

Abstract
Patients with pancreatic ductal adenocarcinoma (PDAC) invariably succumb to metastatic disease, but the underlying mechanisms that regulate PDAC cell movement and metastasis remain little understood. In this study, we investigated the effects of the chemokine gene CXCL12, which is silenced in PDAC tumors yet is sufficient to suppress growth and metastasis when re-expressed. Chemokines like CXCL12 regulate cell movement in a biphasic pattern, with peak migration typically in the low nanomolar concentration range. Herein, we tested the hypothesis that the biphasic cell migration pattern induced by CXCL12 reflected a bias of agonist bioenergetic signaling that might be exploited to interfere with PDAC metastasis. In human and murine PDAC cell models, we observed that non-migratory doses of CXCL12 were sufficient to decrease oxidative phosphorylation and glycolytic capacity and to increase levels of phosphorylated forms of the master metabolic kinase AMPK. Those same doses of CXCL12 locked myosin light chain into a phosphorylated state, thereby decreasing F-actin polymerization and preventing cell migration in a manner dependent upon AMPK and the calcium-dependent kinase CAMKII. Notably, at elevated concentrations of CXCL12 that were insufficient to trigger chemotaxis of PDAC cells, AMPK blockade resulted in increased cell movement. In two preclinical mouse models of PDAC, administration of CXCL12 decreased tumor dissemination, supporting our hypothesis that chemokine-biased agonist signaling may offer a useful therapeutic strategy. Our results offer a mechanistic rationale for further investigation of CXCL12 as a potential therapy to prevent or treat PDAC metastasis.
Keywords: AMPK signaling, CXCL12, CXCR4, biased agonism, functional selectivity
Introduction
With an overall survival of only 6%, pancreatic cancer remains a poorly addressed health problem relative to other commonly diagnosed malignancies (1). The vast majority of patients with pancreatic ductal adenocarcinoma (PDAC) ultimately succumb to metastatic disease (2). Standard chemotherapies for PDAC provide a minimal improvement in survival for patients, particularly those with advanced stages of disease. The inability to chemotherapeutically target advanced PDAC reflects, in part, a current paucity of knowledge regarding the mechanisms driving metastasis. While data from transgenic mice suggested PDAC cell dissemination can occur early in disease progression (3), sequencing and modeling analysis after rapid autopsy of patients revealed that dissemination and metastasis of PDAC occurs subsequent to primary tumor development (4). Though no mechanism is known, human tissue analysis implicates the signaling protein Smad4 in multifocal metastasis of PDAC (5). However the specific extracellular cues that regulate the invasion, migration, and metastatic homing of PDAC cells into distant tissues have yet to be identified.
As soluble/secreted proteins, chemokines, or chemotactic cytokines, direct cell movement in a concentration dependent manner. In normal physiology, chemokines direct cell trafficking of a wide variety of cell types. Dysregulated expression of chemokines, or their cognate G-protein coupled receptors (GPCRs), is associated with pathologic cell migration in many diseases (6). Solid tumor progression has been linked with elevated expression of the chemokine receptor CXCR4 (7). Recently, we have uncovered epigenetic repression of the chemokine ligand CXCL12 in colorectal, mammary, and pancreas cancer, in parallel with increased expression of its cognate receptor CXCR4. Silencing of the ligand enables homing of CXCR4 expressing cancer cells to metastatic sites rich in CXCL12 (8–10). Subsequent histopathological studies have also linked diminished CXCL12 expression with worse prognosis in gastric cancer, osteosarcoma, and breast cancer (11–13). Our more recent study in pancreatic cancer demonstrated that re-expression of autocrine CXCL12 inhibits PDAC cell growth as well as cell migration (10). Chemokines such as CXCL12 stimulate migration in a biphasic manner, with 10 nM optimally stimulating epithelial cell movement and ≥100 nM unable to induce migration (14–18). In contrast, chemokine signaling through the Gαi protein stimulates intracellular calcium flux over a broader concentration range (e.g. 1–1000 nM) (16, 18). Importantly, the 1–1000 nM concentration range is comparable to concentrations of CXCL12 physiologically available in circulation and human tissue (15, 19–21). The disparity between these two downstream signaling effects represents a longstanding conundrum in GPCR function. Lefkowitz and colleagues first observed that distinct agonists of the same β-adrenergic receptor can elicit different signals and functions that are either G-protein dependent or β-arrestin signaling mediated, and termed this functional selectivity as “biased agonism” (22). Thus, as defined by Reiter et al, GPCR agonists can fall into three categories: G-protein biased, β-arrestin biased, or balanced in which both G-proteins and arrestin are recruited and signal. A preliminary report by Lefkowitz et al also showed that the chemokines CCL19 and CCL21 could elicit differential β-arrestin biased agonist signaling when binding to the chemokine receptor CCR7 (23).
Prior reports had shown that, in physiologic solutions, chemokines were capable of reversibly forming multimeric structures, in a concentration dependent manner (14, 24, 25). For CXCL12, at lower concentrations the protein remains monomeric, but as the concentration increases, the presence of dimeric structures predominate (25). These data lead to the hypothesis that the dimeric and monomeric structures may possess functional selectivity for the biphasic migration response observed for CXCL12. Subsequently, in a series of prior reports using engineered variants of CXCL12, we discovered that the unique oligomeric forms of the ligand can function as biased agonists after binding to CXCR4 (26, 27), with monomeric CXCL12 eliciting “balanced” signaling and migration, while dimeric CXCL12 stimulated “G-protein biased” signaling without eliciting migration. However, further investigation into a downstream signaling mechanism to link the lack of migration with the G-protein biased agonism stimulated by dimeric CXCL12 is necessary.
Recent studies of PDAC illustrate that the there is a shift in metabolic flux toward anabolic pathways that facilitate nutrient synthesis (28) and that PDAC cells rely on glutamine metabolism to drive oncogene dependent growth (29–31). However, little is known regarding the metabolic or bioenergetic profile of migrating PDAC cells. Likewise, though hormone dependent calcium signaling and cell polarity regulation have been associated with bioenergetic signaling through the homeostatic regulator AMP-Kinase (AMPK) (32, 33), the potential relationship between cell metabolism and chemotaxis is unexplored. Herein, we establish that the decline in chemotactic migration at elevated CXCL12 concentrations reflects CXCR4-dependent AMPK and myosin light chain phosphorylation (MLC). The lack of migration at high concentrations of CXCL12 was replicated using the engineered CXCL12 dimeric ligand. These are the first data demonstrating that receptor activation by a dimeric chemokine ligand can enhance a specific signaling pathway as a result of biased agonism. The direct stimulation of a non-motile state by the dimeric ligand we herein have termed “ataxis”. Broadly, our work unearths an important energy-signaling mechanism underlying chemotaxis and reveals a viable strategy for pharmaceutical targeting of cancer metastasis.
Materials and Methods
Reagents
AICAR, LPA, AMD3100, Metformin, and Compound C were purchased from Sigma (St. Louis, MO). BAPTA-AM, ionomycin, STO-609, and pertussis toxin were purchased from EMD Biosciences Calbiochem (San Diego, CA). The mitochondrial inhibitor Mito-CP was produced and used as previously published (34). Antibodies against total or phosphorylated AMPK, MLC, LKB1, and MYPT1 were purchased from Cell Signaling Technologies (Danvers, MA). The wild-type, locked monomer, and locked dimer variants of CXCL12 were expressed and purified as previously described (27, 35). The CXCL12 locked dimer (CXCL12-LD) mutations are L36C and A65C and create a dimeric protein. The CXCL12 locked monomer (CXCL12-LM) mutations are L55C and I58C. The engineered locked dimer or locked monomer proteins are structurally indistinguishable from native dimeric and monomeric protein, respectively, and retain full CXCR4 receptor binding capability. Proteins were expressed in E. coli and cells were lysed by french press. Fusion protein was purified through nickel chromatography, refolded by infinite dilution and ULP1 protease was used to cleave the 6XHIS-Sumo tag. Cation exchange and HPLC chromatography was used for final purification.
