

Abstract
Most patients with Ellis-van Creveld syndrome (EvC) are identified with pathogenic changes in EVC or EVC2, however further genetic heterogeneity has been suggested. In this report we describe pathogenic splicing variants in WDR35, encoding retrograde intraflagellar transport protein 121 (IFT121), in three families with a clinical diagnosis of EvC but having a distinctive phenotype. To understand why WDR35 variants result in EvC, we analysed EVC, EVC2 and Smoothened (SMO) in IFT-A deficient cells. We found that the three proteins failed to localize to Wdr35−/− cilia, but not to the cilium of the IFT retrograde motor mutant Dync2h1−/−, indicating that IFT121 is specifically required for their entry into the ciliary compartment. Furthermore expression of Wdr35 disease cDNAs in Wdr35−/− fibroblasts revealed that the newly identified variants lead to Hedgehog signalling defects resembling those of Evc−/− and Evc2−/− mutants. Together our data indicate that splicing variants in WDR35, and possibly in other IFT-A components, underlie a number of EvC cases by disrupting targeting of both the EvC complex and SMO to cilia.
Introduction
Ellis-van Creveld syndrome (EvC; MIM: 225500) is an autosomal recessive disorder which affects development of the skeleton, ectodermal appendages and heart (1,2). Typical manifestations of this condition include short limbs, short ribs, postaxial polydactyly, teeth abnormalities, multiple oral frenulae and nail dysplasia. In addition, two-thirds of patients are identified with congenital heart malformations, mainly in the form of atrioventricular canal defects. Involvement of other organs, including pulmonary and renal abnormalities, hypospadias and cryptorchidism has also been reported in few individuals (3). Phenotypic severity varies in this condition with a proportion of cases resulting in early infant death due to respiratory insufficiency secondary to the thoracic dysplasia and the cardiac malformations. To date patients with EvC have only been described with pathogenic changes in two adjacent genes, EVC and EVC2 (4,5); however, several sequence variant screens have suggested wider genetic heterogeneity (6–8). Approximately, 16% of affected individuals were reported with no disease-causing variants after sequencing all coding exons of EVC and EVC2 and testing for genomic rearrangements (7).
Hedgehog (Hh) signalling is an essential developmental pathway that in vertebrates is coordinated at the primary cilium (9). Previous work involving knockout mice demonstrated that the protein products of Evc and Evc2 form a ciliary transmembrane protein complex (EVC-EVC2) which is required for Hh signal transduction in a tissue-specific manner (10–12). The bulk of the EvC complex localizes to the proximal end of cilia, just above the transition zone, where it physically interacts with the main activator of the pathway, Smoothened (SMO), restricting this protein to this section of the cilium (13). Loss of EVC-EVC2 disrupts key events in Hh signalling. In the presence of the Hh agonist SAG, Evc- and Evc2-mutant cells failed to fully inhibit proteolytic production of GLI3 repressor (GLI3R) due to incomplete dissociation of inhibitory SUFU-GLI3 full length (GLI3FL) complexes, and showed impaired recruitment of GLI3 to cilia tips, hence limiting the induction of Hh-target genes (11,13,14). An additional role of EVC-EVC2 in the generation of GLI transcriptional activator species or their trafficking to the nucleus has also been suggested following the observation that shRNA-depletion of EVC downregulated ligand-independent hyperactivation of Hh signalling in Sufu−/− cells (11). Moreover, the EvC complex is tethered to the cilium base by interacting with a second protein complex containing EFCAB7 and IQCE, proteins which are required for the activation of the transcription factor GLI2 (15). These experiments place EVC-EVC2 at a critical point in vertebrate Hh signalling modulating GLI transcription factor activity downstream of SMO.
Construction and structural maintenance of primary cilia relies on the intraflagellar transport system (IFT). This ancient and highly conserved machinery consists of two multiprotein complexes, IFT-A and IFT-B, which in association with the molecular motors, cytoplasmic dynein 2 and kinesin II respectively, circulate protein cargos along the cilium in retrograde (IFT-A) or anterograde (IFT-B) orientation (16). IFT121 is an IFT-A subunit which is encoded by WDR35. Recessive variants in this gene, as well as in three other IFT-A components, IFT122, IFT43 and WDR19 (IFT144), have been associated with Sensenbrenner syndrome or cranioectodermal dysplasia (CED1-4; MIMs: 218330, 613610, 614099, 614378) (17–20). CED patients are characterized by distinctive craniofacial features including forehead bossing, dolichocephaly and sagital craniosynostosis, short limbs, short ribs, brachydactyly and ectodermal abnormalities involving hair, nails and teeth. Some patients have also been reported with retinal dystrophy, renal and liver disease and brain anomalies (21–23). Additionally, predicted null variants in WDR35 were reported in three fetuses from two families with an unclassified form of short rib polydactyly (SRPS5; MIM: 614091). The two fetuses from one family were described with acromesomelic hypomineralization, campomelia, polysyndactyly, cystic kidneys and laterality defects and the fetus from the other family was found with extreme micromelia, postaxial polydactyly and facial abnormalities (24).
Herein, we show that novel specific variants in WDR35 account for a new form of EvC and mechanistically that IFT121 is specifically required for the proper ciliary localization of the EVC-EVC2 complex and the enrichment of SMO in cilia in response to Hh signalling. Our work supports extended screening of EvC cohorts for pathogenic variants, in particular splicing variants, in IFT-A components.
Results
Clinical description
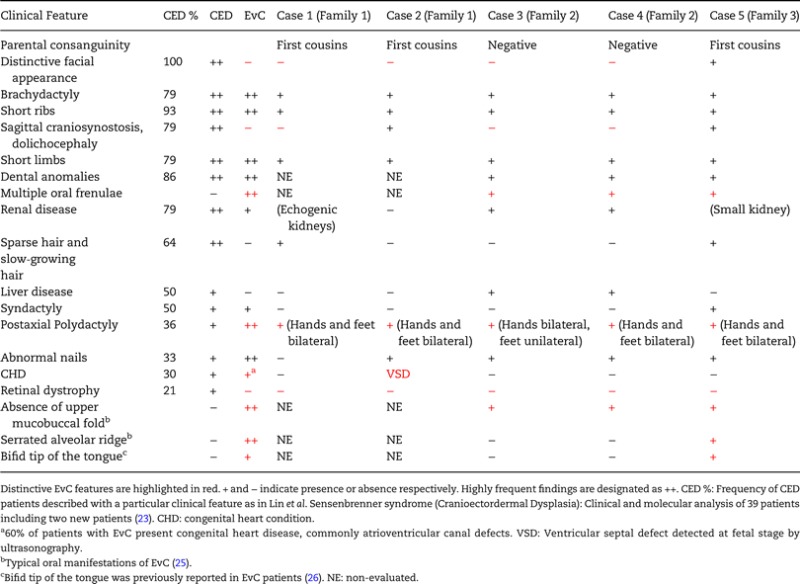
Patients in this study include five individuals (cases 1–5) from three independent families identified with EvC but also having some additional features which are found in CED. Detailed clinical description of these patients is in Supplemental Material and a comparison of their phenotypes with respect to EvC and CED is summarized in Table 1.
Table 1.
