

Abstract
In a closed endocrine loop, 1,25-dihydroxyvitamin D3 (1,25D) induces the expression of fibroblast growth factor-23 (FGF23) in bone, with the phosphaturic peptide in turn acting at kidney to feedback repress CYP27B1 and induce CYP24A1 to limit the levels of 1,25D. In 3T3-L1 differentiated adipocytes, 1,25D represses FGF23 and leptin expression, while not affecting leptin receptor transcription, but inducing C/EBP. Conversely, in UMR-106 osteoblast-like cells, FGF23 mRNA concentrations are upregulated by 1,25D, an effect that is blunted by lysophosphatidic acid, a cell-surface acting ligand. Progressive truncation of the mouse FGF23 proximal promoter linked in luciferase reporter constructs reveals a 1,25D-responsive region between −400 and −200 bp. A 0.6 kb fragment of the mouse FGF23 promoter, linked in a reporter construct, responds to 1,25D with a 4-fold enhancement of transcription in transfected K562 cells. Mutation of either an ETS1 site at −346 bp, or an adjacent candidate VDR/Nurr1-element, in the 0.6 kb reporter construct reduces the transcriptional activity elicited by 1,25D to a level that is not significantly different from a minimal promoter. This composite ETS1-VDR/Nurr1 cis-element may function as a switch between induction (osteocytes) and repression (adipocytes) of FGF23, depending on the cellular setting of transcription factors. Moreover, experiments demonstrate that a 1kb mouse FGF23 promoter-reporter construct, transfected into MC3T3 osteoblast-like cells, responds to high calcium challenge with a statistically significant 1.7–2.0-fold enhancement of transcription. Thus, the FGF23 proximal promoter harbors cis-elements that drive responsiveness to 1,25D and calcium, agents that induce FGF23 to curtail the pathologic consequences of their excess.
Keywords: Gene regulation, Hormone receptors, Adipose, Transcription factors, Vitamin D
INTRODUCTION
The vitamin D receptor (VDR) regulates transcription in response to its 1,25-dihydroxyvitamin D3 (1,25D) ligand by forming a heterodimer with one of the retinoid X receptors (RXRs) and binding to vitamin D responsive elements (VDREs) near or remote to each target gene (Haussler, et al. 2013). Fibroblast growth factor-23 (FGF23) is an osteoblast/osteocyte-elaborated, phosphaturic hormone that feedback represses CYP27B1 and induces CYP24A1 to limit the levels of active 1,25D (Haussler, et al. 2012; Quarles 2012). FGF23 expression is governed by a complex network of hormones, growth factors, and minerals, including 1,25D, PTH, leptin, cortisol, FGF2, iron, calcium, and phosphate (David, et al. 2013; Lanske & Razzaque 2014; Saini, et al. 2013). VDR-null mice possess vanishingly low circulating levels of FGF23 (Yu, et al. 2005), indicating that skeletal secretion of FGF23 is dependent upon the presence of VDR. The precise mechanism by which 1,25D, via the nuclear vitamin D receptor, regulates FGF23 gene transcription is yet to be defined. Kolek and colleagues (Kolek, et al. 2005) originally reported that in osteocyte-like UMR-106 cells, FGF23 mRNA levels are dramatically upregulated by 1,25D. This effect was reproduced, in vivo, with 1,25D-injected mice displaying increased FGF23 mRNA in tibia and calvaria, accompanied by a striking enhancement in circulating immunoreactive FGF23 protein (Kolek et al. 2005). Induction of FGF23 by 1,25D in UMR-106 cells is sensitive to inhibition by cycloheximide (Haussler, et al. 2010; Kolek et al. 2005), suggesting that the transcriptional effect may be secondary and dependent on the induction of an intermediary transcription factor. However, the time course of FGF23 mRNA upregulation by 1,25D in UMR-106 cells, with the first observable effect at 2 hours and the peak effect at 12 hours post 1,25D-treatment, is essentially identical to that of osteopontin mRNA induction by 1,25D (Saini, R. K., unpublished). This observation is puzzling, because the action of 1,25D on osteopontin transcription is insensitive to cycloheximide (Haussler et al. 2010), rendering it a definitive primary effect that occurs in temporal concert with the apparently more complex induction of FGF23.
