

Abstract
KLF1 is an erythroid specific transcription factor that is involved in erythroid lineage commitment, globin switching and terminal red blood cell maturation. Various mutations of KLF1 have been identified in humans, which has led to both benign and pathological phenotypes. The E325K mutation, within the second zinc finger of the KLF1 gene, has been shown to cause a new form of congenital dyserythropoietic anemia (CDA) now labeled as CDA type IV. We report the fourth documented case of this mutation, and propose a clinical diagnostic model to better identify this disease in other patients. Our patient is a Taiwanese child who presented to us at 8 years of age with severe hemolytic anemia, splenomegaly, elevated fetal hemoglobin (HbF), iron overload, and dyserythropoiesis in the bone marrow. KLF1 sequence analysis revealed a G-to-A transition in one allele of exon 3, which resulted in the substitution of a glutamate 325 by a lysine. Flow cytometry analysis revealed decreased protein expression of CD44 on the red blood cells, and decreased red blood cell deformability as measured using an ektacytometer. Blood typing revealed his red blood cells to be Co(a−b−), In(b−), LW(ab−) and Lu(b+), even though DNA testing predicted he would be Co(a+b−) and LW(a+b−ab+). This newly discovered CDA combines features of a hemoglobinopathy, RBC membrane defect and hereditary persistence of HbF (HPFH) which is not seen in previous types of CDA. Increased awareness of this phenotype may improve more prompt and accurate diagnosis of these patients.
Keywords: Congenital dyserythropoietic anemia, KLF-1, E325K mutation, Colton blood group
Introduction
Congenital dyserythropoietic anemias (CDAs) are a rare, inherited form of highly heterogenous disorders characterized by dyserythropoiesis in the bone marrow, anemia, jaundice, splenomegaly and secondary hemochromatosis. While CDAs were originally described based on morphology of the bone marrow, discovery of some of the responsible genes allows for molecular confirmation and paves the way for future genotype–phenotype correlation analyses. There are three major types of CDAs, although rarer variants have been identified. CDA type I is an autosomal recessive disorder caused by a homozygous or compound heterozygous mutation in CDAN1 on chromosome 15q15.1–15.3 [1,2]. The function of the encoded protein, codanin -1, is unknown and the mechanism by which it causes dyserythropoiesis remains to be determined. Patients have moderate to severe anemia, jaundice, hepatosplenomegaly and rarely, skeletal abnormalities. Microscopically, the erythroblasts have chromatin bridging between the nuclei. CDA type II is the most common CDA. Mutations in the gene SEC23B (20p11.23–20p12.1) result in a glycosylation defect of bands 3 and 4.5 [1,3]. CDA II patients have mild to severe anemia, jaundice and hepatosplenomegaly. Their bone marrow is characterized by the presence of binucleated erythroblasts that account for greater than 10% of all erythroid precursors. CDA type III follows an autosomal dominant inheritance pattern and the causative gene has been mapped to chromosome 15q21-q25 but not identified [1,4]. CDA III patients have prominent multi-nucleated erythroblasts containing up to 12 nuclei.
We describe a patient with CDA type IV, of which only three other cases have been reported [5–9]. This patient presented with severe hemolytic anemia, increased RBC fragility, hepatomegaly, elevated fetal hemoglobin, iron overload, and abnormal bone marrow pathology inconsistent with previously described types of CDA. On further study, he was found to have a mutation in KLF1, the gene encoding Erythroid-Krüppel-like factor (EKLF). KLF1 is an erythroid specific transcription factor that is essential for establishing global patterns of erythroid gene expression and plays a critical role in the switch from fetal to adult globin [10]. Various mutations in human KLF1 have been identified with both benign and disease phenotypes. The benign In(Lu) type of Lu(Lu a- and Lu b-) blood group phenotype has been linked with heterozygous mutations in the promoter or coding sequence of KLF1 that lead to haploinsufficiency [11]. Borg et al [12] identified a nonsense mutation in the KLF1 gene, K288X, which caused Hereditary Persistence of Fetal Hemoglobin in a Maltese family.