Main clinical features in the patients of this study compared with EvC and CED
 |
Identification of WDR35 pathogenic variants in patients with EvC
Analysis of EVC and EVC2 in case 1 (consanguineous family 1: Fig. 1A–C) by direct sequencing of coding exons showed no pathogenic changes, pointing to an additional gene. However, as not all types of genetic alterations are detectable with this technique, and the parents were first cousins, we performed SNP array-based homozygosity mapping in the three siblings of the family. Neither of the two affected brothers (cases 1 and 2) were found to be homozygous at the EVC/EVC2 locus, thus proving lack of linkage to the EvC region. To search for the new causative gene we conducted whole-exome sequencing (WES) of case 1. Filtering of WES data assuming a recessive model of inheritance provided putative candidate nucleotide variants in 15 genes (Supplementary Material, Tables S1–S3), with three changes being located within the homozygous regions shared only by the two affected children. Among these three changes we detected a T>A nucleotide transversion in intron 2 of WDR35 at position −18 with respect to exon 3 (c.143−18T>A; Chr2:20182313 A>T; Supplementary Material, Fig. S1A) which was selected as the best candidate because of the known involvement of WDR35 in other skeletal ciliopathies (19,24). The c.143−18T>A variant was embedded in a homozygous block of 12 and 7 Mb in each patient, but not in the normal brother, and was not listed in the 1000 Genomes, the NHLBI Exome Variant Server (EVS) or the Exome Aggregation Consortium (ExAC) browsers (Cambridge, MA, USA; URL: http://exac.broadinstitute.org) [March, 2015)]. Sanger sequencing confirmed the presence of the c.143−18T>A transversion in homozygosis in cases 1 and 2 and in the heterozygous state in the parents. The sequence from the unaffected brother was normal. We anticipated that if the c.143−18T>A change was to be pathogenic it should affect splicing. Due to the death of the affected children, we performed RT-PCR between the 5′-UTR and exon 7 of WDR35 in peripheral blood RNA from both progenitors. As a result, in addition to the expected wild-type RT-PCR product, we amplified and sequenced a smaller cDNA fragment that was shown to correspond to exon 3 skipping (Fig. 1D). Elimination of this exon in cDNA results in the in-frame deletion of 24 amino acids. Real-time RT-PCR showed that the amount of wild-type transcript in each parent was 30 ± 0.05% and 41 ± 0.07% with respect to a normal control (Supplementary Material, Fig. S2). To prove that the −18T>A variant was the change responsible for the aberrant splicing, we generated normal and mutant WDR35 minigenes differing only in the −18T>A substitution and we transfected them into COS-7 cells. RT-PCR followed by sequence analysis of the amplified products revealed no inclusion of exon 3 in the cells treated with the mutant construct, demonstrating that the tested variant was pathogenic (Supplementary Material, Fig. S1B–D). This experiment and the reduced proportion of wild-type transcript in both parents strongly suggest that no wild-type transcript would have been produced in the affected children. However, since tissue from these cases was not available, we cannot be absolutely certain whether a small fraction of normal transcript could still have been synthesized. To ascertain the molecular mechanism behind the −18T>A variant we performed a series of transfections using minigenes generated by site-directed mutagenesis. We found that when the EvC-associated variant in WDR35 was either deleted (−18delA) or altered to other nucleotide (−18A>G), exon 3 was incorporated into the transcript, suggesting that the c.143−18T>A change likely generated an intronic splicing silencer (ISS). Consistent with this, G>A mutagenesis or deletion of surrounding nucleotides at positions −21G or −17G in the context of the −18A construct also resulted in exon 3 inclusion. On the contrary, mutagenesis of −22, −20, −19 and −16 to −13 nucleotide positions did not rescue exon 3 splicing in the −18A minigene. Considering all these data, we proposed the GYYAG nucleotide motif as the putative minimal sequence of the silencer. To test this hypothesis, we generated a similar stretch of nucleotides (GTTTAG) by introducing a −13T>A mutation in the wild-type minigene and obtained exon 3 exclusion (Supplementary Material, Fig. S1C–F).
Figure 1.
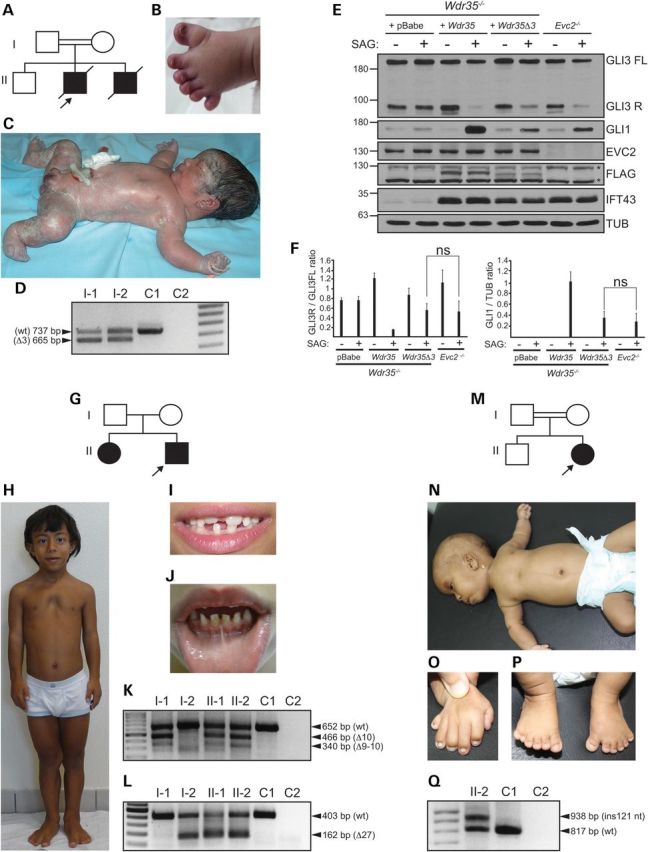
Clinical and molecular findings in patients with a new form of EvC. (A) Pedigree of family 1; the proband (case 1) is designated with an arrow. (B) Left foot of case 2 showing postaxial polydactyly and hypoplastic nails. (C) Full body view of case 1 demonstrating shortening of all four limbs and postaxial polydactyly. The photograph was taken soon after delivery which explains the abnormal shape of the head in this image. (D) RT-PCR analysis of WDR35 (5′-UTR to exon 7) in whole-blood from both parents of family 1 and an unrelated control individual (C1) revealing amplification of a smaller cDNA fragment corresponding to exon 3 exclusion (Δ3) in the parents. (E) Representative immunoblot showing response to SAG (+) or its vehicle (−) of Evc2−/− MEFs in comparison to Wdr35−/− MEFs retrotransduced with the empty vector (+pBabe) or with Wdr35Δ3:FLAG (+Wdr35Δ3), or full length wild-type Wdr35:FLAG (+Wdr35) cDNAs. Total cell extracts probed for anti-EVC2, anti-FLAG (WDR35) and anti-IFT43 are also shown. Asterisks denote non-specific bands. (F) Bar graphs representing GLI3R/GLI3FL ratio (left) and GLI1 levels (right) with respect to tubulin (TUB) from three independent experiments obtained by band densitometry. (G) Pedigree of family 2. The arrow designates the proband (case 3). (H) Full body view of case 3 showing narrow elongated chest, sloping shoulders, pectus excavatum and proximal shortening of limbs. (I and J). Clinical images demonstrating hypodontia, abnormally shaped teeth and enamel hypoplasia in case 3 (I) and multiple labiogingival frenulae in his affected sister (case 4; J). (K and L) RT-PCR analysis of WDR35 between exons 8–15 (K) and 26–28 (L) in whole-blood cDNA from family 2 members and an unrelated control (C1). Transcripts lacking exons 10 (Δ10), 9–10 (Δ9–10) and 27 (Δ27) are indicated. (M) Pedigree of family 3. The arrow designates the proband (case 5). (N) Full body view of case 5 showing dolichocephaly, sparse hair and eyebrows, short upper limbs and narrow chest with pectus excavatum. (O) Right hand of case 5 with brachydactyly postaxial polydactyly and dysplastic nails. (P) Feet of case 5 showing brachydactyly, bilateral postaxial polydactyly with hypoplastic nails and partial soft tissue syndactyly between 2nd and 3rd toes and between 5th and 6th toes bilateral. (Q) RT-PCR analysis of WDR35 from exons 8 to 17 in whole-blood from case 5 and an unrelated control (C1) revealing amplification of a longer transcript (ins 121 nt) in the patient. For D, K, L and Q, C2 is a negative control with no template and wt designates the normal transcript (NM_020779.3).