Employing ROS 17/2.8 osteoblast-like cells, Liu and colleagues (Liu, et al. 2006) independently confirmed the findings of Kolek et al. (Kolek et al. 2005), reporting a substantial induction of FGF23 by 1,25D that was prevented by cotransfection with a dominant negative VDR plasmid (Jurutka, et al. 1997). They also identified a candidate VDRE in the form of a direct repeat with a spacer of 3 nucleotides (DR3) VDRE, AGGTTActgAGTTCC, located at −1124 bp in relation to the transcription start site in the mouse FGF23 gene (Liu et al. 2006). Importantly, site-directed mutagenesis of this VDRE in the context of a mouse FGF23 promoter-reporter construct compromised the ability of 1,25D to stimulate transcription (Liu et al. 2006). The results of Liu et al. (Liu et al. 2006) provide evidence that 1,25D induces FGF23 in a primary fashion, mediated by VDR binding to a VDRE near the proximal promoter of the mouse FGF23 gene. However, it has been reported that the FGF23 gene region in mouse osteocytes is not marked by detectable, 1,25D-dependent VDR/RXR binding sites when ChIP-seq analysis is carried out (St John, et al. 2014). Therefore, the in vivo relevance of VDREs residing in the mouse FGF23 gene remains to be confirmed.
Recently, functional VDREs have been identified in the human FGF23 gene at −35.7 kb (GGGAGAatgAGGGCA), at −16.2 kb (TAACCCtgctttAGTTCA, an everted repeat with a spacer of 6 nucleotides), and at +8.6 kb (AGGGCAggaAGGACA) in relation to the transcription start site (Saini et al. 2013). Notably, these human VDREs are located at some distance from the promoter, but each occurs in a cluster of binding sites for C/EBP and RUNX2 (Saini et al. 2013), indicating they may lie in cis-regulatory modules for control by the vitamin D hormone of osteoblast-expressed genes (Meyer, et al. 2010b). Based upon the observation that FGF23 induction by 1,25D is at least partially cycloheximide sensitive (Haussler et al. 2010), and the fact that 1,25D upregulates ETS1, a transcription factor that cooperates with VDR (Dittmer 2003), we (Saini et al. 2013) previously concluded that 1,25D induces human FGF23 production directly (primarily) via multiple VDREs, and indirectly (secondarily) via stimulation of ETS1 expression, with VDR and ETS1 cooperating in the induction of FGF23 through DNA looping and generation of euchromatin architecture (Saini et al. 2013).
The present communication reports that 1,25D represses FGF23 expression in adipocytes, contrasting with the induction of FGF23 by 1,25D in osteocytes. Thus, the directionality of FGF23 regulation by 1,25D is cell-selective. Evidence also is provided for an ETS1-VDRE/Nurr1 composite element that is conserved across species in the proximal promoter of the FGF23 gene. Combined with our previous report that 1,25D induces ETS1 (Saini et al. 2013), we conclude that this ETS1-VDRE/Nurr1 composite element may play a central role in a combined primary and secondary regulation of FGF23 mRNA by the vitamin D hormone.