Recently, a missense, dominant-negative KLF1 mutation was reported, c.973G>A, which resulted in a previously unidentified type of CDA, currently labeled as CDA type IV [5,6,8,13]. The G-to-A transition in exon 3 of KLF1 results in the substitution of glutamate 325 by a lysine (E325K) in the second zinc finger. The mutated area of the zinc finger is in a sequence essential for binding of KLF1 to its cognate DNA motif and thus leads to a profound dysregulation of globin gene expression. The mutation has a dominant-negative effect on the transcriptional activity of KLF1, thus making the heterozygous patients symptomatic.
Here we describe the full clinical characteristics of this patient, review the cases previously reported, discuss the clinical diagnostic features of this variant of CDA, and propose a diagnostic paradigm as a basis to search for KLF1 mutation in other patients.
Methods
KLF1 Sequence Analysis
We directly sequenced the patient’s DNA, after obtaining informed consent from the mother, by isolating mononuclear cells from peripheral blood using a Qiagen genomic DNA isolation kit. Since the complete KLF1 transcription unit is 3.5 kB, we were able to cover it by the design of only 9 amplimers that overlap its proximal promoter, 5′ UTR, introns, exons, and 3′ UTR (sequences available upon request).
Flow Cytometry Analysis of Erythrocyte Cell Markers
After receiving approval by the institutional review board, peripheral RBCs of our patient and a control were washed with PBS supplemented with 1% BSA. The samples were then stained with Band3-FITC (NYBC), Kell-FITC (NYBC), GPC-APC (NYBC), CD47-FITC (BD Pharmingen), CD59-FITC (NYBC), Duffy-FITC (NYBC), RhAG-FITC (NYBC), CD44-APC (eBioscience, San Diego, USA), CD36-FITC (BD Pharmingen, San Jose, USA), CD71-PE (BD Pharmingen), β1-integrin-PE (Millipore, Billerica, USA), α5-integrin-PE (Millipore, Billerica, USA), α4-integrin-APC (MiltenyiBiotec, Auburn, USA) and GPA-PE (BD Pharmingen). Cell populations were isolated by using FACS Flow cytometer running FACSDiva software.
RBC Membrane Deformability Analysis
25ml of blood were suspended in 4 ml of 3% polyvinypyrrolidone and subjected to an increased shear stress (from 0 to 150 dynes cm−2) in an ektacytometer. The plot of deformability index (DI) versus applied shear stress was analyzed to quantitate membrane deformability [14].
Results
Patient Clinical Description
The patient is an 8 year old male Taiwanese immigrant found to have hyperbilirubinemia and anemia at birth. During his first 3 years of life in Taiwan, he received blood transfusions every 2 months. Upon presentation to our center he was a developmentally normal child with short stature, height in the10th centile and weight in the 25th centile. He had hepatomegaly, massive splenomegaly palpable to his suprapubic area and frontal and maxillary bossing consistent with prominent extra medullary hematopoiesis. His hemoglobin was 7–9 g/dL, MCV 83–87 fL, MCHC 30 g/dL, RDW 18–22% and absolute reticulocyte count was 420–490 ×103/μL. Blood smear showed marked anisopoikilocytosis, schistocytes, mild polychromasia and nucleated red blood cells (NRBCs), up to 15–30 NRBCs/100 white blood cells, many with double nuclei (Figure 1a). He had persistent hyperbilirubinemia with an average total bilirubin of 2.9 mg/dL, indirect of 2.6 mg/dL and direct bilirubin of 0.3 mg/dL, an elevated LDH of 1581 u/L and an undetectable haptoglobin level. He also had an elevated ferritin level of 500–800 ng/mL, even though he had only received 2 blood transfusions in the previous 5 years. T2* MRI of his liver at that time equaled 6.88 mg/g per dry weight of liver, which is consistent with moderate iron overload.
Fig. 1. Patient Bone Marrow Pathology.

(a) Peripheral blood morphology was notable for binucleate RBCs. (b–c) Bone marrow aspirate revealed dyserythropoiesis including (b) internuclear bridging and (c) abnormal nuclei. (d) Electron microscopy analysis of the bone marrow showed immature erythroid cells with marked heterochromatin, invagination of nuclear membrane, intranuclear precipitated material and nuclear blebbing.