To understand why the newly identified WDR35 variant leads to an EvC phenotype we compared the output of Hh signalling of Evc2−/− mouse embryonic fibroblasts (MEFs) and Wdr35-null cells (Wdr35yet/yet) (24) stably expressing a Wdr35:FLAG cDNA fusion that mimicked case 1 variant (WDR35Δ3). Since human and mouse WDR35 exon 3 are 91% identical and code for the same number of amino acids, we generated the murine variant by deleting exon 3 in the mouse Wdr35 cDNA and introduced it into Wdr35−/− fibroblasts by retroviral delivery. Wdr35−/− MEFs infected with equal number of viral particles carrying the empty vector (pBabe) or a full length wild-type Wdr35:FLAG (Wdr35-wt) construct served as controls. We analysed the pathway by measuring the protein levels of the Hh-transcriptional target GLI1 and the GLI3R/GLI3FL ratio and found the response to SAG of Evc2−/− and Wdr35Δ3 MEFs to be remarkably similar. Compared with control cells, in the presence of the agonist, both cultures showed partial inhibition of the proteolytic processing of GLI3FL into its repressor form GLI3R and expressed low levels of GLI1, thus providing a molecular explanation for the convergence of phenotype associated with WDR35Δ3 and EVC/EVC2 variants. In contrast, Wdr35−/− MEFs infected with pBabe were unresponsive to SAG, indicating that Wdr35Δ3 acts as a hypomorphic allele and that WDR35 is essential for appropriate response to Hh signalling (Fig. 1E and F). Indeed evaluation of WDR35 expression by anti-FLAG immunoblotting in the restrotransduced cultures revealed lower levels of WDR35Δ3 with respect to full length WDR35-wt, suggesting that the WDR35Δ3 variant was thermodynamically unstable. Consistent with this, the amount of WDR35Δ3 increased following incubation with the proteasome inhibitor MG132 (Supplementary Material, Fig. S3).
In view of these results, we sequenced WDR35 in a panel of 10 EvC patients previously excluded for EVC and EVC2 disease-causing variants on the basis of exon sequencing (Supplementary Material, Table S4). Two of these cases had also been tested for large deletions/duplications in the EvC region (7). One proband (case 3; non-consanguineous family 2: Fig. 1G–J) was found to be a compound heterozygote for a donor splice site variant in intron 10 (c.1194+1G>A; Chr2:20166484 C>T) and a translationally silent nucleotide change in exon 27 (c.3378G>A; Chr2:20113815 C>T), involving a position which according to bioinformatic predictions (Rescue-ESE; http://genes.mit.edu/burgelab/rescue-ese/) (27,28) is part of a putative exonic splicing enhancer (ESE; ATTGGA), that is no longer detected in the presence of the c.3378G>A variant (Supplementary Material, Fig. S4A). Segregation analysis confirmed the presence of both variants in the affected sister (case 4) and identified the changes in intron 10 and exon 27 as being paternally and maternally inherited respectively. Neither c.1194+1G>A nor c.3378G>A were present in the 1000 Genomes or EVS databases and interrogation of the ExAC browser which contains data from disease-specific and population genetic studies revealed an allele frequency of 0.000008256 (1/121128 chromosomes) and 0.00002473 (3/121314 chromosomes) for the c.1194+1G>A and c.3378G>A variants respectively. This is similar to other previously reported WDR35 pathogenic variants such as p.Leu641Ter (c.1922T>G) (29), p.Arg545Ter (c.1633C>T) (24) or p.Tyr1068Cys (c.3203A>G) (23) which are listed in the ExAC browser with allele frequencies of 0.0001801 (8/44424 chromosomes), 0.00002473 (3/121306 chromosomes) and 0.000008260 (1/121068 chromosomes). To determine the splicing defects caused by the variants identified in family 2 in vivo we performed RT-PCR in all members of the family using whole-blood as a source of RNA. This showed that the c.1194+1G>A variant was associated with exon 10 skipping and to a lesser extent with simultaneous skipping of exons 9 and 10 in the father and children, whilst the carriers of the c.3378G>A variant had a proportion of transcripts lacking exon 27 (Fig. 1K and L). Exclusion of exon 10 or exons 9–10, both cause in-frame deletions of 62 or 104 amino acids respectively, while loss of exon 27 predicts the replacement of the last 130 terminal amino acids by 35 new residues. The negative effect of c.3378G>A on exon 27 splicing was further verified via minigene analysis. Exon 27 was not spliced in a high proportion of transcripts from the c.3378G>A minigene demonstrating that this change either disrupts an ESE or creates an exonic splicing silencer (ESS). Control minigenes in which the corresponding c.3378G position was erased or altered to T did not fully rescued exon 27 splicing, further proving that a G at position 3378 is essential for total inclusion of this exon (Supplementary Material, Fig. S4B–D). Additionally, we assessed whether a proportion of wild-type WDR35 transcript could still be synthesized in the affected children and to do this we performed whole-blood RT-PCR in case 4 and an unrelated control using primers located in exons 10 and 27 which are deleted in the paternal and maternal transcripts respectively. This experiment showed no amplification of the corresponding fragment in the patient indicating absence of normal WDR35 mRNA (Supplementary Material, Fig. S5).
Lastly we characterized the causative variant in another Egyptian child of consanguineous parents (first cousins) having features common to both EvC and CED (case 5; consanguineous family 3: Fig. 1M–P). Similar to case 1, homozygosity analysis by SNP arrays discarded linkage to EVC/EVC2, but two CED loci, IFT122 and WDR35, were inside large homozygous blocks of 39.5 and 23.2 Mb respectively. Since sequencing of the coding regions of these genes identified no pathogenic variants, we moved on to WDR35 cDNA analysis. RT-PCR from exon 8 to 17 yielded two PCR products in the patient, one of which was larger than the expected fragment (Fig. 1Q). Subcloning and sequencing of these products demonstrated that the abnormal splice transcript had incorporated 121 extra nucleotides from intron 13 between exons 13 and 14, while the other PCR fragment was normal (Supplementary Material, Fig. S6A). Quantification of the wild-type mRNA levels in whole-blood by qRT-PCR revealed a 84.6% fall in case 5 with respect to a normal control (Supplementary Material, Fig. S2). Insertion of these extra 121 nucleotides introduces a frameshift that runs into a premature stop 7 codons later and thus the resultant transcript would likely be subjected to nonsense-mediated RNA decay. Direct sequencing of the corresponding genomic region in case 5 identified a homozygous G>T substitution two base pairs downstream of the 121 nucleotides (c.1434−684G>T; Chr2:20151929 C>A) which creates a new donor splice site (GGTAGT>GTTAGT) that is recognized with a high score (0.98; scale: 0–1) by the Fruit Fly and Human Splice Predictor programme (http://www.fruitfly.org/seq_tools/splice.html) (30) and is responsible for the activation of a cryptic exon (Supplementary Material, Fig. S6B and C). Sequencing of 139 normal controls, 45 of which were Egyptians, failed to detect the c.1434−684G>T variant, while analysis of the normal sibling identified the change in the heterozygous state. The c.1434−684G>T variant was also not listed in the 1000 Genomes database which provides allele frequencies for intronic variants. Hence, screening of WDR35 in EvC patients lacking genetic defects in EVC or EVC2 only yield single-nucleotide changes affecting splicing.