Cells
The human pancreatic carcinoma cells Panc1 (CRL-1469) and MiaPaCa2 (CRL-1420) were purchased from the American Type Culture Collection (ATCC, Rockville, MD). Patient derived pancreatic ductal adenocarcinoma cells MCW512, corresponding to MCW-4 from our prior report, were obtained from the Medical College of Wisconsin Surgical Oncology Biobank using IRB approved protocols and cultured as previously published (10). The cell lines K8282 and K8484 were derived from the original KRasLSL.G12D/+-p53R172H/+-PdxCre (KPC) mice on the mixed 129/SvJae/C57BL/6 background and were the kind gift of Dr. Kenneth Olive (Columbia University, NY). FC1199, FC1242, FC1245, and DT10022 cell lines were derived from KPC mice in which each of the founder mutant mice had been backcrossed to the C57BL/6 genetic background. KPC cells were maintained in high glucose DMEM with 10% (v/v) FBS (Life Technologies Inc., Grand Island, NY). The Pan-02 cell lines were provided by the National Cancer Institute Cell Repository (Bethesda, MD) and maintained in RPMI-1640 with 10% (v/v) FBS.
Orthotopic xenograft model
Severe combined immunodeficiency mice (cr-Prdkcscid, Charles Rivers Laboratories, Wilmington, MA) were anesthetized and orthotopically implanted with either 106 Panc1 or MiaPaCa2 cells stably expressing firefly luciferase and tumor formation tracked in vivo by bioluminescent imaging (Lumina IVIS 100, Perkin Elmer, Alameda, CA) using our previously published technique (10). At 7 days post-implantation, mice were sorted into vehicle or treatment groups with equivalent average luminescence and treated twice-weekly thereafter with 200 μL intra-peritoneal injections of phosphate-buffered saline or 5 μM recombinant CXCL12 protein. Mice in the Panc1 model were allowed to survive until humanely euthanized when morbid, in accordance with an IACUC approved protocol, while MiaPaCa2 xenografted mice were sacrificed on day 70. Ex vivo analysis was performed with individual luminescence measurements of the liver, lung, and adjacent lymph nodes. The peritoneal cavity was visually inspected and imaged post-organ harvest to detect potential peritoneal movement of tumor cells.
Energetic flux assay
Changes in bioenergetic flux in pancreatic cancer cells were measured using Seahorse Bioscience XF96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA). MiaPaCa2 cells were first plated overnight in Seahorse plates, and then equilibrated in unbuffered, serum-free medium containing only 5.5 mM glucose and 4 mM L-glutamine for 3 hours. Prior to the injection of chemokine into each well, eight baseline measurements of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were taken and averaged as a time zero energy measurement. Chemokines were then automatically dispensed into wells, with 8 measurements taken over 1 hour to determine basal energetic flux, after which glycolytic and oxidative stress tests were employed using the inhibitors, oligomycin, dinitrophenol (DNP), and Antimycin A. Stress test inhibitors were injected sequentially with 3 measurements taken after each individual treatment. Oligomycin was used to measure ATP-linked OCR and reserve ECAR, DNP was used to measure reserve OCR, and Antimycin A was used to correct for non-specific flux.
Immunoblotting
Cells were plated to 80% confluency in 60 mm dishes and then starved 24 hours for transfected cells or 5 hours for stimulated cells. Stimulations were performed in serum-free medium and inhibitors placed on cells 1 hour before stimulation. After stimulation, cells were washed twice in cold PBS and lysed using a modified RIPA buffer. Lysates were normalized for protein concentration, size separated using reducing SDS-PAGE, electro-transferred to PVDF membranes (Millipore) and then probed using primary and horseradish peroxidase-conjugated secondary antibodies. Proteins were visualized by chemiluminescence with auto-exposure and quantified by densitometric analysis using the FluorChem HD2 from Cell Biosciences (Santa Clara, CA). The optical densities of the proteins in immunoblots from independent experiments were obtained by densitometry.
Flow cytometry
Cells are grown to 80% confluency then lifted using Enzyme-Free Dissociation Buffer (Invitrogen) and washed twice using cold PBS. Cells were incubated in a 1% (w/v) BSA blocking solution, incubated for 1 hour in primary antibody on ice, washed twice and then incubated for 1 hour in fluorophore-conjugated secondary antibody. Cells were then fixed in 2% (w/v) paraformaldehyde and fluorescence intensity measured on a BD-LSR II flow cytometer and analyzed by FlowJo software (BD Biosciences).
Immunofluorescence
Immunofluorescence analysis was performed as published previously (36). Briefly, cells were plated overnight at 50% confluency on glass coverslips, serum-starved 2 hours and then treated. After treatment, cells were fixed using paraformaldehyde, blocked and permeabilized, and then incubated overnight at 4°C with a phosphor-specific antibody for MLC. Cells were then washed and incubated with a goat-anti-rabbit AlexaFluor-488-conjugated antibody and phalloidin-AlexaFluor-594 for two hours at room temperature, counterstained with DAPI for 15 minutes, and coverslipped. Microscopy was performed and fluorescence intensity measured using the Nikon Eclipse Ti microscope and software at 600x magnification under oil immersion. Percent of cells with stress fiber formation was quantified with images blinded for treatment and then scored by a separate investigator for presence of stress fiber or cortical actin.
RT-PCR
RNA was isolated using the RNA Easy kit from Qiagen and treated with DNAse I to remove genomic DNA contaminants. Conversion to cDNA was performed by priming with random hexamers and the SuperScript II cDNA synthesis kit (Life Technologies). PCR products were separated by agarose gel electrophoresis and visualized by ethidium bromide staining and ultraviolet imaging. Primers for the promoter region of the Mannose-Binding Lectin (MBL) gene were used to test for genomic DNA contaminant, while primers for the GAPDH transcript were used as a loading control. Amplification of chemokine receptors and chemokine were done using the following primers (5′-3′): Mouse CXCL12 Forward – ACCTCGGTGGTCCTCTTGCTGTCC, Mouse CXCL12 Reverse – GTTGGCTCTGGCGATGTGGCTCTC, Mouse CXCR4 Forward – TTGTCCACGCCACCAACAGTCA, Mouse CXCR4 Reverse – TGAAACACCACCATCCACAGGC, Mouse CXCR7 Forward – GGAGCCTGCAGCGCTCACCG, and Mouse CXCR7 Reverse - CTTAGCCTGGATATTCACCC.
CXCR4 depletion
shRNA sequences complementary to CXCR4 transcript, sequences 4052 (#1) and 4055 (#2), were purchased from the Open Biosystems dataset (Huntsville, AL) pre-inserted into the LL3.7-Puro vector. Mock, Scramble, or CXCR4 shRNAs were cloned into competent E. coli. Lentiviral particles were constructed by transfecting each plasmid with viral accessory plasmids, pVSVG, pREV, and pRRE into HEK-293T cells using Mirus reagent (Mirus, Madison, WI). After overnight transfection, medium was exchanged for harvest media and cells cultured for 48 hours. Harvested viral particles were then collected, filtered, and stored for later use. MiaPaCa2 cells were grown to ~50% confluency and then transduced with harvested Mock, Scramble, or CXCR4-encoding lentiviral particles using polybrene for 24 hours. Transduced cells were then selected using puromycin for seven days, after which stable protein depletion was measured by both immunoblotting and flow cytometry.
Calcium Flux
Calcium mobilization was measured using the Fluo-4-AM cell permeable dye (Invitrogen) according to manufacturer’s directions. Cells were plated to 90% confluency in 96-well plates and then serum-starved for 2 hours the next day. Cells were washed with Calcium/Magnesium-free Dulbecco’s PBS, loaded with Fluo-4-AM for 30 minutes at 37°C and then 10 minutes at room temperature. Stimulants, diluted in calcium/magnesium-free HBSS were then loaded into auto-injectors of the Victor Wallac (Waltham, MA). Background readings were taken before stimulant injection (3 readings), and then 30 readings every 5 seconds were taken post-injection with ionomycin as a positive control.
Transwell migration
Cells were grown to confluence in transwell inserts (Corning Costar (Corning, NY). The upper well of the transwell membrane was coated with Collagen I (15 μg/mL) then incubated in 2% (w/v) BSA/PBS until cells were ready for plating. Cells were lifted using enzyme-free buffer, washed, and plated to the upper well, with chemoattractants added to the bottom well in serum-free media. MiaPaCa2 and MCW512 cell migration (105 cells plated) was measured after 6 and 4 hours, respectively. Transwell migration was enumerated in a representative set of images taken using fluorescence microscopy after the upper side of each well was swabbed, fixed using 4% (w/v) paraformaldehyde, and stained with DAPI. Any functional inhibitors were mixed with cells plated in the top of transwell chamber, with a pre-treatment incubation time of 30 minutes.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 4 (San Diego, CA). Paired analyses were calculated using a student’s t-test. Multiple comparisons were analyzed using a one-way ANOVA and either a Dunnett post-hoc analysis or a Tukey post-hoc analysis to identify pair-wise differences between distinct experimental groups. Statistical significance was defined as P≤0.05.
