) or 1 (CD reference gene) and the predictors were our gene features summarized in Table 1. Model parameter estimation and additional permutation analyses were performed using the glm function in the statistical software R (45). We reported the prioritization score as log [probability of being CD related genes/(1−probability of being CD related genes)].
GO enrichment analyses
The Gorilla web-based tool (http://cbl-gorilla.cs.technion.ac.il) was used to identify enriched GO terms (17). We provided an unranked list of genes of interest as our target set and an unranked list of all human genes as the background set, then tested to see if there were biological process GO terms significantly enriched for in the target set. We reported FDR for each enrichment test.
M1–M2 data and gene expression analysis
PB mononuclear cells were isolated from individuals with informed consent as approved by the Yale University Institutional Review Board using the Ficoll-Hypaque gradient. Monocyte-derived-macrophages were generated as in Pena et al. (18) with slight modifications. Specifically, after PBMC isolation, 5 × 106 cells in serum-free RPMI 1640 were seeded in each well of a 6-well plate and incubated at 37°C for an hour. The media was then replaced with fresh complete RPMI media containing M-CSF (10 ng/ml) (Shenandoah Biotechnology, Warwick, PA, USA). Cells were cultured for 7 days and media were changed every 2 days. On day 7, 5 × 106 cells from 1 well were harvested in QIAzol (Qiagen) and designated M0 cells (cells before macrophage polarization). The remaining cells were stimulated in complete RPMI with 100 ng/ml LPS (Sigma-Aldrich, St Louis, MO, USA) and 20 ng/ml IFN-γ (R&D Systems, Minneapolis, MN, USA) for M1 polarization or 20 ng/ml IL-4 (R&D Systems) for M2 polarization. Cells were harvested in QIAzol at 6 and 24 h post-stimulation. Total RNA was extracted using the Qiagen miRNAeasy mini kit (Qiagen) according to the manufacturer's protocol. Microarrays were performed using Illumina HumanHT-12 v3 Expression BeadChips (Illumina, San Diego, CA, USA). In total, seven healthy controls and six CD cases were included. The microarray expression data were analyzed by the R bio-conductor packages lumi (46) and gplots (47,48). We reported a gene to be DEX between M1 and M2 if it had an FDR < 0.01 at either the 6 or 24 h time point.
Data access
The RNA-Seq data have been deposited at GEO (http://www.ncbi.nlm.nih.gov/geo/; accession ID: GSE57945). GWAS data are available at dbGAP phs000130.v1.p1 and through the WTCCC www.wtcc.org.uk website.
Supplementary Material
Funding
This work was supported by grants U01 DK62429, U01 DK062422, R01 DK092235, SUCCESS and the Sanford J. Grossman Charitable Trust. We give special thanks to the patients for participating in this research.
Supplementary Material
Acknowledgements
We thank Dr Ken Hui and Dr Mingfeng Li for helpful discussions on the data analyses, Dr Nenad Sestan for providing brain RNA sequencing data and Yale Center for Genomic Analyses. We gratefully acknowledge the PRO-KIIDS investigators, Drs Robert Baldassano, Susan Baker, Wallace Crandall, Jonah Essers, Anne Griffiths, Melvin Heyman, Richard Kellermayer, James Markowitz and Ashish Patel.
Conflict of Interest statement. None declared.