WDR35 mediates cilial localization of the EvC complex and SMO
Given the partial overlap of IFT-A components with EVC at the base of cilia (Fig. 2A), we asked whether loss of WDR35 could affect the expression or localization of EVC-EVC2. To address this we analysed EVC and EVC2 in Wdr35−/− fibroblasts by immunoblotting and immunofluorescence. Owing to defective IFT, Wdr35−/− cultures have a low proportion of ciliated cells which exhibit very short primary cilia (24). The retrograde motor mutant Dync2h1pol/pol, a newly developed ENU null allele (P. Mill, unpublished data), which also have abnormal short cilia due to disrupted retrograde IFT and Wdr35+/− cells were used as controls. Western blot analysis showed no changes in the total protein levels of EVC and EVC2 between all genotypes, however by immunofluorescence we failed to detect either protein only in Wdr35−/− cilia. Furthermore in response to SAG, which drives SMO into the ciliary compartment (31), only Wdr35−/− cilia remained negative for SMO immunofluorescence (Fig. 2B–D). These results indicate that additional IFT-A dependent functions requiring IFT121, but not retrograde transport, are specifically required for the correct ciliary entry of EVC, EVC2 and SMO.
Figure 2.

Cilial localization of EVC, EVC2 and SMO requires WDR35. (A) Representative immunofluorescence image showing partial overlapping localization between EVC and IFT43 at the base of cilia (arrows) in normal MEFs. (B) Compartmentalization of Wdr35−/− cilia was examined by immunofluorescence of the IFT-B component IFT88 as well as transition zone markers NPHP1 and CEP290 in MEFs. Cilia are oriented with the base to the left and tip to the right. Arrows point to the NPHP1 and CEP290 signals at the transition zone. (C) EVC and EVC2 immunobloting demonstrating unchanged total levels of EVC and EVC2 in Wdr35−/−, Wdr35+/− and Dync2h1−/− MEFs. (D) Immunofluorescence images revealing absence of EVC, EVC2 and SMO signals in mutant axonemes of Wdr35−/− MEFs indicating a defect in trafficking of these proteins into the ciliary compartment as opposed to cytoplasmic destabilization. SMO was visualized after activation of the Hh pathway with SAG. Shifted-overlay images are shown for IFT88, EVC, EVC2 and SMO. Nuclei were stained with DAPI. Scale bars: 10 µm.
WDR35 disease variants cause different clinical phenotypes depending on their effect on cilia
Since pathogenic changes in WDR35 lead to both EvC and CED phenotypes, the latter being milder, we investigated the correlation between the molecular defects caused by WDR35 variants and the clinical manifestations in patients. To test this we conducted retroviral-mediated cDNA rescue experiments in immortalized Wdr35−/− fibroblasts using Wdr35:FLAG cDNA species that mimic EvC (Wdr35Δ3, Wdr35Δ10 and Wdr35Δ27) or previously reported CED variants (p.Ser168Arg; c.504T>A and p.Glu626Gly; C.1877A>G) (19,29). Until now all the reported CED variants in WDR35 include missense changes in homozygosis or in combination with truncating variants (19,23,29,32). Similar to Wdr35Δ3, exclusion of exon10 (Wdr35Δ10) or exon 27 (Wdr35Δ27) in the mouse cDNA result in protein effects comparable to those caused by the human variants as do the selected CED changes which involved residues that are conserved in mouse. The resulting cultures were analysed for both cilia structure (assembly and length) and function (localization of EVC-EVC2 and SMO, and response to Hh-signalling). Supporting the different severity of the two conditions the cells retrotransduced with EvC-associated Wdr35 cDNAs were less capable to restore the Wdr35−/− ciliary alterations and less responsive to Hh signalling than the cells carrying CED variants (Figs. 3 and 4A–D and 1E and F). However, among EvC variants the clinical phenotypes did not align with the functional data. Specifically, patients 3–4 had a milder phenotype than patients 1–2 and yet Wdr35Δ10 and Wdr35Δ27 functioned as near null alleles, whereas Wdr35Δ3 behaved as a hypomorphic variant. We believe that this is likely due to the presence of residual amounts of normally spliced WDR35 in patients 3–4. Although whole-blood RT-PCR using primers from exon 10 to 27 did not show any normal cDNA in these patients, given that the splicing of exons regulated by ESE/ESS depends on the levels of different splicing factors, some of which are tissue-specific, we cannot exclude that the c.3378G>A variant allows synthesis of some normal mRNA in certain tissues (33). Indeed, in the minigene assay corresponding to this variant, exon 27 was correctly spliced into a low proportion of transcripts (Supplementary Material, Fig. S4D). Finally, we examined the impact of WDR35 variants in the integrity of WDR35-IFT43 IFT-A subcomplexes (34). We assessed this by co-immunoprecipitation making use of the FLAG epitope incorporated into the different Wdr35 cDNAs and by inmunofluorescence (anti-FLAG and anti-IFT43). We first noted that the levels of IFT43 depend on WDR35 since the amount of this protein was strongly reduced in Wdr35−/− fibroblasts (Fig. 4E). Immunoblotting in the cell lines expressing Wdr35 variants also showed lower levels of WDR35 and IFT43 except in Wdr35Δ10 and Glu626Gly cultures, suggesting that the majority of pathogenic variants impair the stability of WDR35 and by extension of IFT43. In spite of this, IFT43 co-immunoprecipated with WDR35 in all cell lines except Wdr35Δ27, thus indicating that the C-terminal of WDR35 is an important region for WDR35-IFT43 complex formation (Fig. 4F–H). On the other hand by immunofluorescence we detected IFT43 and WDR35 in the primary cilium of nearly all the cell lines except in Wdr35Δ27 and Wdr35Δ10 cultures, albeit the fluorescence intensity of both proteins varied from very low values in Wdr35Δ3 fibroblasts to levels similar to normal in Glu626Gly cells (Fig. 3A and H–J). Loss of WDR35 and IFT43 immunofluorescence signals in Wdr35Δ27 cilia can be explained by reduced stability of both proteins outside the complex, whereas in the case of Wdr35Δ10 cells, indicates that exon 10 may be critical for targeting WDR35-IFT43 complexes into the ciliary compartment, since on Western blots total WDR35Δ10 and IFT43 protein levels were normal (Fig. 4).
Figure 3.
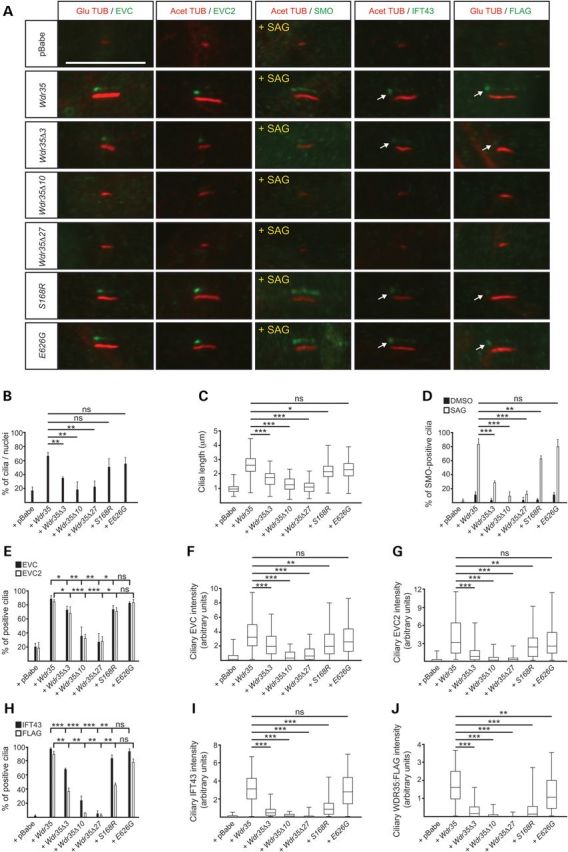
Primary cilia defects resulting from EvC versus CED WDR35 variants. (A) Representative images (shifted-overlay) corresponding to immunofluorescence analysis of EVC, EVC2, SMO, IFT43 and FLAG (WDR35) in primary cilia of Wdr35−/−MEFs stably expressing the Wdr35:FLAG variants indicated on the left. Cilia are oriented with the base to the left and tip to the right. Arrows point to the specific staining of the pool of IFT43 and FLAG (WDR35) at the base of cilia. Acet TUB: Acetylated tubulin and Glu TUB: detyrosinated tubulin. (B) Frequency of ciliated cells in Wdr35−/− cultures stably expressing Wdr35:FLAG variants or retrotransduced with the empty vector (+pBabe) or full length wild-type Wdr35:FLAG (+Wdr35) from three independent experiments. At least 500 cells were analysed per cell line. (C) Box and whiskers plot representing average primary cilia length. Three independent experiments were performed and a minimum number of 120 cilia were measured in each cell line. (D) Percentage of cilia positive for SMO staining after treatment with SAG (+) or its vehicle (−), n ≥ 120 cilia. (E) Percentage of cells with EVC- and EVC2-positive cilia (n ≥ 180 cilia). (F and G) Average fluorescence intensity in the cilium of EVC (F) and EVC2 (G) represented as a box and whiskers plot (n ≥ 50 cilia). Note that although EVC and EVC2 are detected in WDR35Δ3 cilia their corresponding fluorescence intensities are greatly diminished. (H) Frequency of cilia with positive staining for IFT43 (n ≥ 110 cilia) and FLAG (WDR35) (n ≥ 60 cilia) in the different Wdr35:FLAG cell lines. (I and J) Box and whiskers plots of the average fluorescence intensity at the ciliary base of IFT43 (I) and FLAG (WDR35) (J), n ≥ 40 cilia.