Results
Treatment with recombinant chemokine blocks pancreatic cancer metastasis
We recently demonstrated that stable re-expression of CXCL12 in human PDAC cells has a tumor suppressive effect by slowing cancer cell proliferation and mobility (10). Based on those finding, we hypothesized that, as a secreted protein, CXCL12 would function as a biologic response modifier of PDAC malignancy. Combining our previously established pancreatic orthotopic xenograft models with a therapeutic regimen and window defined in our previous studies using the protein in vivo for colorectal cancer and melanoma (10, 27, 37), we found that mice treated twice-weekly with CXCL12 had less dissemination, relative to vehicle treatment (Figure 1A–B). Ex vivo luminescence of metastatic destination organs, including liver, lung, & proximal lymph nodes, showed that CXCL12 treatment significantly lowered overall tumor dissemination, compared to controls (Figure 1C–D). Notably, decreased metastasis was separately observed in both the MiaPaCa2 and Panc1 xenograft models, without any concomitant increase in peritoneal cavity dissemination. These data on the anti-metastatic effects of CXCL12 mirror our prior report in colorectal cancer and melanoma (27).
Figure 1. Systemic treatment with recombinant CXCL12 blocks pancreatic cancer metastasis in orthotopic xenograft mouse models.

(A) Panc1 and MiaPaCa2 firefly luciferase transfected cells were orthotopically implanted into the pancreata of immunocompromised mice. At day 7 post-implantation, mice were sorted into vehicle or 5 μM CXCL12 treatment groups and were injected intraperitoneally twice-weekly and thereafter until study end. Tumor growth and dissemination were tracked weekly using bioluminescence imaging. The MiaPaCa2 model was ended at 70 days while the Panc1 model was performed as a survival study with animal removal at signs of morbidity. (B) Representative mice with Panc1 implants are shown, with CXCL12 treated mice exhibiting less tumor dissemination at day 112. (C) Tumor dissemination and metastasis was analyzed ex vivo and showed that, in both MiaPaCa2 and Panc1 models, CXCL12 treatment significantly decreased pancreatic cancer cell metastasis. (D) Representative bioluminescence image and heat-map of excised livers from Panc1 engrafted vehicle and CXCL12-treated groups, reflecting extent of metastasis. (*) denotes P≤0.05. Values = mean ± SE. n=5–6.
CXCL12 alters pancreatic cancer bioenergetic rates and AMPK signaling
One explanation for the anti-metastatic effect of CXCL12 in vivo is that exogenous chemokine disrupts the concentration gradients necessary for chemotactic migration of cancer cells. Based on our observation that CXCL12 regulates in vitro pancreatic cell migration in a biphasic manner, wherein low nanomolar doses (≥10 nM) stimulate migration while higher doses (≥100 nM) are “ataxic” and unable to evoke migration, we hypothesized that the change from CXCL12 from a pro- to an anti-metastatic ligand reflected functional selectivity or biased agonist signaling through the CXCR4 receptor (10). The mechanisms for functional selectivity of CXCL12 through CXCR4 are unknown. Using a Seahorse-XF analyzer we first tested the notion that the biphasic migratory response to CXCL12 reflected changes in energy metabolism. A declining, dose-dependent trend in basal oxygen consumption and glycolysis rates in MiaPaCa2 cells stimulated with CXCL12was measured (Figure S1, Figure 2A–B). Significant decreases in ATP-linked oxidative phosphorylation, basal glycolysis, and reserve glycolytic capacity were observed in MiaPaCa2 cells stimulated with the ataxic 1000 nM dose of CXCL12, compared to vehicle control (Figure 2A–B). When oxygen consumption and glycolytic rates were visualized together, an overall declining trend in cellular energy was observed as the concentration of CXCL12 increased (Figure 2C).
Figure 2. Ataxic doses of CXCL12 alter pancreatic cancer cell bioenergy levels and stimulate prolonged AMP-Kinase activity.

(A–C) MiaPaCa2 cells were serum-starved and treated with a concentration curve ranging from chemotactic (10 nM) to ataxic (1000 nM) doses of CXCL12. Upon treatment, basal oxygen consumption and extracellular acidification rates (OCR & ECAR) were measured using a Seahorse-XF analyzer continuously for 1 hour. At 1 hour, energetic stress tests were employed sequentially, with oligomycin first used to measure ATP-linked OCR or ECAR reserve, then dinitrophenol used second to measure OCR Reserve. (*) denotes P≤0.05, (**) denotes P≤0.01. Values = mean ± SE, n=5. (D–E) Human MiaPaCa2 or patient-derived MCW512 (MCW-4), or murine KRasLSL.G12D/+-p53R172H/+-PdxCretg/+ (KPC) derived FC1199 cells were stimulated for either 15 minutes or 1 hour with 1, 10, 100, or 1000 nM recombinant CXCL12. At each time point, protein lysates were collected, separated on a reducing SDS-PAGE, and transferred to a membrane. Membranes were then blotted for T172-phophorylated AMP-Kinase (pAMPK) and total AMPK. (*) denotes P≤0.05, (**) denotes P≤0.01, (***) denotes P≤0.001. n=3–4.
We next predicted that CXCL12 dependent changes in cell energetics may be linked to altered activity of AMPK, a known regulator of metabolic stress. Immunoblot analysis demonstrated that CXCL12 dose-dependently stimulated phosphorylation at the activating threonine-172 site of AMPK (Figure 2D). Non-motile 100 and 1000 nM doses of CXCL12 stimulated significant and sustained AMPK phosphorylation through 1 hour (Figure 2D). CXCL12 regulation of migration and AMPK activity was next tested in a previously characterized patient-derived PDAC cell line (10). As predicted, patient-derived MCW512 cells migrated in response to exogenous CXCL12 stimulation in a biphasic fashion, with peak chemotaxis at 10 nM CXCL12 (Figure S1C). As with MiaPaCa2 cells, 1000 nM CXCL12 stimulated significant increase in AMPK phosphorylation in patient-derived cells (Figure S2D). Together, these data demonstrate that ataxic doses of CXCL12 stimulate prolonged AMPK activity in human PDAC cells.
To address whether CXCL12-induced AMPK activity was species dependent, we next acquired a battery of mouse-derived PDAC cell lines and first analyzed chemokine and receptor expression. RT-PCR analysis showed that 4 of 7 mouse lines lacked appreciable CXCL12 transcript, 4 of 7 mouse lines expressed CXCR4 transcript, and each line expressed CXCR7 mRNA (Figure S2A). Immunoblot and flow cytometric analyses confirmed protein expression of CXCR4 (Figure S2B–D). Of the seven cell lines, FC1199 and Pan-02 most closely mimicked the expression pattern of CXCL12, CXCR4, and CXCR7 that we noted in human PDAC (10). Given its novel isolation from the primary tumor of C57BL/6 KRasLSL.G12D/+-p53R172H/+-PdxCretg/+ (KPC) mouse model of PDAC, we identified the FC1199 cell line as a suitable model for further study. FC1199 cells had increased AMPK activity in a dose-dependent fashion after CXCL12 stimulation (Figure 2E). Consistent with the human cell lines, CXCL12 stimulated AMPK phosphorylation was significantly increased 1 hour after stimulation with 100 or 1000nM, but not 1 or 10 nM doses, in FC1199 cells (Figure 2E). These data indicate that CXCL12-induced AMPK activity is a species-independent event.
In sum, our observations indicate that CXCL12 regulates bioenergy consumption rates as well as energy-sensing signaling in a dose-dependent manner. The optimal cell migratory dose of CXCL12 lead to transient AMPK activity and minimal change in glycolysis or oxidative phosphorylation. In contrast, doses exceeding 100 nM CXCL12, which do not evoke cell migration, lead to sustained (≥ 1 hour) AMPK activity and decreased glycolysis and oxidative phosphorylation.