Morphology, Fragility and RBC Enzyme Analysis
The marrow aspirate was hypercellular from erythroid hyperplasia and dyserythropoiesis including binucleate forms, nuclear budding and rare karyorrhexis (Figure 1b and c). There were 4% pronormoblasts, 61% normoblasts and 35% myeloid precursors resulting in an abnormal ME ratio (1:3) secondary to erythroid expansion. Electron microscopy (EM) analysis of the bone marrow showed rare immature erythroid cells with marked heterochromatin although not diagnostic of CDA type I, II or III (Figure 1d). Several cells showed a peripheral double cytoplasmic membrane and there was rare invagination of nuclear membrane with intranuclear precipitated material. Hemoglobin electrophoresis revealed an elevated fetal hemoglobin level of 42%, and no other abnormal or embryonic hemoglobins. Gene analysis for alpha or beta globin mutations was negative, and gene mapping showed the presence of 4 α and 2 β globin genes. Red cells had mildly increased osmotic fragility. RBC enzyme testing revealed a mild decrease in adenylate kinase (97 U/g) and phophofructokinase values (2.3 U/g) which are likely nonspecific findings, along with a markedly increased adenosine deaminase value (6.4 U/g) presumably due to the elevated reticulocyte count. RBC levels of G-6-PD (15.9 U/g), pyruvate kinase (60.8 U/g), glucose phosphate isomerase (95.5 U/g), phosphoglycerate kinase (262 U/g), triosephosphateisomerase (2821 U/g) and hexokinase (4.3 U/g) were all mildly increased and were attributed to the elevated reticulocyte count. Pyrimidine 5′ nucleotidase was normal.
EKLF/KLF1 Sequence Analysis
Due to the unique combination of severe hemolytic anemia, elevated fetal hemoglobin, and bone marrow morphology suggestive of, but not fully diagnostic for CDA I, II or III, we tested this patient for a KLF1 gene mutation. All of the primers pairs gave rise to single PCR products of the expected size. Sequencing revealed a G-to-A transition in one allele of exon 3 of KLF1, a change that results in the substitution of glutamate 325 by a lysine (E325K) in the second zinc finger (Figure 2) [15].
Fig. 2. Genomic Sequencing Result of Patient.

Genomic sequencing revealed a G-to-A transition in one allele, visible as overlapping peaks (ineither direction) as analyzed by Vector NTI alignment software, leading to expression of the EKLF/E325K mutant.
Since mutations leading to haploinsufficiency of KLF1 result in the rare In(Lu) blood group, it is not surprising that our patient’s RBCs revealed absence of the In(Lu) blood group phenotype. Serological testing showed his RBCs to be Co(a−b−), In(b−) and LW(ab−) and Lu(b+). This is consistent with the findings of Parsons et al [7] who reported the same blood type in a patient who had a CDA that was many years later determined to be caused by a KLF1 E325K mutation [6]. DNA from the patient was tested on the HEA BeadChip array and these results predicted that our patient would actually be Co(a+b−) and LW(a+b−ab+). This apparent discrepancy between DNA and serologic testing is an effect of the mutated KLF1 gene that does not allow for expression of the proteins displaying these antigens. It may also explain why this patient and other reported patients have not made anti-Co(a) and LW antibodies: since EKLF expression is erythroid-specific, the antigens may be expressed on non-erythroid tissues. The patient’s two siblings and parents have normal hemoglobin levels, peripheral blood smears and hemoglobin electrophoresis. They also did not have the In(Lu) nor the Co(a−b−), In(b−) and LW(ab−) blood group phenotypes. Since the E325K mutation is dominant-negative, his phenotypically normal family members were not tested for the mutation.
RBC Membrane Surface Markers and Fragility Analysis
We examined the patient’s erythrocyte membrane proteins by flow cytometric analysis and ektacytometry on his peripheral blood six weeks after his most recent blood transfusion. His blood smear and lab testing at that time were consistent with extensive hemolysis. A similar picture has been reported in the KLF1 deficient mouse due to defects in erythrocyte membrane proteins. Nilson [16] reported decreased erythrocyte membrane protein expression of β-spectrin, ankyrin, and band 3 in KLF1 deficient mice. Heterozygous neonatal anemia (Nan/+) mice also have decreased levels of band 3, proteins 4.1, 4.2 and 4.9, β-adducin and p55 when compared to wild type mice [17].
Flow Cytometry
As shown in Figure 3, left panel, the patient had decreased protein expression of CD44. He had normal levels of expression of Kell, GPC, CD47, CD59, Duffy, RhAG, CD36, CD71, Band3, β1-integrin, α5-integrin, α4-integrin and GPA. It has been reported that RBCs from patients with the KLF1 E325K mutation are negative for CD44 expression. The mixed phenotype is most likely due to the still-circulating transfused blood.