Figure 4.
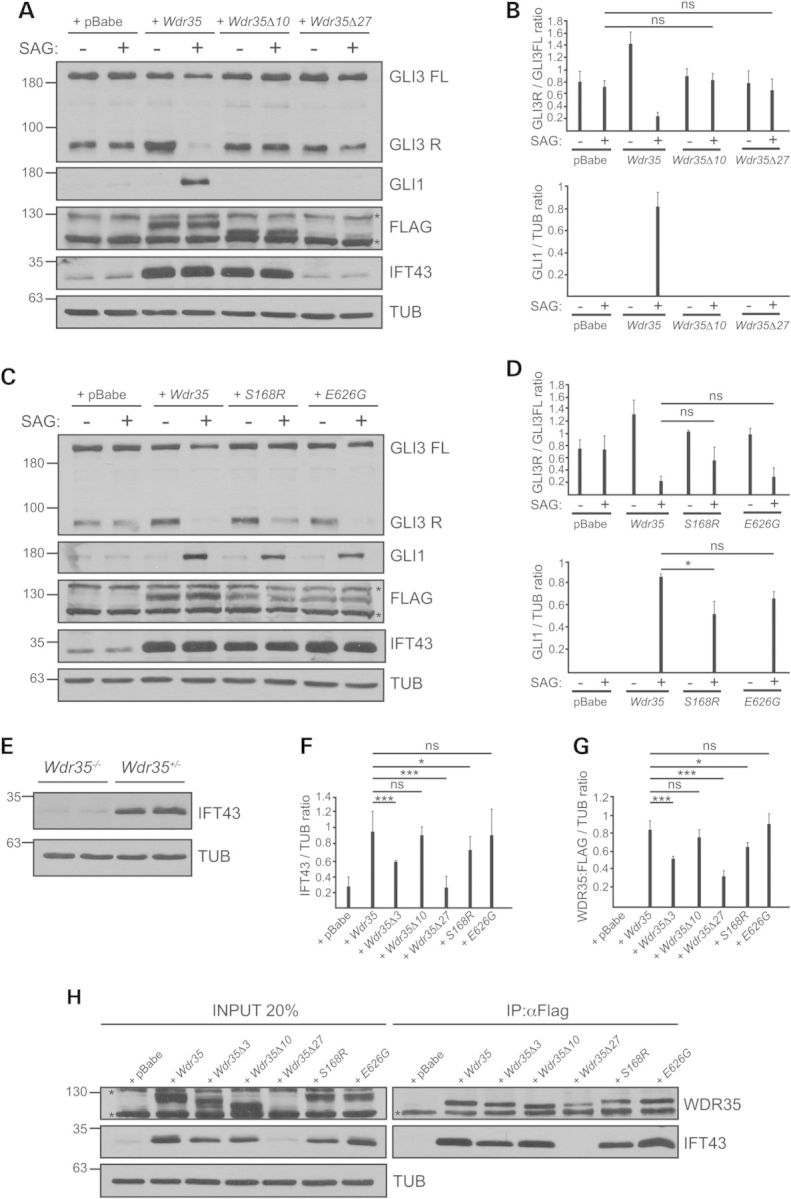
Biochemical characterization of WDR35 variants causing EvC and CED. (A–D) Representative immunoblots (n = 3) showing levels of GLI3FL, GLI3R and GLI1 after incubation with SAG or its vehicle in cultures carrying Wdr35:FLAG variants corresponding to cases 3–4 (A) or CED variants (C) with respect to control cultures retrotransduced with empty vector (+pBabe) or full length Wdr35:FLAG (+Wdr35). Densitometry analysis of GLI3R/GLI3FL and relative levels of GLI1 corresponding to panels A and B are shown in B and D respectively. (E) Representative immunoblot demonstrating lower levels of IFT43 in Wdr35−/− cells with respect to Wdr35+/− control MEFs. (F and -G) Quantification of IFT43 (F) and WDR35:FLAG (G) total protein levels by densitometric analysis of Western blots corresponding to cell extracts of Wdr35−/−cell lines retrotransduced with the indicated Wdr35:FLAG variants, n = 2. Generally the EvC-associated variants are more disruptive to the formation and stability of the IFT43-WDR35 complexes than the CED variants. (H) Representative immunoblot showing endogenous levels of IFT43 in the 20% input cellular extract and anti-FLAG immunoprecipitates of Wdr35−/− cells retrotransduced with the indicated FLAG-tagged variants, n = 2. WDR35 was detected with anti-FLAG (left blot) or anti-WDR35 (right blot). Asterisks denote non-specific bands.
Discussion
In summary we have identified WDR35 variants affecting splicing in patients classed as EvC that showed no linkage to the EvC region or were negative for EVC/EVC2 pathogenic variants. Considering the five patients as a whole, our data describe a new distinctive form of EvC characterized by a combination of clinical features overlapping with both EvC and CED. The patients from this study in addition to having short ribs, mesomelic shortening of limbs and teeth and nail dysplasia had very characteristic findings of EvC such as postaxial polydactyly of all limbs, absence of upper mucobuccal fold and multiple oral frenulae that are rare in CED (25), and, importantly except case 5, lacked the distinctive CED facial appearance which is reported in 100% of CED patients with known genetic alteration (23). On the other hand cases 2 and 5 had dolicocephaly and cases 3 and 4 renal failure and hepatic fibrosis, which are features more common in CED than EvC (23).
Sequencing of the coding region of WDR35 did not explain 9 cases of EvC previously excluded for EVC/EVC2 disease-causing variants on the basis of exon sequencing. There are several possibilities that could account for these cases such as deep intronic variants or genomic reorganizations in EVC, EVC2 or WDR35, or genetic alterations in other IFT-A genes previously implicated in CED (IFT122, IFT43 and WDR19). Notably, all patients described in this report had WDR35 pathogenic variants that were either leaky (case 5), or result in a functionally hypomorphic allele (cases 1–2), or depend on the proportion of regulatory splicing factors (cases 3–4) and so, although according to our functional analysis they are more damaging than the CED changes, do not represent classical null alleles. This is probably mandatory for WDR35 variants to be able to cause an EvC phenotype, since they would need to phenocopy the diminished response to Hh signalling characteristic of Evc−/− and Evc2−/− cells (11). Our functional analysis of EvC and CED variants show a genotype-phenotype correlation between WDR35 variants and their effect on cilia, including their effect in the localization and function of EVC-EVC2 and SMO. These studies were performed in mouse fibroblast lines due to the unavailability of patient fibroblasts. However, owing to the possibility of residual amounts of wild-type transcript in some patients, the corresponding cell lines might not perfectly replicate the human condition. This is the case of family 1 in which the total absence of wild-type transcript could not be confirmed in cases 1–2.