CXCL12 stimulates AMPK through CXCR4 and Gαi calcium signaling
As our previous studies demonstrated that CXCL12 induced biphasic migration is mediated through CXCR4 binding (27), rather than CXCR7, we asked if the AMPK phosphorylation stimulated by CXCL12 is dependent on CXCR4 signaling. As a first approach, immunoblot and flow cytometric analyses confirmed depletion of CXCR4 in two MiaPaCa2 clones (Figure S3A–B). Transwell migration assays then showed that the control and scramble-sequence expressing cells migrated towards 10 nM CXCL12, while CXCR4-knockdown clones were unable to migrate in response to chemokine stimulation (Figure S3C–D). We subsequently assessed AMPK activity in those CXCR4-knockdown cells. As shown in Supplemental Figure 3E–F, at the short time point of 15 minutes, CXCL12 stimulated little if any AMPK phosphorylation in CXCR4-depleted clones while the scramble transduced cells ably responded to non-motility-inducing doses of chemokine. We next tested whether AMPK activation was due to Gαi protein coupled signaling. Consistent with elevated CXCL12 signaling in a biased agonist fashion, immunoblot analysis showed that AMPK phosphorylation was decreased after pretreatment with either pertussis toxin (Figure S3G) or the small molecule CXCR4 antagonist AMD3100 (Figure S3H). These data demonstrate that AMPK phosphorylation in response to the ataxic ≥100 nM doses of CXCL12 reflect activation of the canonical CXCR4-Gαi signaling mechanism.
In previous studies, we and others established that, in contrast to the biphasic functional migration response, CXCL12 stimulates intracellular calcium flux in a saturable concentration-dependent fashion (25–27). In MiaPaCa2 cells, CXCL12 doses exceeding 10 nM stimulated equivalent maximal levels of intracellular calcium flux with and without extracellular calcium ions (Figure 3A–D). In patient-derived PDAC cells, 1000 nM CXCL12 stimulated a significant and sustained increase in calcium (Figure S4A–B). Given the similarity of concentration dependent change in AMPK activity and calcium mobilization in response to CXCL12, we predicted that increased AMPK phosphorylation was dependent on calcium mobilization. Subsequently, the cell permeable calcium ion chelator BAPTA-AM abrogated CXCL12 or ionomycin-induced AMPK phosphorylation in both MiaPaCa2 and patient-derived cells (Figure S4C–D).
Figure 3. CXCL12 stimulated AMPK activity is calmodulin-kinase-kinase-2 dependent.

Calcium mobilization was probed using the Fluo-4 membrane permeable fluorescent dye and measured on a Victor-Wallac plate reader continuously for 180 seconds following stimulation with 1, 10, 100, or 1000 nM CXCL12 and the ionomycin (IM) positive control. (A–B) MiaPaCa2 cells were pre-incubated in ion-free buffer to assess intracellular calcium mobilization and a representative time curve (A) and measurement of maximal flux achieved (B) shown. (**) denotes P≤0.01, (***) denotes P≤0.001 in comparison to vehicle control. n=4. (C–D) The experiment was repeated with cells pre-incubated in buffer containing calcium ions to assess total calcium intracellular and extracellular mobilization and a representative time curve (C) and measurement of maximal flux achieved (D) shown. (**) denotes P≤0.01, (***) denotes P≤0.001 in comparison to vehicle control. Values = mean ± SE. n=5. (E–F) MiaPaCa2 or FC1199 cells were pre-treated with the Calmodulin-Kinase-Kinase-2 inhibitor STO-609 and then stimulated with 10 or 1000 nM CXCL12 for 15 minutes. Protein lysates were then taken and immunoblot analysis performed to assess AMPK activity. Densitometric analysis revealed that 1000 nM CXCL12 or, as detected on a separate gel, ionomycin, induced AMPK phosphorylation was significantly inhibited using STO-609 in both MiaPaCa2 and FC1199 cells. (*) denotes P≤0.05 comparison ± STO-609. Values = mean ± SE. n=3.
Prior reports exploring the regulation of AMPK activity have shown that T172 can be phosphorylated by either the cell polarity regulator LKB1 or calcium dependent calmodulin-kinase-kinase-2 (CamKK2). While LKB1 activity was not increased by stimulation with CXCL12 (Figure S4E), the CamKK2 specific inhibitor STO-609 reversed AMPK phosphorylation induced by 1000 nM CXCL12 (Figure 3E–F). In total, our experiments examining CXCL12 downstream signaling in human and murine PDAC cells demonstrate that AMPK activity driven by CXCL12 is mediated through receptor dependent intracellular calcium flux and CamKK2 activity.
AMPK regulates the CXCL12 biphasic migration response
RAPTOR, the established regulator mTOR (mTORC1) signaling, is a key downstream target for AMPK-mediated phosphorylation. While AMPK is conventionally thought to regulate mTOR activity in the context of cell growth, we did not detect any altered RAPTOR signaling (data not shown), consistent with other studies demonstrating non-growth related AMPK signaling (33). The repeated cycling of myosin light chain (MLC) phosphorylation and subsequent de-phosphorylation is essential for the perpetual lever-like action that myosin exhibits on F-actin fibers, allowing cells to contract, relax, and then migrate (38, 39). Activation of AMPK has been proposed to affect MLC activity by preventing the de-phosphorylation that occurs on the regulatory unit of MLC (40, 41). Mirroring our previous studies in intestinal epithelial cells (36), immunoblot analysis of MiaPaC2 cells showed that the chemotactic dose lead to peak phosphorylation of MLC at 15 minutes, which then returned to baseline levels by 60 minutes; a similar temporal pattern in MLC phosphorylation was observed with the migration positive control (Figure 4A). In contrast, both ataxic CXCL12 stimulation and the AICAR positive control evoked a sustained increase in MLC phosphorylation over 60 minutes (Figure 4B).
Figure 4. Elevated doses of CXCL12 induce AMPK dependent locking of MLC-actin migration machinery through sustained phosphorylation of MLC and MYPT1.

(A–B) MiaPaCa2 cells were stimulated with chemotactic (10 nM) and ataxic (1000 nM) doses of CXCL12, along with 1 μg/mL Lysophosphatidic Acid (LPA) or 100 nM AICAR as positive controls, over a time course of 5, 15, 30, and 60 minutes. Lysates were then collected and probed for phosphorylated and total levels of myosin light chain (MLC) protein. 10 nM CXCL12 and the migration control LPA stimulated pMLC in a biphasic time dependent manner, peaking within 15 minutes. 1000 nM CXCL12 mimicked AICAR, stimulating high pMLC that plateaued through 60 minutes. (C–D) Pre-treatment with the AMPK inhibitor compound C (Com. C) abrogated phosphorylation of both MLC and MYPT1 (T853) stimulated by 1000 nM CXCL12 or AICAR at 30 minutes, confirmed by densitometric analysis. (*) denotes P≤0.05 in comparison of stimulation ± pretreatment with compound C. n=5. (E–G) MiaPaCa2s, pre-treated with the AMPK inhibitor compound C (CC), were stimulated for 1 hour with 10 nM and 1000 nM CXCL12. LPA and AICAR were controls. Cells were stained for pMLC (green) and F-actin (red) using fluorophore-conjugated antibodies. Analysis was limited to contact uninhibited cells that had potential to migrate. (A) Microscopy revealed that cells treated with 1000 nM CXCL12 or AICAR were rounded in shape with primarily cortical actin and minimal stress-fiber actin filaments. 10 nM CXCL12 or LPA stimulated more filopodia, and more actin stress fibers. AMPK inhibition lead to increased filopodia and stress fiber formation at 1000 nM CXCL12, but did not alter vehicle or 10 nM CXCL12 stimulated phenotype. (F) pMLC levels, measured on a single cell basis using FITC fluorescence intensity per micron, increased upon 1000 nM CXCL12 or AICAR stimulation, which was reversed with compound C. (*) denotes P≤0.05 compared to vehicle; (#) denotes P≤0.05 compared to1000 nM CXCL12. (G) Blinded quantification of actin filament accumulation showed significantly increased percentage of cells with actin stress fibers after stimulation with 10 nM CXCL12 or positive control LPA compared to control cells. (***) denotes P≤0.001 compared to vehicle; (###) denotes P≤0.001 compared to1000 nM CXCL12 + Compound C. Values = mean ± SE. A minimum of 6 cells was analyzed per treatment. n=4 biological replicates.
We hypothesized that differential MLC phosphorylation in response to CXCL12 was due to AMPK downstream signaling. The AMPK inhibitor Compound C reversed MLC phosphorylation induced by 1000 nM CXCL12 (Figure 4C). Previous studies indicated that AMPK regulates MLC by inhibitory phosphorylation of myosin phosphatase 1 (MYPT1) (42, 43), an enzyme responsible for de-phosphorylating MLC. We thus predicted that ataxic doses of CXCL12 would activate AMPK and in turn inhibit MYPT1 function. In a time course, CXCL12 lead to inhibitory phosphorylation of T853 of MYPT1 between 15 and 60 minutes (Figure S5). We then examined the AMPK dependence of CXCL12 regulation of MYPT1 and showed that compound C significantly reversed the ataxic CXCL12 or AICAR-induced inhibitory phosphorylation of MYPT1 (Figure 4D) suggesting that elevated levels of CXCL12 disrupt MLC activity through AMPK.