Fig. 3. Flow Cytometry Analysis of RBC Membrane Proteins.
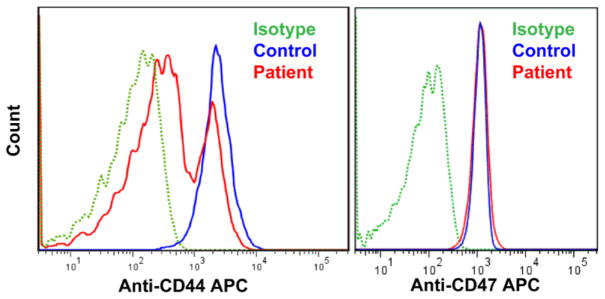
The patient sample showed decreased protein expression of anti-CD44 when compared to the control, as shown on the left panel. The mixed phenotype is most likely due to the still-circulating transfused blood from 6 weeks prior. For all other proteins tested, for example CD47 as shown on the right panel, there was no difference seen in the patient when compared to the control.
Membrane Deformability
RBC membrane deformability was then evaluated by using an ektacytometer. Representative data for membrane mechanical stability of the patient and control, measured as deformability index (DI) are shown in Figure 4. The patient had decreased membrane deformability (maximum DI 0.45) when compared to a control (maximum DI 0.75). The slope of DI versus shear stress curve was also lower for the patient than control, indicating decreased membrane deformability (increased membrane rigidity).
Fig. 4. Deformability Index Results.

Deformability Index (DI) of the patient’s RBC membrane was compared to control using an ektacytometer. The slope of DI versus shear stress is lower for the patient than the control. Also, the maximum DI is dramatically less in the patient, indicating decreased membrane deformability and increased membrane rigidity.
Patient Outcome
Since diagnosis of this patient with an E325K mutation, he has received blood transfusions every 4–6 weeks. After being on chronic transfusion therapy for four months, his growth improved to the 25thcentile, and his liver and spleen are palpable just under the costophrenic angle. The modest elevation in ferritin level, which is discrepant with the T2* of his liver showing moderate iron overload, is similar to what has been reported in patients with thalassemia intermedia [18]. Also, iron loading in thalassemia intermedia can occur even in the absence of prolonged transfusion therapy and responds well to iron chelation therapy [19]. Thus our patient was started on iron chelation therapy with close monitoring.
Conclusions
Clinical picture of EKLF/KLF1 E325K
Three other patients have been reported with an E325K mutation in the KLF1 gene [5–9]. All patients had severe hemolytic anemia, elevated fetal hemoglobin in the 40% range, hyperbilirubinemia, a marrow examination showing moderate dyserythropoiesis, splenomegaly, growth delay, mild increase in red cell osmotic fragility, and nucleated RBCs in blood smear with striking binucleate erythroid forms [Table 1]. These patients’ erythrocytes had low CD44 and water channel AQP1 expression, both of which are regulated by KLF1. The patient described by Singleton et al [5]/Parsons et al [7]/Wickramasinghe et al [9] and Arnaud [6] also had persistent expression of embryonic globins (not seen in our patient) and increased electrophoretic mobility of Band 3 (not assessed in our patient). The patient described by Arnaud et al [6], also had multiple congenital anomalies: hypertrophic cardiomyopathy, micropenis, hypospadias, enlarged anterior fontanel and hypertelorism. Ravindranath et al’s [8] report also described congenital anomalies, which included female internal and external genitalia with 46XY genotyping.
Table I.
Phenotypic comparison of all 4 documented cases of CDA due to KLF1 E325K mutation.