The c.1194+1G>A variant warrants comment since apart from the expected skipping of exon 10 also leads to the simultaneous exclusion of exons 9 and 10 in a minority of transcripts. Two-exons skipping has already been reported in a considerable number of genes associated with both acceptor and donor splice site variants (35–40). In the majority of these cases the exclusion of two contiguous exons was shown to be due to the order of intron removal. Based on these reports skipping of exons 9–10 in individuals carrying the c.1194+1G>A variant could be explained if, at least in a proportion of transcripts, intron 9 was removed faster than introns 8 and 10 resulting in the rapid fusion of exons 9–10. Consequently, in these transcripts, the inactivation of the 5′-splice site of intron 10 would lead to the exclusion of a DNA fragment comprising intron 8- the fused exon (9-10)- and intron 10. Alternatively, simultaneous skipping of exons 9–10 could occur if in the presence of the c.1194+1G>A variant the combination of the 5′-splice site of intron 9 and the acceptor site of intron 10 was not strongly recognized by the splicesome, thus resulting in the alternative usage of the donor splice site of intron 8. However, this is less likely given that the six nucleotides corresponding to the consensus sequence of the 5′splice sites of intron 8 and intron 9 are identical.
This work also provides novel molecular insight into EVC-EVC2 biology by demonstrating that the presence of the EvC complex and Hh-driven SMO enrichment in the cilium depend on IFT121. Failure of these proteins to enter Wdr35−/− cilia appears to be specific since transition zone proteins like NPHP1 and CEP290 are normally found in Wdr35−/− cilia and similarly the anterograde IFT component IFT88 accumulates along Wdr35−/− axonemes (Fig. 2B). Moreover, in cells mutant for purely retrograde IFT (Dync2h1−/−) EVC and EVC2 were localized to the ciliary compartment. Since EVC, EVC2 and SMO are membrane proteins with one, two and seven transmembrane domains respectively (10), our findings are in line with the emerging role of IFT-A in the translocation of membrane proteins into the cilium. As similarly demonstrated for the small membrane associated protein (ARL13B), adenylate cyclase III (ACIII) and SMO in mutant mice for the IFT-A subunit IFT144 (41), we now show that the EvC complex is part of a growing class of diverse membrane proteins that rely on IFT-A for entry into the ciliary membrane. In conclusion, our work sheds new light into the genetics and molecular heterogeneity of the skeletal dysplasia field by defining a new form of EvC and provides a mechanism for the convergence of phenotypes resulting from pathogenic changes in EVC/EVC2 and WDR35 splicing variants.
Material and Methods
Whole-exome sequencing and array hybridization
Whole-exome sequencing and data analysis was provided by EdgeBio. Agilent SureSelect Human All Exon 50Mb kit was used for exome capture and exome libraries were sequenced on the Life Technologies SOLiD v.4 platform with 50 × 35 paired end reads. The filtering pipeline used to analyse variants is in Supplemental Material, Table S1. High density SNP arrays were performed as described earlier (42).
Sequence variant screening and nomenclature
WDR35 coding exons and at least 100 bp of the adjacent introns were amplified by standard PCR from peripheral blood genomic DNA. PCR products were enzymatically treated with alkaline phosphatase and exonuclease I (ExoSapit; GE Healthcare) prior to be sequenced using a dye terminator cycle sequencing kit and run on an ABI 3730 sequencer (Applied Biosystems). Sequencing of IFT122 was conducted using an in-house next generation sequencing (NGS) panel for detection of pathogenic variants in skeletal dysplasias comprising 315 genes. There are two mRNA isoforms annotated in NCBI for WDR35, NM_001006657.1 and NM_020779.3, which differ in the presence of the 33-nucleotide alternative exon 11, however we only detected the shorter transcript (NM_020779.3) in blood cDNA from both patients and controls. Nevertheless, to be consistent with the previous WDR35 literature, variants were named using NG_021212.1 and NM_001006657.1 as reference sequences and checked with Mutalyzer software (http://www.mutalyzer.nl/) (43). Chromosomal positions are referred to the GRCh37/hg19 genome assembly.
Minigene assays and RT-PCR
For each splicing assay, a PCR fragment spanning the corresponding exon and adjacent intronic sequences was cloned into the plasmid pSLP3. Both single-point and deletion mutants were generated by PCR-mutagenesis or using the Quick-change site-directed mutagenesis kit (Agilent technologies). All constructs were sequenced to verify the absence of any other nucleotide changes. Minigenes were introduced into COS-7 cells with Lipofectamine 2000 (Life Technologies) and 24 h after transfection, RNA was extracted with TriReagent (Ambion). For whole-blood RT-PCR, blood samples were collected in PAXgene RNA tubes and total RNA isolated with the PAXgene RNA blood kit (PreAnalytiX) following the manufacturer's instructions. cDNAs were synthesized with random primers from 5 µg of total RNA using SuperScript III First Strand kit (Life technologies).
Real-time RT-PCR
Whole-blood RNA isolated from PAXgene tubes was retrotranscribed with the High Capacity cDNA Reverse Transcription Kit from Applied Biosystems using random primers. qRT-PCR was performed using Power SYBR Green PCR Master Mix on a 7900HT Fast Real-Time PCR System apparatus (Applied Biosystems). Following PCR, dissociation curves were generated to ensure amplification of the desired product. Samples were run in triplicates and normalized against GUSB expression. For each primer pair fold difference was calculated by the 2−ΔΔCt method. The location and sequence of the primers are indicated in Supplementary Material, Figure S2.
Retroviral constructs, titration and transduction
Humanized Wdr35 cDNAs were engineered by site-directed mutagenesis as described in the minigene assays section using the IMAGE clone 5704780 (BC056925.1) as template. This clone contains a p.Tyr634His (c.1900T>C) change with respect to the reference sequence (NM_172470.3) which was corrected by site-directed mutagenesis. A FLAG-tag (DYKDDDDK) was added in-frame to the C-terminus of each Wdr35 cDNA variant by PCR amplification and the resulting constructs were cloned into the retroviral vector pBabe and verified by sequencing. Retroviruses were produced in HEK293T cells by co-transfecting the packaging plasmid pCL-Eco with the different retroviral Wdr35:FLAG constructs using calcium phosphate. After 48 h, supernatants were collected, filtered and centrifuged at 53 000 g for 2 h at 4°C. Pellets containing retroviruses were resuspended in phosphate buffer saline (PBS) overnight at 4°C. For retroviral titration, MEFs were infected with the same amount of each retroviral stock for 10 h and then seeded at low-cell concentration. Following puromycin selection (2 μg/ml, 72 h) survival colonies were counted and the concentration of competent retroviral particles (c.r.p.) obtained assuming that each cell colony was generated from one cell infected by one retroviral particle. For experimental purposes 3 × 105 MEFs (p60) were treated with 3.5 × 104 c.r.p. using polybrene and subjected to three successive infections. Positive-infected cells were selected with puromycin as indicated above before being used in experimental procedures.