We next questioned if the CXCL12 stimulated change in MLC phosphorylation significantly altered the assembly of cell migration machinery through AMPK. Immunofluorescence microscopy revealed that after stimulation with ataxic doses of CXCL12 cells were rounded with few cytoplasmic projections, consistent with the non-migratory phenotype (Figure 5E). By comparison, the chemotactic dose of CXCL12 led to more cell spreading, with abundant filopodia and lamellopodia formation (Figure 5E). Consistent with our immunoblot analysis, after one-hour stimulation, immunofluorescence microscopy revealed that ataxic, and not the chemotactic, dose significantly increased levels of phosphorylated MLC in treated cells (Figure 5F). In parallel, PDAC cells stimulated with 10 nM CXCL12 had a higher percentage of actin stress fibers, while actin in 1000 nM CXCL12-treated cells was organized primarily in cortical bundles (Figure 5G). Finally, compound C pretreatment of cells stimulated with 1000 nM CXCL12 reversed MLC phosphorylation, and in turn increased formation of actin stress fibers and cell spreading (Figure 5E–G).
Figure 5. Pancreatic cancer cell migration is dependent on metabolic activity and CXCL12 biphasic regulation of migration is AMPK dependent.

(A–B) In 0.5% serum containing media, both basal and 10 nM CXCL12 induced Panc1 migration were decreased in a dose-dependent fashion by a combination of a mitchondrial inhibitor Mito-CP and the glycolytic inhibitor 2-deoxyglucose (2-DG), mixed with cells in the upper chamber of the transwell 30 minutes prior to migration (A). A combination of metformin and 2-DG significantly decreased 10 nM CXCL12 induced migration of MiaPaCa2 cells (B). n=3–4. (*) denotes P≤0.05, (**) denotes P≤0.01 compared to unstimulated (−) control, and (#) denotes P≤0.05 compared 10 nM CXCL12 stimulated cells. (C–D) MiaPaCa2 cells were stimulated with 1, 10, 100, or 1000 nM doses of CXCL12 with or without pretreatment with the AMPK inhibitor compound C. Biphasic chemotactic transwell migration stimulated by CXCL12 was abolished in cells pretreated with compound C for 30 minutes. The 10% fetal bovine serum (FBS) used as a positive control stimulated equal cell migration irrespective of compound C pretreatment. Representative images of high-powered fields of transwell membranes from each condition. (***) denotes P≤0.001 in comparison to vehicle control; (###) denotes P≤0.001 in comparison between 100 and 1000 nM CXCL12 ± Compound C. Values = mean ± SE. n = 5.
We then sought to establish a link between metabolism and migration. We first showed in a transwell migration assay, blocking glycolysis with 2-deoxyglucose (2-DG) and inhibiting mitochondria function with our previously established inhibitor Mito-CP (34), that abrogation of metabolic activity decreased both basal migration chemokine-directed migration of MiaPaCa2 cells in a concentration dependent manner (Figure 5A). We then duplicated this result using dual inhibition of metabolic flux with 2-DG and metformin, an established inhibitor of pancreatic cancer cell metabolism (44) (Figure 5B). Last, we tested the notion that chemotaxis regulated by CXCL12 is dependent on AMPK activity in a MiaPaCa2 transwell migration assay stimulated with a concentration curve of CXCL12 in the presence or absence of the AMPK inhibitor (Figure 5C–D). In the absence of AMPK inhibition, CXCL12 stimulated a biphasic pattern of migration consistent with our previously published data (10). However, pre-treatment with compound C completely reversed the biphasic pattern and lead to saturable CXCL12-induced migration. Importantly, the introduction of the AMPK inhibitor significantly increased migration at the previously ataxic doses of 100 and 1000 nM CXCL12, while continuing to permit chemotaxis at the optimal dose of 10 nM CXCL12 (Figure 5C–D).
Our prior work established not only that CXCL12 exists primarily as a monomer at low concentrations and as a dimer at elevated concentrations, but that an engineered locked dimer CXCL12 protein effectively interfered with colorectal cancer and melanoma metastasis, with increased stability in vivo compared with wild-type protein (27, 37). We tested whether the biophysical dimeric chemokine mechanism might explain CXCL12-induced ataxis through activation of the AMPK bioenergetic signaling mechanism. Using our previously established engineered mutants of CXCL12 locked in either the monomeric or dimeric state (27, 35), we first showed that 10 nM CXCL12-LM stimulated migration of MiaPaCa2 cells equivalent to 10 nM wild-type protein (Figure 6A). As predicted, 10 nM CXCL12-LD did not elicit significant transwell migration of MiaPaCa2 cells but did stimulate equivalent calcium mobilization compared to monomer or wild-type protein in both MiaPaCa2 and FC1199 cells (Figure 6B). Reflecting our observations using 1000nM of the wild-type protein, CXCL12-LD stimulated prolonged AMPK activity compared to CXCL12-LM. Further, 1 and 2 hour stimulation with locked dimer CXCL12 resulted in substantially more phosphorylation of AMPK compared to monomeric CXCL12 (Figure 6C). Finally, we tested the notion that the lack of migration observed in CXCL12-LD treated cells is dependent on AMPK activity. Using a transwell assay of MiaPaCa2 cells, we found that pre-treatment with the AMPK inhibitor Compound C reversed the ataxis stimulated by 10 nM CXCL12-LD. Instead, in the presence of Compound C the CXCL12-LD stimulated a strong migration that was equivalent to that evoked by the same concentration of wild-type CXCL12 or CXLC12-LM (Figure 6D).
Figure 6. Locked dimeric CXCL12 preferentially stimulates ataxis and prolonged AMPK phosphorylation relative to locked monomeric CXCL12 in pancreatic cancer cells.
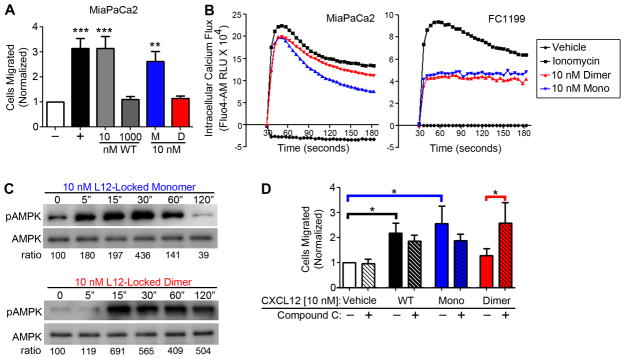
(A) Transwell migration of MiaPaCa2 cells shows that CXCL12 locked monomer (M) stimulates migration similar to 10 nM wild-type CXCL12 (WT) and CXCL12 locked dimer (D) recapitulates ataxis stimulated by 1000 nM wild-type CXCL12. (**) denotes P≤0.01 and (***) denotes P≤0.001 in comparison to vehicle control. n=4. (B) Peak intracellular calcium mobilization, measured using Fluo-4, was similar for 10 nM locked monomer or locked dimer CXCL12 variants in both MiaPaCa2 and FC1199 over a 3 minute time course. Plots are representative of 3 biological replicates. (C) Using immunoblot analysis, AMPK phosphorylation was measured over time in FC1199 cells stimulated with locked monomeric CXCL12 at 5, 15, 30, 60, and 120 minutes. Densometric analysis, shown below, confirmed that CXCL12 locked dimer maintained AMPK phosphorylation at 60 and 120 minutes compared to CXCL12 locked monomer. (D) In a transwell migration assay, MiaPaCa2 cells were pretreated ± AMPK inhibitor Compound C for 30 minutes and then allowed to migrate towards vehicle or 10 nM of wild-type CXCL12 (WT), CXCL12 locked monomer (Mono), or CXCL12 locked dimer (Dimer). 10 nM wild-type and monomer proteins stimulated significant migration, while Compound C reversed dimeric ataxis in pair-wise analyses. (*) denotes P≤0.05. Values = mean ± SE. n=6.
Taken together, these data suggest that the concentration-dependent decline of chemotactic migration is mediated in part by AMPK. Ataxic doses of CXCL12 prevented dephosphorylation of MLC by inhibiting MYPT1 function. This loss in MLC dephosphorylation then prevented the uncoupling of MLC to F-actin, leading to the accumulation of cortical actin bundles. Thus, concentrations of CXCL12 exceeding 100 nM require AMPK activity in order to actively prevent migration. These data indicate that the decline in migration is the result of biased agonism of CXCL12 binding to its cognate receptor CXCR4. Prolonged AMPK activity was similarly observed with stimulation by locked dimeric CXCL12, suggesting that this engineered protein has significant potential as a more stable in vivo regulator of PDAC metabolism and metastasis.