MATERIALS and METHODS
Cell culture
Five mammalian cell lines (from the American Type Culture Collection) were employed in this study. Rat osteoblast/osteocyte-like (UMR-106) cells were cultured in DMEM/F12 (Hyclone) supplemented with 2.5 mM L-glutamine, and human embryonic kidney (HEK-293) cells were grown also in DMEM (Hyclone). Mouse embryonic pre-adipose (3T3-L1), preosteoblast (3T3-E1) and human K562 myelogenous leukemia cells were cultured with DMEM/high glucose (Hyclone). All cells were passaged in the above indicated media supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin (pen-strep) under a humidified atmosphere of 5% CO2 in air at 37°C. K562 cells were maintained as suspensions but received treatments identical to those of adherent cultures. Mouse 3T3-L1 cells were differentiated to adipocytes as follows: attached cells were incubated with Differentiation Medium A (DMEM/high glucose/10% FBS/Pen-Strep + 1 μg/mL insulin + 0.25 μM dexamethasone + 0.5 mM isobutylmethylxanthine) for three days, followed by four days (with medium change after the first two days) in Differentiation Medium B (DMEM/high glucose/10% FBS-pen/strep + 1 μg/mL insulin). On day 7, cells were treated with hormone or ethanol vehicle, followed by harvest on day 8.
Oil red O staining
3T3-L1 cells or adipocytes were washed twice with phosphate-buffered saline (PBS), fixed in 10% formalin for 10 min at RT, and washed again twice with PBS. Cells were then stained with a filtered oil red O solution (in 60% isopropanol) for 30 min at RT. Cells were washed twice with PBS and visualized (Fig. 1).
Figure 1. Oil Red O staining shows differentiation of MC3T3-L1 cells into adipocytes.
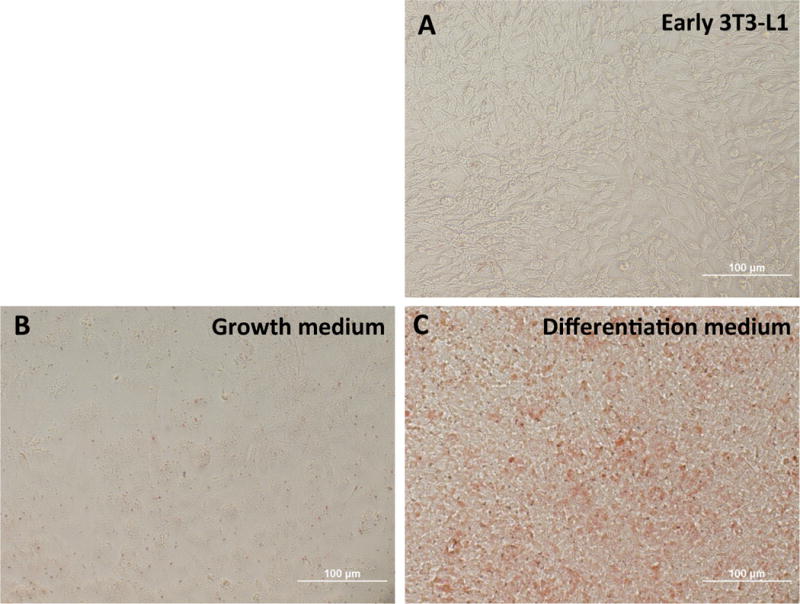
(A) Undifferentiated 3T3-L1 cells cultured in standard medium. (B) 3T3-L1 cells maintained in growth medium. (C) 3T3-L1 cells cultured in differentiation medium for seven days and stained with Oil Red O as described in Methods.
Plasmid constructs
The 1.0 kb mouse FGF23 promoter and truncations thereof (0.6 kb, 0.4 kb, 0.2 kb and 0.06 kb) in the reporter construct pGL3-Basic were a kind gift of Drs. Mikiko Ito and Ken-ichi Miyamoto, Tokushima University, Tokushima City, Japan (Ito, et al. 2005). Mutations of the VDRE and ETS-1 binding site in the 0.6 kb mouse FGF23 promoter construct were carried out using the following oligonucleotides to introduce the mutations: to mutate the ETS1 site, 5′-gtactgctggctgccttcacaCTAAATgatggaagtggggac-3′ (ETS1 site capitalized, mutated bases underlined); to mutate the VDRE/Nurr1, 5′cctgatggaAGTGGGgacTTGTCAacaaatgacccagg-3′ (ETS1 site capitalized, mutated bases underlined). To create single mutations, the indicated oligonucleotide and its complement were utilized along with the 0.6 kb mouse FGF23 reporter construct and a Quick Change II XL kit (Agilent Technologies, Santa Clara CA) according to the manufacturer’s instructions. To construct the double mutation, the MutETS1 plasmid was further mutagenized using the VDRE oligonucleotide pair described above. Successful mutation in each case was verified by DNA sequencing of the MutETS1, MutVDRE and double (2× Mut) plasmids.