Cell culture
All cells were cultured in growth medium [DMEM with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 µg/ml streptomycin and 0.25 µg/ml amphotericin B (Life Technologies)]. For SAG treatment, cells were seeded at a density of 4.5 × 105 cells/p60 dishes and 24 h later changed to low serum medium (DMEM with 0.5% FBS and antibiotics) containing 100 nM SAG (Calbiochem) or its vehicle (DMSO) and maintained for another additional 24 h. For proteasome inhibition, cells were treated with 40 µM of MG132 (Calbiochem) for 7 h before lysis. The same concentration of MG132 was maintained in the lysis buffer to avoid proteasome activity during cell lysis. MEFs were isolated from E11.5 or E13.5 littermates following standard protocols and immortalized using retroviruses carrying a genomic fragment of the SV40-adenovirus containing the Large T and small T antigen.
Antibody production
The entire coding sequence of the mouse IFT43 protein (NM_001199843.1) was fused to a 6 × His-tag and purified from E. coli by Ni2+-affinity chromatography (Novagen) to immunise rabbits. Anti-IFT43 antibodies were affinity purified by passing the serum through a column containing the antigen coupled to CNBr-activated Sepharose 4B (GE Healthcare).
Immunofluorescence
For immunofluorescence cells were seeded on cover glasses and maintained for 24 h in low serum medium to promote ciliation. Subsequently cells were fixed in either 4% PFA/PBS for 5 min at room temperature (for EVC, EVC2, SMO, NPHP1, IFT43 and IFT88 staining) or chilled methanol for 3 min on ice (for FLAG and CEP290 detection). After permeabilization (for PFA-fixed cells) and blocking, cover glasses were incubated overnight in a wet-chamber with the corresponding antibodies. Primary antibodies: anti-EVC, anti-EVC2, SMO antiserum and anti-NPHP1 (1:1000) (11,44), anti-IFT43 (1:2000; this work), anti-IFT88 (1:1000; Proteintech, 13967-AP), anti-FLAG (1:50; SIGMA F1804) and anti-CEP290 (1:100; Abcam, Ab84870). Ciliary axonemes were labelled with anti-acetilated tubulin (SIGMA, 1:6000) or anti-detyrosinated tubulin (MILLIPORE, 1:2000) and anti-γ-tubulin (SIGMA, 1:2000) stained the basal bodies. Fluorescence-conjugated secondary antibodies (1:1000, 1 h at room temperature) were from Molecular Probes and nuclei were stained with DAPI.
Immunoprecipitation and western blotting
Cells were homogenized in lysis buffer [20 mm Hepes pH 7.4, 100 mm NaCl, 1,5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 0,1% NP40 and protease and phosphatase inhibitors (SIGMA)] and the cleared lysates were incubated end-over-end with anti-FLAG M2 magnetic beads (SIGMA) overnight at 4°C. Immunocomplexes were washed extensively in lysis buffer and eluted from beads. Western blotting was performed as described earlier (11). Primary antibodies: GLI1 (1:1000; Cell Signalling, 2643), GLI3 (0,4 µg/ml; R&D, AF3690), TUB (1:100000; SIGMA, T9026), WDR35 [1:1000, (24)]; FLAG (1:2000; SIGMA, F1804); IFT43 (1:5000, this work), EVC [0,9 µg/ml, (11)]; EVC2 [1:2000, (11)] and anti-UBIQUITIN (1:1000; Cell signalling, 3936). Horseradish peroxidase-conjugated secondary antibodies (1:10000) were purchased to Jackson ImmunoResearch.
Data analysis
Quantification of total protein levels was performed by band densitometry with ImageJ, cilia length was obtained with NIS-Elements software and to calculate the fluorescence intensity at the ciliary region, we measured for each cilium the average intensity of the specific signal and subtracted the intensity of an adjacent region without specific stain using ImageJ. Statistical analysis were assessed by unpaired Student's t-test using GraphPad software. P-values ≥ 0.05 were considered non-significant (n.s.); ***P < 0.001; **P < 0.01; *P < 0.05. Bar graphs are expressed as mean ± SD.
Supplementary Material
Funding
This work was supported by the Spanish Ministry of Economy and Competitiveness (SAF2013-43365-R) and by funding from the Italian Ministry of Health (RF-2010-2310935, RC-2014 to A.D.L.).
Supplementary Material
Acknowledgements
We would like to thank the patients and their families for their participation in this study. Informed consent was obtained from all patients' guardians and the study was approved by the CSIC Institutional Review Board. We thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010). We also thank Dr Bernhard Schermer for the kind gift of the anti-NPHP1 antibody, Dr Ignacio Palmero for helping with the retroviral vector containing Large T and small T antigen and the Center of Biological Resources of Reunion (CRB) where DNA samples were stored.
Conflict of Interest statement. None declared.
References
- 1.Ellis R.W., van Creveld S. (1940) A syndrome characterized by ectodermal dysplasia, polydactyly, chondro-dysplasia and congenital morbus cordis: report of three cases. Arch. Dis. Child., 15, 65–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKusick V.A., Egeland J.A., Eldridge R., Krusen D.E. (1964) Dwarfism in the Amish I. The Ellis-Van Creveld syndrome. Bull. Johns Hopkins Hosp., 115, 306–336. [PubMed] [Google Scholar]
- 3.Baujat G., Le Merrer M. (2007) Ellis-van Creveld syndrome. Orphanet J. Rare Dis., 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruiz-Perez V.L., Ide S.E., Strom T.M., Lorenz B., Wilson D., Woods K., King L., Francomano C., Freisinger P., Spranger S., et al. (2000) Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet., 24, 283–286. [DOI] [PubMed] [Google Scholar]
- 5.Ruiz-Perez V.L., Tompson S.W., Blair H.J., Espinoza-Valdez C., Lapunzina P., Silva E.O., Hamel B., Gibbs J.L., Young I.D., Wright M.J., et al. (2003) Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am. J. Hum. Genet., 72, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ali B.R., Akawi N.A., Chedid F., Bakir M., Ur Rehman M., Rahmani A., Al-Gazali L. (2010) Molecular and clinical analysis of Ellis-van Creveld syndrome in the United Arab Emirates. BMC Med. Genet., 11, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Asdia M.C., Torrente I., Consoli F., Ferese R., Magliozzi M., Bernardini L., Guida V., Digilio M.C., Marino B., Dallapiccola B., et al. (2013) Novel and recurrent EVC and EVC2 mutations in Ellis-van Creveld syndrome and Weyers acrofacial dyostosis. Eur. J. Med. Genet., 56, 80–87. [DOI] [PubMed] [Google Scholar]
- 8.Tompson S.W., Ruiz-Perez V.L., Blair H.J., Barton S., Navarro V., Robson J.L., Wright M.J., Goodship J.A. (2007) Sequencing EVC and EVC2 identifies mutations in two-thirds of Ellis-van Creveld syndrome patients. Hum. Genet., 120, 663–670. [DOI] [PubMed] [Google Scholar]
- 9.Ingham P.W., Nakano Y., Seger C. (2011) Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet., 12, 393–406. [DOI] [PubMed] [Google Scholar]
- 10.Blair H.J., Tompson S., Liu Y.N., Campbell J., MacArthur K., Ponting C.P., Ruiz-Perez V.L., Goodship J.A. (2011) Evc2 is a positive modulator of Hedgehog signalling that interacts with Evc at the cilia membrane and is also found in the nucleus. BMC Biol., 9, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caparros-Martin J.A., Valencia M., Reytor E., Pacheco M., Fernandez M., Perez-Aytes A., Gean E., Lapunzina P., Peters H., Goodship J.A., et al. (2013) The ciliary Evc/Evc2 complex interacts with Smo and controls Hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum. Mol. Genet., 22, 124–139. [DOI] [PubMed] [Google Scholar]
- 12.Ruiz-Perez V.L., Blair H.J., Rodriguez-Andres M.E., Blanco M.J., Wilson A., Liu Y.N., Miles C., Peters H., Goodship J.A. (2007) Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development, 134, 2903–2912. [DOI] [PubMed] [Google Scholar]