Discussion
The CXCL12-CXCR4 chemokine axis has been implicated in numerous cancers, including highly metastatic PDAC, under the conventional model that elevated receptor expression correlates with malignancy (7). However, this model has not translated to successful clinical development of CXCR4-targeted anti-cancer therapies, suggesting a clear need for a novel pharmacological approach. In particular, the mechanism underlying the ability of chemokines to control directional cellular migration at different doses is not known. The conventional model for chemokine involvement in cancer metastasis has been over-expression of CXCR4 leading to increased sensitization and passive migration towards distant tissue sources of CXCL12 such as the liver (45). This current dogma ignores the potential active role that the ligand CXCL12 plays in modulating cancer. Our work in highly malignant PDAC extends the current understanding of chemokines in cancer in two important ways. First, while other recent PDAC studies have focused on the role of chemokines in the primary tumor (46, 47), our data herein and in a previous study (10), identify an ataxic, protective role for CXCL12-CXCR4 in metastasis of pancreatic cancer. Second, though numerous studies have defined shifts in metabolism that occur during pancreatic cancer cell growth (29–31), our report is the first to observe the changes in bioenergetic metabolism that occur during pancreatic cancer cell migration. Our data indicate that CXCL12 can function as a balanced or biased agonist for its cognate receptor CXCR4, depending on the chemokine concentration. Elevated doses of CXCL12 effectively activate an AMPK-mediated bioenergetic brake that shifts pancreatic cancer cells into a non-migratory response. We additionally demonstrate in a preclinical model that CXCL12 inhibits pancreatic cancer metastasis in vivo.
While canonical chemokine signaling occurs through G-protein dependent calcium mobilization, the precise biochemical mechanisms downstream of chemokine ligand-receptor binding are still emerging. In this study of PDAC, we show that 10 nM CXCL12 stimulates maximal migration (chemotactic) while concentrations >100 nM are unable to stimulate migration (ataxic). As we and others have shown for a number of chemokine axes (14, 16–18), over a concentration curve, CXCL12 stimulates migration in a biphasic fashion, while eliciting intracellular calcium mobilization in a saturable manner. The decline in chemokine-induced migration at elevated doses has commonly been attributed to receptor desensitization, but often without a mechanistic explanation (48). Our data reveal a novel mechanism that explains the lack of migration at elevated concentrations of chemokines through a biased agonist signaling cascade. Here, we show that CXCL12 dose-dependently alters glycolysis, oxidative phosphorylation, and promotes activity of the homeostatic metabolism regulator AMPK. We confirmed that this dose-dependent effect on bioenergetic signaling was CXCR4-Gαi-calcium signaling dependent. While others have demonstrated that small molecule GPCR agonists are capable of inducing AMPK activity (33) in a calcium dependent manner, our study is the first to link chemokines, as soluble proteins, with downstream AMPK signaling (Figure 7). Further, chemotactic CXCL12 concentrations induced cycling of phosphorylation of MLC, facilitating the perpetual motion of stress-fiber actin (Figure 7A) and in turn allowing directional movement (Figure 7C). In contrast, cells stimulated with ataxic doses of CXCL12 stimulated the AMPK dependent increase of MYPT1 inhibitory phosphorylation, which blocks MLC phosphorylation:dephosphorylation cycling (Figure 7B), effectively locking the migration machinery, and preventing cell movement (Figure 7D). While the role of the alternative CXCL12 receptor CXCR7 was not explored, we predict that since CXCR7 signaling is G-protein independent (27) it will not affect AMPK activity. Additionally, as activity of AKT, a known downstream effector of chemokines, has recently been linked to inhibition of AMPK (33), further study into the interplay between AKT and AMPK in chemokine receptor signaling is necessary. Cumulatively, these data support a model wherein elevated concentrations of chemokine ligand, such as might be found once cells have arrived at their chemotactic destination, activate a bioenergetic molecular brake to stop moving.
Figure 7. Model for CXCL12 biased agonist regulation of migration through AMPK-MLC signaling.

(A) In PDAC cells stimulated with chemotactic doses of CXCL12 (≥10 nM), signaling leads to a balanced cycling of MLC phosphorylation. MLC phosphorylation by MLCK and subsequent de-phosphorylation by MYPT1 allows MLC to first bind filamentous actin (F-actin), move the fiber with a lever-like action, and then re-attach to allow the process to begin again for further movement. (B) In PDAC cells stimulated with ataxic biased doses of CXCL12 (≥100 nM), extended AMPK signaling inhibits the activity of MYPT1, leading to imbalanced cycling of MLC phosphorylation. Buildup of phosphorylated MLC prevents the lever-like action of MLC and subsequent re-attachment to f-actin; as a result, f-actin fibers remain immobile. (C) At the cellular level, by 60 minutes of stimulation, chemotactic doses of CXCL12 induce direction movement through balanced MLC phosphorylation cycling, subsequent stress fiber actin formation, and filopodia formation. (D) Ataxic doses of CXCL12 cause unbalanced and high levels of phospho-MLC binding, perimembranous localization of actin in cortical fashion, and prevent polarized cell contraction and relaxation necessary for directional movement.
Lefkowitz and colleagues first posited the concept of biased agonism wherein different ligands were capable of distinct signaling through a single GPCR (22). Importantly, their more recent study has shown that β-arrestin, a critical regulator of GPCR biased agonism, also modulates MLC phosphorylation downstream of angiotensin signaling (49). Our recent work identified chemokine oligomerization as a biophysical mechanism for changes in migration potential during chemotaxis, and suggested that ligand concentration-dependent biased agonism could explain the associated changes in signaling (25–27). At low nanomolar concentrations, CXCL12 preferentially exists as a monomeric protein, while at high nanomolar and micromolar concentrations and in the presence of binding partners, the chemokine forms dimeric structures (25, 26, 50). Using engineered mutants locked into each form, we determined that monomeric CXCL12 was equipotent to the native protein and thus functions as a balanced agonist. In contrast, dimeric CXCL12 was incapable of stimulating migration but retained the equipotent G-protein-biased calcium signaling response as wild-type or monomeric forms of the ligand (27). As CXCL12 dimer-treated cells were able to migrate in response to CXCR4-independent stimulation, we first termed the dimer effect as “cellular idling” (27). We additionally showed that oligomers of CXCL12 differentially recruited β-arrestin and stimulated phosphorylation of ERK1/2 (27). In this study, we used engineered oligomeric mutants of CXCL12 to demonstrate that the ataxis of pancreatic cancer cells elicited by elevated concentrations of CXCL12 is mirrored with locked dimeric CXCL12. In addition, similar to ataxic, i.e. >100nM, doses of wild-type CXCL12, dimeric CXCL12 elicited more prolonged AMPK activity compared with a monomeric variant of the ligand. Thus, these new data establish a mechanistic link between the observed biphasic regulation of migration by CXCL12 and biased agonism elicited by distinct concentrations or oligomeric states of the chemokine. These data are the first to identify a mechanism by which enhanced signaling drives the arrest of cellular migration, which we have termed “ataxis”, at elevated physiologic concentrations of the chemokine ligand.