| Patient | RBC Indices | Bone Marrow | HbEP | Clinical features |
|---|---|---|---|---|
| The Patient |
Hgb: 7–9 g/dL MCV: 83 fL MCHC: 30 pg RDW: 22% Retic: 16% Anisopoikilocytosis, schistocytes, polychromasia, nucleated RBCs |
Pathology: Hypercellular marrow, erythroid hyperplasia, dyserythropoiesis. EM: Immature erythroid cells with heterochromatin, peripheral double membrane of the cytoplasm, invagination of nuclear membrane |
Hb A: 56% Hb A2: 2% Fetal: 42% |
Splenomegaly, Short stature, Thalassemic faces |
| Arnaud6 |
Hgb: 5–7.5 g/dL MCV: 85 fL MCHC: 33 pg RDW: 20% Anisopoikilocytosis, schistocytes, nucleated RBCs |
Pathology: Erythroid hyperplasia, dysplastic changes in erythroblasts EM: Atypical cytoplasmic inclusions, enlarged nuclear pores, endoreplication of the nuclear membrane |
Hb A: 56% Hb A2: 1.2% Fetal: 37% Hb Portland: 3% |
Hydrops Fetalis, Intrauterine transfusion, Splenomegaly, Short stature |
| Ravindranath8 | Normoblastemia Spherocytes | Not documented | Fetal: 31% | Hydrops Fetalis, Intrauterine transfusion, Splenomegaly, 46XY with internal/external female genitalia |
| Singleton/Parson/Wickramasinghe5,7,9 |
Hgb: 7.4–9.5 g/dl Retic: 5–12% Normochromic, normocytic anemia |
Pathology: Normoblastic erythroid hyperplasia, basophilic stippling of polychromatic erythroblasts and erythrocytes EM: Polychromatic erythroblasts, erythrocytes with inclusions, phagocytosed erythroblasts |
Fetal: 40–50% | Severe anemia at birth. Regular transfusions first year of life. |
Because KLF1 is both a master regulator of the switch from fetal to adult hemoglobin as well as a transcription regulator of many RBC membrane proteins, this CDA presents a picture that combines features of a hemoglobinopathy, RBC membrane defect and hereditary persistence of Hb F (HPFH), making it possible to distinguish it from these conditions. Thalassemia major is characterized by microcytosis, ineffective erythropoiesis, marrow expansion and iron overload, while the RBC membrane disorders are characterized by hemolysis and splenomegaly. HPFH, conversely, has minimal effect on RBC indices, no hemolysis and is characterized by a pronounced elevation in fetal hemoglobin. Patients with heterozygous KLF-1 E325K exhibit a combination of these effects. They have symptoms of both ineffective erythropoiesis and a RBC membrane defect, resulting in iron overload, medullary expansion, intravascular hemolysis and severe splenomegaly [Table 2]. Further, serologic typing of the RBCs in these patients would be expected to be Co(a−b−), In(b−) and LW(ab−) and not of the In (Lu) phenotype. Increased awareness of this clinical combination should prompt diagnostic testing for the KLF1 E325K mutation as this entity may be more prevalent than previously recognized.
Table II.
Clinical manifestations of heterozygous KLF1 E325K mutation are a unique combination of hemoglobinopathy, RBC membrane defect and hereditary persistence of HbF.
| Disease | Hb F | Ineffective Erythropoiesis | Hemolysis | Membrane Defect |
|---|---|---|---|---|
| HPFH Heterozygous | ++ | − | − | − |
| βThalassemia | + | +++ | + | − |
| Hereditary Spherocytosis | − | − | ++/+++ | +++ |
| KLF1 E325K | ++ | +++ | +++ | ++ |
Acknowledgments
The authors would like to thank Dr. Melissa Cushing, Dr. Connie Westhoff and Christine Lomas-Francis for the serology testing, genotyping and clinical discussions regarding the blood group testing. We would also like to thank the Flow Cytometry Core lab at the New York Blood Center for use of their facilities and Dr. Mohandas Narla for the use of RBC antibodies. This work was supported by PHS grant R21 CA133608 and NYSTEM contract CO26435 to James J Bieker.