- 13.Dorn K.V., Hughes C.E., Rohatgi R. (2012) A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev. Cell., 23, 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang C., Chen W., Chen Y., Jiang J. (2012) Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res., 22, 1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pusapati G.V., Hughes C.E., Dorn K.V., Zhang D., Sugianto P., Aravind L., Rohatgi R. (2014) EFCAB7 and IQCE regulate hedgehog signaling by tethering the EVC-EVC2 complex to the base of primary cilia . Dev. Cell., 28, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedersen L.B., Rosenbaum J.L. (2008) Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol., 85, 23–61. [DOI] [PubMed] [Google Scholar]
- 17.Arts H.H., Bongers E.M., Mans D.A., van Beersum S.E., Oud M.M., Bolat E., Spruijt L., Cornelissen E.A., Schuurs-Hoeijmakers J.H., de Leeuw N., et al. (2011) C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J. Med. Genet., 48, 390–395. [DOI] [PubMed] [Google Scholar]
- 18.Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbo M., Filhol E., Bole-Feysot C., et al. (2011) Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet., 89, 634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilissen C., Arts H.H., Hoischen A., Spruijt L., Mans D.A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S.G., et al. (2010) Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet., 87, 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D.A., Emma F., Klingenberg C., Hennekam R.C., Torre G., Garshasbi M., Tzschach A., et al. (2010) Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet., 86, 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amar M.J., Sutphen R., Kousseff B.G. (1997) Expanded phenotype of cranioectodermal dysplasia (Sensenbrenner syndrome). Am. J. Med. Genet., 70, 349–352. [DOI] [PubMed] [Google Scholar]
- 22.Arts H., Knoers N. (2013) Cranioectodermal dysplasia. In Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Dolan C.R., Fong C.T., Smith R.J.H., Stephens K. (eds). GeneReviews® [Internet], University of Washington, Seattle, WA, pp. 1993–2015, http://www.ncbi.nlm.nih.gov/books/NBK154653/. [PubMed] [Google Scholar]
- 23.Lin A.E., Traum A.Z., Sahai I., Keppler-Noreuil K., Kukolich M.K., Adam M.P., Westra S.J., Arts H.H. (2013) Sensenbrenner syndrome (Cranioectodermal dysplasia): clinical and molecular analyses of 39 patients including two new patients. Am. J. Med. Genet. A, 161A, 2762–2776. [DOI] [PubMed] [Google Scholar]
- 24.Mill P., Lockhart P.J., Fitzpatrick E., Mountford H.S., Hall E.A., Reijns M.A., Keighren M., Bahlo M., Bromhead C.J., Budd P., et al. (2011) Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am. J. Hum. Genet., 88, 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hennekam R.C.M., Allanson J., Krantz I. (2010) Ellis-van Creveld syndrome(chondroectodermal dysplasia). In Gorlin's Syndromes of the Head and Neck, 5th edn Oxford University press, New York, NY, USA, pp. 346–351. [Google Scholar]
- 26.Temtamy S.A., et al. (2008) Long interspersed nuclear element-1 (LINE1)-mediated deletion of EVC, EVC2, C4orf6, and STK32B in Ellis-van Creveld syndrome with borderline intelligence. Hum. Mutat. 29, 931–938. [DOI] [PubMed] [Google Scholar]
- 27.Fairbrother W.G., Yeh R.F., Sharp P.A., Burge C.B. (2002) Predictive identification of exonic splicing enhancers in human genes. Science, 297, 1007–1013. [DOI] [PubMed] [Google Scholar]
- 28.Yeo G., Hoon S., Venkatesh B., Burge C.B. (2004) Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc. Natl. Acad. Sci. USA, 101, 15700–15705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffer J.L., Fryssira H., Konstantinidou A.E., Ropers H.H., Tzschach A. (2013) Novel WDR35 mutations in patients with cranioectodermal dysplasia (Sensenbrenner syndrome). Clin. Genet., 83, 92–95. [DOI] [PubMed] [Google Scholar]
- 30.Reese M.G., Eeckman F.H., Kulp D., Haussler D. (1997) Improved splice site detection in Genie. J. Comput. Biol., 4, 311–323. [DOI] [PubMed] [Google Scholar]
- 31.Rohatgi R., Milenkovic L., Scott M.P. (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science, 317, 372–376. [DOI] [PubMed] [Google Scholar]
- 32.Bacino C.A., Dhar S.U., Brunetti-Pierri N., Lee B., Bonnen P.E. (2012) WDR35 mutation in siblings with Sensenbrenner syndrome: a ciliopathy with variable phenotype. Am. J. Med. Genet. A, 158A, 2917–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen M., Manley J.L. (2009) Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol., 10, 741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Behal R.H., Miller M.S., Qin H., Lucker B.F., Jones A., Cole D.G. (2012) Subunit interactions and organization of the Chlamydomonas reinhardtii intraflagellar transport complex A proteins. J. Biol. Chem., 287, 11689–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang L.J., Simard M.J., Vidaud D., Assouline B., Lemieux B., Vidaud M., Chabot B., Thirion J.P. (2001) A novel mutation in the neurofibromatosis type 1 (NF1) gene promotes skipping of two exons by preventing exon definition. J. Mol. Biol., 307, 1261–1270. [DOI] [PubMed] [Google Scholar]
- 36.Hori T., Fukao T., Murase K., Sakaguchi N., Harding C.O., Kondo N. (2013) Molecular basis of two-exon skipping (exons 12 and 13) by c.1248+5g>a in OXCT1 gene: study on intermediates of OXCT1 transcripts in fibroblasts. Hum. Mutat., 34, 473–480. [DOI] [PubMed] [Google Scholar]
- 37.Lehner S., Gahle M., Dierks C., Stelter R., Gerber J., Brehm R., Distl O. (2013) Two-exon skipping within MLPH is associated with coat color dilution in rabbits. PLoS One, 8, e84525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider S., Wildhardt G., Ludwig R., Royer-Pokora B. (1993) Exon skipping due to a mutation in a donor splice site in the WT-1 gene is associated with Wilms’ tumor and severe genital malformations. Hum. Genet., 91, 599–604. [DOI] [PubMed] [Google Scholar]
- 39.Symoens S., Malfait F., Vlummens P., Hermanns-Le T., Syx D., De Paepe A. (2011) A novel splice variant in the N-propeptide of COL5A1 causes an EDS phenotype with severe kyphoscoliosis and eye involvement. PLoS One, 6, e20121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahara K., Schwarze U., Imamura Y., Hoffman G.G., Toriello H., Smith L.T., Byers P.H., Greenspan D.S. (2002) Order of intron removal influences multiple splice outcomes, including a two-exon skip, in a COL5A1 acceptor-site mutation that results in abnormal pro-alpha1(V) N-propeptides and Ehlers-Danlos syndrome type I. Am. J. Hum. Genet., 71, 451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liem K.F., Jr, Ashe A., He M., Satir P., Moran J., Beier D., Wicking C., Anderson K.V. (2012) The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J. Cell Biol., 197, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martínez-Glez V., Valencia M., Caparrós-Martín J.A., Aglan M., Temtamy S., Tenorio J., Pulido V., Lindert U., Rohrbach M., Eyre D., et al. (2012) Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum. Mut., 33, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wildeman M., van Ophuizen E., den Dunnen J.T., Taschner P.E. (2008) Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mut., 29, 6–13. [DOI] [PubMed] [Google Scholar]
- 44.Liebau M.C., Hopker K., Muller R.U., Schmedding I., Zank S., Schairer B., Fabretti F., Hohne M., Bartram M.P., Dafinger C., et al. (2011) Nephrocystin-4 regulates Pyk2-induced tyrosine phosphorylation of nephrocystin-1 to control targeting to monocilia. J. Biol. Chem., 286, 14237–14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.