References
- 1.Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., Lee J.C., Schumm L.P., Sharma Y., Anderson C.A., et al. (2012) Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. (2010) Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet., 6, e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He X., Fuller C.K., Song Y., Meng Q., Zhang B., Yang X., Li H. (2013) Sherlock: detecting gene-disease associations by matching patterns of expression QTL and GWAS. Am. J. Hum. Genet., 92, 667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nica A.C., Parts L., Glass D., Nisbet J., Barrett A., Sekowska M., Travers M., Potter S., Grundberg E., Small K., et al. (2011) The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet., 7, e1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu J., Wolfs M.G., Deelen P., Westra H.J., Fehrmann R.S., Te Meerman G.J., Buurman W.A., Rensen S.S., Groen H.J., Weersma R.K., et al. (2012) Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genet., 8, e1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fairfax B.P., Humburg P., Makino S., Naranbhai V., Wong D., Lau E., Jostins L., Plant K., Andrews R., McGee C., et al. (2014) Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science, 343, 1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duerr R.H., Taylor K.D., Brant S.R., Rioux J.D., Silverberg M.S., Daly M.J., Steinhart A.H., Abraham C., Regueiro M., Griffiths A., et al. (2006) A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science, 314, 1461–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cargill M., Schrodi S.J., Chang M., Garcia V.E., Brandon R., Callis K.P., Matsunami N., Ardlie K.G., Civello D., Catanese J.J., et al. (2007) A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am. J. Hum. Genet., 80, 273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Australo-Anglo-American Spondyloarthritis C., Reveille J.D., Sims A.M., Danoy P., Evans D.M., Leo P., Pointon J.J., Jin R., Zhou X., Bradbury L.A., et al. (2010) Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat. Genet., 42, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wellcome Trust Case Control C., Australo-Anglo-American Spondylitis C., Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., et al. (2007) Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet., 39, 1329–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weaver C.T., Hatton R.D. (2009) Interplay between the TH17 and TReg cell lineages: a (co-)evolutionary perspective. Nat. Rev. Immunol., 9, 883–889. [DOI] [PubMed] [Google Scholar]
- 12.Spits H., Artis D., Colonna M., Diefenbach A., Di Santo J.P., Eberl G., Koyasu S., Locksley R.M., McKenzie A.N., Mebius R.E., et al. (2013) Innate lymphoid cells—a proposal for uniform nomenclature. Nat. Rev. Immunol., 13, 145–149. [DOI] [PubMed] [Google Scholar]
- 13.Cotsapas C., Voight B.F., Rossin E., Lage K., Neale B.M., Wallace C., Abecasis G.R., Barrett J.C., Behrens T., Cho J., et al. (2011) Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet., 7, e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J.Z., McRae A.F., Nyholt D.R., Medland S.E., Wray N.R., Brown K.M., Investigators A., Hayward N.K., Montgomery G.W., Visscher P.M., et al. (2010) A versatile gene-based test for genome-wide association studies. Am. J. Hum. Genet., 87, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W., Ferguson J., Ng S.M., Hui K., Goh G., Lin A., Esplugues E., Flavell R.A., Abraham C., Zhao H., et al. (2012) Effector CD4+ T cell expression signatures and immune-mediated disease associated genes. PloS One, 7, e38510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.C., Muller M. (2011) pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics, 12, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics, 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pena O.M., Pistolic J., Raj D., Fjell C.D., Hancock R.E. (2011) Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J. Immunol., 186, 7243–7254. [DOI] [PubMed] [Google Scholar]
- 19.Uhlig H.H. (2013) Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut, 62, 1795–1805. [DOI] [PubMed] [Google Scholar]
- 20.Ogura Y., Bonen D.K., Inohara N., Nicolae D.L., Chen F.F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R.H., et al. (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature, 411, 603–606. [DOI] [PubMed] [Google Scholar]
- 21.Murthy A., Li Y., Peng I., Reichelt M., Katakam A.K., Noubade R., Roose-Girma M., DeVoss J., Diehl L., Graham R.R., et al. (2014) A Crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature, 506, 456–462. [DOI] [PubMed] [Google Scholar]
- 22.Glocker E.O., Frede N., Perro M., Sebire N., Elawad M., Shah N., Grimbacher B. (2010) Infant colitis—it's in the genes. Lancet, 376, 1272. [DOI] [PubMed] [Google Scholar]
- 23.Glocker E.O., Kotlarz D., Boztug K., Gertz E.M., Schaffer A.A., Noyan F., Perro M., Diestelhorst J., Allroth A., Murugan D., et al. (2009) Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med., 361, 2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pigneur B., Escher J., Elawad M., Lima R., Buderus S., Kierkus J., Guariso G., Canioni D., Lambot K., Talbotec C., et al. (2013) Phenotypic characterization of very early-onset IBD due to mutations in the IL10, IL10 receptor alpha or beta gene: a survey of the Genius Working Group. Inflamm. Bowel Dis., 19, 2820–2828. [DOI] [PubMed] [Google Scholar]
- 25.Zeissig Y., Petersen B.S., Milutinovic S., Bosse E., Mayr G., Peuker K., Hartwig J., Keller A., Kohl M., Laass M.W., et al. (2014) XIAP variants in male Crohn's disease. Gut, 64, 66–76. [DOI] [PubMed] [Google Scholar]
- 26.Rivas M.A., Beaudoin M., Gardet A., Stevens C., Sharma Y., Zhang C.K., Boucher G., Ripke S., Ellinghaus D., Burtt N., et al. (2011) Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet., 43, 1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raychaudhuri S., Plenge R.M., Rossin E.J., Ng A.C., International Schizophrenia C., Purcell S.M., Sklar P., Scolnick E.M., Xavier R.J., Altshuler D., et al. (2009) Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet., 5, e1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rossin E.J., Lage K., Raychaudhuri S., Xavier R.J., Tatar D., Benita Y., International Inflammatory Bowel Disease Genetics C., Cotsapas C., Daly M.J. (2011) Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet., 7, e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farh K.K., Marson A., Zhu J., Kleinewietfeld M., Housley W.J., Beik S., Shoresh N., Whitton H., Ryan R.J., Shishkin A.A., et al. (2015) Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature, 518, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abraham C., Cho J.H. (2009) Inflammatory bowel disease. N. Engl. J. Med., 361, 2066–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rioux J.D., Daly M.J., Silverberg M.S., Lindblad K., Steinhart H., Cohen Z., Delmonte T., Kocher K., Miller K., Guschwan S., et al. (2001) Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat. Genet., 29, 223–228. [DOI] [PubMed] [Google Scholar]
- 32. Illumina (2011) Illumina Human BodyMap Project. Available from: http://www.ebi.ac.uk/arrayexpress. (query ID: EMTAB-513).