Reexamining the role for CXCL12-CXCR4 in the context of biased signaling sheds important light on chemokine function in malignancy. Under the conventional model, some have suggested that CXCR4 is a pro-metastatic factor for pancreatic cancer (51), while others have shown that CXCL12 can stimulate increased activity of the cell survival factor AKT, implying that the chemokine is a pro-growth factor (52). However, nearly all of those studies rely upon a single, typically migratory, dose of CXCL12 to examine signaling outputs and ignore potentially significant functional effects of higher concentrations of CXCL12 previously observed in vivo (20). Along with our previous study (10), these data suggest a model for CXCL12-CXCR4 in pancreatic cancer wherein low, chemotactic concentrations elicit cancer cell movement to distant tissues but locally increased, ataxic concentrations of the ligand CXCL12 abrogate cancer cell movement through bioenergetic signaling and cytoskeletal disregulation. Translating this novel signaling mechanism, we found that systemic treatment with wild-type CXCL12 elicits a similar anti-metastatic effect against malignant pancreatic cancer cells. Some have suggested that inhibition of CXCR4 is a strategy for blocking PDAC malignancy (46, 47). However, administration of small molecule antagonists of CXCR4, such as AMD3100, passively blocks cell movement through competitive inhibition, primarily by blocking 1 of the 2 ligand binding sites on the CXCR4 receptor. Given the CXCR4 antagonist binds only one of two necessary binding sites with micromolar binding affinity, it is perhaps not surprising that AMD3100 failed to provide clinical benefit in long term treatment of HIV patients (53). Our report suggests that the CXCL12 protein is an effective alternative to CXCR4 antagonists to inhibit PDAC metastasis. By specifically eliciting the anti-migratory downstream signaling components, biased agonist variants of CXCL12 have potential to actively block pancreatic cancer cell movement. Further study is needed to fully uncover the pharmacokinetics/pharmacodynamics of this biologic response modifying drug, though extended biologic therapy for >75 days in our two models of pancreatic cancer herein showed few signs of toxicity. Future studies must also examine how CXCL12 biologic therapy will modulate the immune surveillance in the pancreatic primary tumor environment. Lastly, as pancreatic tumors are rich in GAGs (54, 55), which are known to alter chemokine binding, further investigation into the in vivo interaction between recombinant chemokine and extracellular components is necessary.
In sum, this work uncovers a biochemical mechanism for CXCL12 biphasic regulation of migration that links our previous reports in oligomeric chemokine biased agonism (27) and CXCL12 chemotactic signaling (36). We show that ataxic doses of CXCL12 lead to incomplete turnover of migratory signaling and subsequent locking of migration machinery that is bioenergetic signaling dependent. The cumulative findings of our recent reports underscore the anti-metastatic potential of exploiting chemokine biased agonist signaling, address the dire need for identifying strategies to target metastasis, and delineate the importance of future comprehensive pre-clinical studies of chemokine-ligand specific therapies for pancreatic cancer.
Supplementary Material
Acknowledgments
Supported in part by CA178960 (MBD, BK), AI058072 (BFV), as well as a grant from the MCW Cancer Center and philanthropic donations from the Bobbie Nick Voss Charitable Foundation (MBD). Ishan Roy is a member of the Medical Scientist Training Program at MCW, which is partially supported by an NIGMS training grant (T32GM080202).
Footnotes
Conflicts of Interest: Michael Dwinell and Brian Volkman are founders of Protein Foundry, LLC which produces ultra-pure recombinant chemokines for research (proteinfoundry.com).
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: A Cancer Journal for Clinicians. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Katz MHG, Pisters PWT, Evans DB, Sun CC, Lee JE, Fleming JB, et al. Borderline Resectable Pancreatic Cancer: The Importance of This Emerging Stage of Disease. J Am Coll Surg. 2008;206(5):833–46. doi: 10.1016/j.jamcollsurg.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rhim A, Mirek E, Aiello N, Maitra A, Bailey J, McAllister F, et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell. 2012 Jan 20;148(1–2):349–61. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haeno H, Gonen M, Davis M, Herman J, Iacobuzio-Donahue C, Michor F. Computational Modeling of Pancreatic Cancer Reveals Kinetics of Metastasis Suggesting Optimum Treatment Strategies. Cell. 2012 Jan 20;148(1–2):362–75. doi: 10.1016/j.cell.2011.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009 Apr 10;27(11):1806–13. doi: 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luster AD. Chemokines — Chemotactic Cytokines That Mediate Inflammation. N Engl J Med. 1998 Feb 12;338(7):436–45. doi: 10.1056/NEJM199802123380706. 2012/11. [DOI] [PubMed] [Google Scholar]
- 7.Zlotnik A. Chemokines and cancer. Int J Cancer. 2006 Nov 1;119(9):2026–9. doi: 10.1002/ijc.22024. [DOI] [PubMed] [Google Scholar]
- 8.Wendt MK, Johanesen PA, Kang-Decker N, Binion DG, Shah V, Dwinell MB. Silencing of epithelial CXCL12 expression by DNA hypermethylation promotes colonic carcinoma metastasis. Oncogene. 2006 Aug 17;25(36):4986–97. doi: 10.1038/sj.onc.1209505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wendt MK, Cooper AN, Dwinell MB. Epigenetic silencing of CXCL12 increases the metastatic potential of mammary carcinoma cells. Oncogene. 2008 Feb 28;27(10):1461–71. doi: 10.1038/sj.onc.1210751. [DOI] [PubMed] [Google Scholar]
- 10.Roy I, Zimmerman NP, Mackinnon AC, Tsai S, Evans DB, Dwinell MB. CXCL12 Chemokine Expression Suppresses Human Pancreatic Cancer Growth and Metastasis. PLoS ONE. 2014 Mar 04;9(3):e90400. doi: 10.1371/journal.pone.0090400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhi Y, Chen J, Zhang S, Chang X, Ma J, Dai D. Down-regulation of CXCL12 by DNA hypermethylation and its involvement in gastric cancer metastatic progression. Dig Dis Sci. 2012 Mar;57(3):650–9. doi: 10.1007/s10620-011-1922-5. [DOI] [PubMed] [Google Scholar]
- 12.Baumhoer D, Smida J, Zillmer S, Rosemann M, Atkinson MJ, Nelson PJ, et al. Strong expression of CXCL12 is associated with a favorable outcome in osteosarcoma. Mod Pathol. 2012;25(4):522–8. doi: 10.1038/modpathol.2011.193. print. [DOI] [PubMed] [Google Scholar]
- 13.Ramos E, Camargo A, Braun K, Slowik R, Cavalli I, Ribeiro E, et al. Simultaneous CXCL12 and ESR1 CpG island hypermethylation correlates with poor prognosis in sporadic breast cancer. BMC Cancer. 2010;10(1):23. doi: 10.1186/1471-2407-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajarathnam K, Sykes BD, Kay CM, Dewald B, Geiser T, Baggiolini M, et al. Neutrophil activation by monomeric interleukin-8. Science. 1994 Apr 1;264(5155):90–2. doi: 10.1126/science.8140420. [DOI] [PubMed] [Google Scholar]
- 15.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996 Aug 29;382(6594):829–33. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 16.Paavola CD, Hemmerich S, Grunberger D, Polsky I, Bloom A, Freedman R, et al. Monomeric Monocyte Chemoattractant Protein-1 (MCP-1) Binds and Activates the MCP-1 Receptor CCR2B. Journal of Biological Chemistry. 1998 Dec 11;273(50):33157–65. doi: 10.1074/jbc.273.50.33157. [DOI] [PubMed] [Google Scholar]
- 17.Smith JM, Johanesen PA, Wendt MK, Binion DG, Dwinell MB. CXCL12 activation of CXCR4 regulates mucosal host defense through stimulation of epithelial cell migration and promotion of intestinal barrier integrity. Am J Physiol Gastrointest Liver Physiol. 2005 Feb;288(2):G316–26. doi: 10.1152/ajpgi.00208.2004. [DOI] [PubMed] [Google Scholar]
- 18.Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC, 3rd, Basnet H, et al. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008 Sep 16;1(37):ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derdeyn CA, Costello C, Kilby JM, Sfakianos G, Saag MS, Kaslow R, et al. Correlation between Circulating Stromal Cell-Derived Factor 1 Levels and CD4+ Cell Count in Human Immunodeficiency Virus Type 1-Infected Individuals. AIDS Res Hum Retroviruses. 1999 Aug 10;15(12):1063–71. doi: 10.1089/088922299310359. 2015/02. [DOI] [PubMed] [Google Scholar]
- 20.Poznansky MC, Olszak IT, Foxall R, Evans RH, Luster AD, Scadden DT. Active movement of T cells away from a chemokine. Nat Med. 2000;6(5):543–8. doi: 10.1038/75022. print. [DOI] [PubMed] [Google Scholar]