Transfection and Dual Luciferase Reporter (DLR) assay
ExpressIN (Thermo Scientific, Lafayette CO) transfection reagent was utilized to transfect HEK-293 cell lines in 24-well plates (plated at 60,000 cells/well) according to the manufacturer’s protocol. Briefly, each well was transfected with 2.0 μl ExpressIN Reagent, 250 ng of the VDRE-containing pLUC-MCS plasmids to be tested, 25 ng of pSG5-hVDR (plasmid expressing human VDR), 20 ng of pRL-null (Renilla luciferase reporter) and 1 μl of 100X sodium pyruvate. Transfection of UMR-106 cells was similar; however, FuGene HD Transfection Reagent (Roche Applied Science) was used in this cell line. Transfection of K562 cells in suspension was performed by plating 2 × 105 cells per well in 24 well plates, then using PolyJet reagent (SignaGen Laboratories, Gaithersburg MD) according to the manufacturer’s instructions. Briefly, each well received 2.0 μl PolyJet reagent, 500 ng of the mouse FGF23 promoter constructs to be tested, 100 ng of pSG5-hVDR (plasmid expressing human VDR) and 20 ng of pRL-null (Renilla luciferase reporter). After transfection, each well was treated with either 1,25D (final concentrations of 1, 10 or 100 nM) or ethanol vehicle for 20 hours at 37°C. Phosphate or calcium-treated cells were treated with the appropriate dilution of 1 M sodium phosphate or 4.2 M CaCl2 in water. After incubation, cells were washed with PBS and lysed in 1X passive lysis buffer (Promega). Lysates were harvested and analyzed sequentially for Firefly luciferase and Renilla luciferase activity using a Dual Luciferase assay kit (Promega) and a Sirius Luminometer (Zylux Corp.) according to the manufacturers’ protocols.
Quantitative real-time PCR
HEK-293 cells were plated at 1 × 106 cells in 60 mm dish. UMR-106 cells were plated at 5 × 105 cells/well in a 6-well plate, allowed to attach overnight, then treated with lysophosphatidic acid (LPA) and analogues, 1,25D or ethanol vehicle in media containing 1% charcoal/dextran stripped fetal bovine serum for 24 h. LPA and Ki16425 were obtained from Cayman Chemical Co. (Ann Arbor, MI), OMPT was obtained from Avanti Polar Lipids (Alabaster, AL), dissolved in ethanol, and used as described in the figure legends.