Footnotes
Disclosure:
The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Iolascon A, Russo R, Delauncy J. Congenital dyserthropoietic anemias. Curr Opin Hematol. 2011;18:146–151. doi: 10.1097/MOH.0b013e32834521b0. [DOI] [PubMed] [Google Scholar]
- 2.Tamary H, Dgany O. GeneReviews™ [Internet] Seattle, WA: Congenital Dyserythropoietic Anemia Type I, 2009 [Updated 2011] Available from http://www.ncbi.nlm.nih.giv/books/NBK2313. [Google Scholar]
- 3.Schwartz K, Iolascon A, Verissimo F, et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet. 2009;41:936–940. doi: 10.1038/ng.405. [DOI] [PubMed] [Google Scholar]
- 4.Lind L, Sandström H, Wahlin A, et al. Localization of the gene for congenital dyserythropoietic anemia type III, CDAN3, to chromosome 15q21-q25. Hum Mol Genet. 1995;1:109–112. doi: 10.1093/hmg/4.1.109. [DOI] [PubMed] [Google Scholar]
- 5.Singleton B, Fairweather V, Lau W, et al. A Novel EKLF Mutation in a Patient with Dyserythropoietic Anemia: The First Association of EKLF with Disease in Man, ASH Abstracts. 2009. [Google Scholar]
- 6.Arnaud L, Saison C, Helias V, et al. A Dominant Mutation in the Gene Encoding the Erythroid Transcription Factor KLF1 Causes a Congenital Dyserythropoietic Anemia. Am J of Hum Genet. 2010;87:721–727. doi: 10.1016/j.ajhg.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsons S, Jones J, Anstee D, et al. A Novel Form of Congenital Dyserythropoietic Anemia Associated With Deficiency of Erythroid CD44 and a Unique Blood Group Phenotype [In(a−b−), Co(a−b−] Blood. 1994;83:860–868. [PubMed] [Google Scholar]
- 8.Ravindranath Y, Goyette G, Buck S, et al. A New Case of KLFI G973A Mutation and Congenital Dyserythropoietic Anemia (CDA)-Further Definition of Emerging New Syndrome and Possible Association with Gonadal Dysgenesis, ASH Abstracts. 2011. [Google Scholar]
- 9.Wickramasinghe SN, Illum N, Wimberley PD. Congenital dyserythropoietic anaemia with novel intra-erythroblastic and intra-erythrocytic inclusions. Br J Haematol. 1991;79:322–330. doi: 10.1111/j.1365-2141.1991.tb04541.x. [DOI] [PubMed] [Google Scholar]
- 10.Siatecka M, Bieker JJ. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood. 2011;118:2044–2054. doi: 10.1182/blood-2011-03-331371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singleton B, Burton N, Green C, et al. Mutations in EKLF/KLF1 form the molecular basis of the rare blood group In(Lu) phenotype. Blood. 2008;112:2081–2088. doi: 10.1182/blood-2008-03-145672. [DOI] [PubMed] [Google Scholar]
- 12.Borg J, Papadopoulos P, Georgitsi M, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes Hereditary Persistence of Fetal Hemoglobin. Nat Genet. 2010;42:801–805. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singleton BK, Lau W, Fairweather VS, et al. Mutations in the second zinc finger of human EKLF reduce promoter affinity but give rise to benign and disease phenotypes. Blood. 2011;118:3137–3145. doi: 10.1182/blood-2011-04-349985. [DOI] [PubMed] [Google Scholar]
- 14.Chasis J, Agre P, Mohandas N. Decreased Membrane Mechanical Stability and in Vivo Loss of Surface Area Reflect Spectrin Deficiencies in Hereditary Spherocytosis. J Clin Invest. 1988;82:617–623. doi: 10.1172/JCI113640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bieker JJ. Isolation, genomic structure, and expression of human erythroid Krüppel-like growth factor (EKLF) DNA Cell Biol. 1996;15:347–352. doi: 10.1089/dna.1996.15.347. [DOI] [PubMed] [Google Scholar]
- 16.Nilson D, Denise S, Bodine D, Gallagher P. Major erythrocyte membrane protein genes in EKLF-deficient mice. Experimental Hem. 2006;34:705–712. doi: 10.1016/j.exphem.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 17.Siatecka M, Sahr K, Anderson S, et al. Severe anemia in the Nan mutant mouse caused by sequence-selective disruption of erythroid Krüppel-like factor. PNAS. 2010;107:15151–15156. doi: 10.1073/pnas.1004996107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tony S, Daar S, Elshinawy M, et al. T2* MRI in Regularly Transfused Children with Thalassemia Intermedia: Serum Ferritin Does Not Reflect Liver Iron Stores. Pediatr Hematol Oncol. 2012;29:579–584. doi: 10.3109/08880018.2012.708891. [DOI] [PubMed] [Google Scholar]
- 19.Taher A, Porter J, Viprakasit V, et al. Deferasirox reduces iron overload significantly in nontransfusion-dependant thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120:970–977. doi: 10.1182/blood-2012-02-412692. [DOI] [PubMed] [Google Scholar]