- 21.Soriano A, Martínez C, García F, Plana M, Palou E, Lejeune M, et al. Plasma Stromal Cell–Derived Factor (SDF)-1 Levels, SDF1–3′A Genotype, and Expression of CXCR4 on T Lymphocytes: Their Impact on Resistance to Human Immunodeficiency Virus Type 1 Infection and Its Progression. Journal of Infectious Diseases. 2002 Oct 1;186(7):922–31. doi: 10.1086/343741. [DOI] [PubMed] [Google Scholar]
- 22.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular Mechanism of β-Arrestin-Biased Agonism at Seven-Transmembrane Receptors. Annu Rev Pharmacol Toxicol. 2012 Feb 10;52(1):179–97. doi: 10.1146/annurev.pharmtox.010909.105800. 2014/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zidar DA, Violin JD, Whalen EJ, Lefkowitz RJ. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proceedings of the National Academy of Sciences. 2009 Jun 16;106(24):9649–54. doi: 10.1073/pnas.0904361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crump MP, Gong J, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, et al. EMBO J. 23. Vol. 16. EMBO Press; 1997. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1; pp. 6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veldkamp CT, Peterson FC, Pelzek AJ, Volkman BF. The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci. 2005 Apr;14(4):1071–81. doi: 10.1110/ps.041219505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veldkamp CT, Ziarek JJ, Su J, Basnet H, Lennertz R, Weiner JJ, et al. Monomeric structure of the cardioprotective chemokine SDF-1/CXCL12. Protein Sci. 2009 Jul;18(7):1359–69. doi: 10.1002/pro.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drury LJ, Ziarek JJ, Gravel S, Veldkamp CT, Takekoshi T, Hwang ST, et al. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci U S A. 2011 Oct 25;108(43):17655–60. doi: 10.1073/pnas.1101133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science. 2009 May 22;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ying H, Kimmelman A, Lyssiotis C, Hua S, Chu G, Fletcher-Sananikone E, et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell. 2012 Apr 27;149(3):656–70. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013 Apr 04;496(7443):101–5. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng G, Zielonka J, McAllister D, Tsai S, Dwinell MB, Kalyanaraman B. Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation. Br J Cancer. 2014 Jul 01;111(1):85–93. doi: 10.1038/bjc.2014.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006 Jul 03;116(7):1776–83. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hardie DG, Ashford MLJ. Physiology. 2. Vol. 29. American Physiological Society; 2014. AMPK: Regulating Energy Balance at the Cellular and Whole Body Levels; pp. 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng G, Zielonka J, Dranka BP, McAllister D, Mackinnon AC, Joseph J, et al. Mitochondria-Targeted Drugs Synergize with 2-Deoxyglucose to Trigger Breast Cancer Cell Death. Cancer Research. 2012 May 15;72(10):2634–44. doi: 10.1158/0008-5472.CAN-11-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziarek JJ, Getschman AE, Butler SJ, Taleski D, Stephens B, Kufareva I, et al. Sulfopeptide Probes of the CXCR4/CXCL12 Interface Reveal Oligomer-Specific Contacts and Chemokine Allostery. ACS Chem Biol. 2013 Sep 20;8(9):1955–63. doi: 10.1021/cb400274z. 2015/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moyer RA, Wendt MK, Johanesen PA, Turner JR, Dwinell MB. Rho activation regulates CXCL12 chemokine stimulated actin rearrangement and restitution in model intestinal epithelia. Lab Invest. 2007 Aug;87(8):807–17. doi: 10.1038/labinvest.3700595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takekoshi T, Ziarek JJ, Volkman BF, Hwang ST. A Locked, Dimeric CXCL12 Variant Effectively Inhibits Pulmonary Metastasis of CXCR4-Expressing Melanoma Cells Due to Enhanced Serum Stability. Molecular Cancer Therapeutics. 2012 Nov 01;11(11):2516–25. doi: 10.1158/1535-7163.MCT-12-0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.SOMLYO AP, SOMLYO AV. Physiol Rev. 4. Vol. 83. American Physiological Society; 2003. Ca2+ Sensitivity of Smooth Muscle and Nonmuscle Myosin II: Modulated by G Proteins, Kinases, and Myosin Phosphatase; pp. 1325–58. [DOI] [PubMed] [Google Scholar]
- 39.Ratz PH. Inhibitor κB Kinase: Another Node in the Cell Signaling Network Regulating Smooth Muscle Contraction. Circulation Research. 2013 Aug 16;113(5):484–6. doi: 10.1161/CIRCRESAHA.113.301952. [DOI] [PubMed] [Google Scholar]
- 40.Wang S, Liang B, Viollet B, Zou M. Inhibition of the AMP-Activated Protein Kinase-a2 Accentuates Agonist-Induced Vascular Smooth Muscle Contraction and High Blood Pressure in Mice. Hypertension. 2011 May 01;57(5):1010–7. doi: 10.1161/HYPERTENSIONAHA.110.168906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banko M, Allen J, Schaffer B, Wilker E, Tsou P, White J, et al. Chemical Genetic Screen for AMPKα2 Substrates Uncovers a Network of Proteins Involved in Mitosis. Mol Cell. 2011 Dec 23;44(6):878–92. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zagorska A, Deak M, Campbell DG, Banerjee S, Hirano M, Aizawa S, et al. New Roles for the LKB1-NUAK Pathway in Controlling Myosin Phosphatase Complexes and Cell Adhesion. Sci Signal. 2010 Mar 30;3(115):ra25. doi: 10.1126/scisignal.2000616. [DOI] [PubMed] [Google Scholar]
- 43.Ramachandran C, Patil RV, Combrink K, Sharif NA, Srinivas SP. Rho-Rho kinase pathway in the actomyosin contraction and cell-matrix adhesion in immortalized human trabecular meshwork cells. Molecular Vision. 2011;17:1877–90. [PMC free article] [PubMed] [Google Scholar]
- 44.Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin Disrupts Crosstalk between G Protein–Coupled Receptor and Insulin Receptor Signaling Systems and Inhibits Pancreatic Cancer Growth. Cancer Research. 2009 Aug 15;69(16):6539–45. doi: 10.1158/0008-5472.CAN-09-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006 Mar 1;107(5):1761–7. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 46.Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proceedings of the National Academy of Sciences. 2013 Dec 10;110(50):20212–7. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma Y, Hwang RF, Logsdon CD, Ullrich SE. Dynamic Mast Cell–Stromal Cell Interactions Promote Growth of Pancreatic Cancer. Cancer Research. 2013 Jul 01;73(13):3927–37. doi: 10.1158/0008-5472.CAN-12-4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier J, Arenzana-Seisdedos F, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. 1996 Aug 29;382(6594):833–5. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 49.Simard E, Kovacs JJ, Miller WE, Kim J, Grandbois M, Lefkowitz RJ. β-Arrestin Regulation of Myosin Light Chain Phosphorylation Promotes AT1aR-mediated Cell Contraction and Migration. PLoS ONE. 2013 Nov 08;8(11):e80532. doi: 10.1371/journal.pone.0080532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ziarek JJ, Veldkamp CT, Zhang F, Murray NJ, Kartz GA, Liang X, et al. Heparin Oligosaccharides Inhibit Chemokine (CXC Motif) Ligand 12 (CXCL12) Cardioprotection by Binding Orthogonal to the Dimerization Interface, Promoting Oligomerization, and Competing with the Chemokine (CXC Motif) Receptor 4 (CXCR4) N Terminus. Journal of Biological Chemistry. 2013 Jan 04;288(1):737–46. doi: 10.1074/jbc.M112.394064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marchesi F, Monti P, Leone BE, Zerbi A, Vecchi A, Piemonti L, et al. Increased survival, proliferation, and migration in metastatic human pancreatic tumor cells expressing functional CXCR4. Cancer Res. 2004 Nov 15;64(22):8420–7. doi: 10.1158/0008-5472.CAN-04-1343. [DOI] [PubMed] [Google Scholar]
- 52.Singh AP, Arora S, Bhardwaj A, Srivastava SK, Kadakia MP, Wang B, et al. CXCL12/CXCR4 Protein Signaling Axis Induces Sonic Hedgehog Expression in Pancreatic Cancer Cells via Extracellular Regulated Kinase- and Akt Kinase-mediated Activation of Nuclear Factor κB: IMPLICATIONS FOR BIDIRECTIONAL TUMOR-STROMAL INTERACTIONS. Journal of Biological Chemistry. 2012 Nov 9;287(46):39115–24. doi: 10.1074/jbc.M112.409581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hendrix CW, Collier AC, Lederman MM, Schols D, Pollard RB, Brown S, et al. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr. 2004 Oct 1;37(2):1253–62. doi: 10.1097/01.qai.0000137371.80695.ef. [DOI] [PubMed] [Google Scholar]
- 54.Yonezawa S, Sato E. Expression of mucin antigens in human cancers and its relationship with malignancy potential. Pathol Int. 1997 Dec;47(12):813–30. doi: 10.1111/j.1440-1827.1997.tb03713.x. [DOI] [PubMed] [Google Scholar]
- 55.Hruban RH, Iacobuzio-Donahue C, Wilentz RE, Goggins M, Kern SE. Molecular pathology of pancreatic cancer. Cancer J. 2001 Jul-Aug;7(4):251–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.