- 33.Sipos F., Galamb O., Wichmann B., Krenacs T., Toth K., Leiszter K., Muzes G., Zagoni T., Tulassay Z., Molnar B. (2011) Peripheral blood based discrimination of ulcerative colitis and Crohn's disease from non-IBD colitis by genome-wide gene expression profiling. Dis. Markers, 30, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc., 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trapnell C., Pachter L., Salzberg S.L. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics (Oxford, England), 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anders S., Huber W. (2010) Differential expression analysis for sequence count data. Genome Biol., 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franke A., McGovern D.P., Barrett J.C., Wang K., Radford-Smith G.L., Ahmad T., Lees C.W., Balschun T., Lee J., Roberts R., et al. (2010) Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet., 42, 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Devlin B., Roeder K. (1999) Genomic control for association studies. Biometrics, 55, 997–1004. [DOI] [PubMed] [Google Scholar]
- 39.Gaffney D.J., Veyrieras J.B., Degner J.F., Pique-Regi R., Pai A.A., Crawford G.E., Stephens M., Gilad Y., Pritchard J.K. (2012) Dissecting the regulatory architecture of gene expression QTLs. Genome Biol., 13, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dixon A.L., Liang L., Moffatt M.F., Chen W., Heath S., Wong K.C., Taylor J., Burnett E., Gut I., Farrall M., et al. (2007) A genome-wide association study of global gene expression. Nat. Genet., 39, 1202–1207. [DOI] [PubMed] [Google Scholar]
- 41.Greenawalt D.M., Dobrin R., Chudin E., Hatoum I.J., Suver C., Beaulaurier J., Zhang B., Castro V., Zhu J., Sieberts S.K., et al. (2011) A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome Res., 21, 1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kabakchiev B., Silverberg M.S. (2013) Expression quantitative trait loci analysis identifies associations between genotype and gene expression in human intestine. Gastroenterology, 144, 1488–1496, 1496.e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hindorff L.A., Morales M.J., Morales J., Junkins H.A., Hall P.N., Klemm A.K., Manolio T.A. A Catalog of Published Genome-Wide Association Studies. Available from: www.genome.gov/gwastudies Accessed in 2013. [Google Scholar]
- 44.Rosenbloom K.R., Sloan C.A., Malladi V.S., Dreszer T.R., Learned K., Kirkup V.M., Wong M.C., Maddren M., Fang R., Heitner S.G., et al. (2013) ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res., 41, D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.R_Core_Team. (2014) R: A Language and Environment for Statistical computing. Available from: http://www.R-project.org.
- 46.Du P., Kibbe W.A., Lin S.M. (2008) lumi: a pipeline for processing Illumina microarray. Bioinformatics, 24, 1547–1548. [DOI] [PubMed] [Google Scholar]
- 47.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol., 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Warnes, G.R., Bolker, B., Bonebakker, L., Gentleman, R., Liaw, W.H.A., Lumley, T., Maechler, M., Magnusson, A., Moeller, S., Schwartz, M., Venables, B. (2014) gplots: Various R Programming Tools for Plotting Data. Available from: http://CRAN.R-project.org/package=gplots .
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.