Cells were harvested and total cellular RNA was isolated utilizing an Aurum Total RNA Mini kit (Bio-Rad, Hercules, CA, USA). The RNA obtained was quantified using A260/280 spectrophotometry. DNase-treated RNA (1 μg) was reverse transcribed using the iScript cDNA Synthesis kit (Bio-Rad). The obtained cDNA was used in 20 μl PCR reactions containing 10 μl FastStart Universal SYBR green Master (Roche) and primers. Reactions were performed in 96 well PCR plates on an ABI 7500 Fast instrument. Data were analyzed using the comparative Ct method as means of relative quantitation, normalized to an endogenous reference (GAPDH) and relative to a calibrator (normalized Ct value from vehicle-treated cells) and expressed as 2−ΔΔCt according to Applied Biosystems’ User Bulletin 2: Rev B, “Relative Quantitation of Gene Expression.” Primer sets for real-time PCR were as follows:
Rat FGF23 (forward 5′-ACGGAACACCCCATCAGACTATC-3′, reverse 5′-TATCACTACGGAGCCAGCATCCTC-3′);
Rat CYP24A1 (forward 5′-GATCACCTTTCCAAGAAGGAACT-3′, reverse 5′-AGAGAATCCACATCAAGCTGTTC-3′);
Rat GAPDH (forward 5′-AGGTCGGTGTGAACGGATTTG-3′, reverse 5′-CATTCTCAGCCTTGACTGTGCC-3′);
Human GAPDH (forward 5′-ACAACTTTGGTATCGTGGAAGGAC-3′, reverse 5′-CAGGGATGATGTTCTGGAGAG-3′); or (forward 5′-TGACAACTTTGGTATCGTGGAAGG-3′, reverse 5′-AGGGATGATGTTCTGGAGAGCC-3′);
Mouse GAPDH (forward 5′-TTCCGTGTTCCTACCCCCAATG-3′, reverse 5′-TGCCTGCTTCACCACCTTCTT-3′);
Human CYP24A1 (forward 5′-CAGCGAACTGAACAAATGGTCG-3′, reverse 5′-TCTCTTCTCATACAACACGAGGCAG-3′);
Mouse Cyp24a1 (forward 5′-CGTTCTGGGTGAATACACGCTAC-3′, reverse 5′-TTCGGGTCTAAACTTGTCAGCATC-3′);
Mouse Leptin (forward 5′-GTGCCTATCCAGAAAGTCCAG-3′, reverse 5′-TGAAGCCCAGGAATGAAGTC-3′);
Mouse Leptin Receptor (ObR) (forward 5′-GCATGCAGAATCAGTGATATTTGG-3′, reverse 5′-CAAGCTGTATCGACACTGATTTCTTC-3′);
Human FGF23 (forward 5′-TGCTGGCTTTGTGGTGATTA-3′; reverse 5′-TTCTCCGGGTCGAAATAGTG-3′)
Mouse FGF23 forward 5′-GGTGATAACAGGAGCCATGAC-3′
Reverse 5′-TGCTTCTGCGACAAGTAGAC-3′
Mouse C/EBPβ forward 5′-AGCGGCTGCAGAAGAAGGT-3′,
Reverse 5′-GGCAGCTGCTTGAACAAGTTC-3′;
Mouse PPARγ forward 5′-CGCTGATGCACTGCCTATGA-3′;
Reverse 5′-AGAGGTCCACAGAGCTGATTCC-3′;
Mouse resistin forward 5′-TCAACTCCCTGTTTCCAAATGC-3′
Reverse 5′-TCTTCACGAATGTCCCACGA-3′
Mouse aP2 forward 5′-CATGGCCAAGCCCAACAT-3′;
Reverse 5′-CGCCCAGTTTGAAGGAAATC-3′
Statistical analysis
Data are expressed as means ± SD. Statistical differences between two groups were determined by a two-sided Student’s t test. Differences among multiple groups were analyzed by ANOVA. A p value of less 0.05 was considered significant.
RESULTS
1,25D represses the expression of FGF23 in 3T3-L1 differentiated adipocytes
Previously, only upregulation of FGF23 mRNA expression by 1,25D has been reported in mammalian cells (Haussler et al. 2012). However, as illustrated in Figure 2, in differentiated 3T3-L1 mouse adipocytes, FGF23 is significantly and dose-dependently repressed by 1,25D. When undifferentiated 3T3-L1cells are transferred from growth medium (GM) to differentiation medium (DM), FGF23 expression is reduced by 50%, followed by a further suppression of FGF23 mRNA by 1,25D that is detectable at a hormone concentration as low as 0.1 nM, but first becomes statistically significant at 1.0 nM 1,25D (Fig. 2). The inset to Fig. 2 depicts the average of three independent experiments like that illustrated in the main body of the figure, verifying the 1,25D dose-dependency of FGF23 suppression in differentiated 3T3-L1 adipocytes.
Figure 2. FGF23 mRNA repression by 1,25D in 3T3-L1 adipocytes.
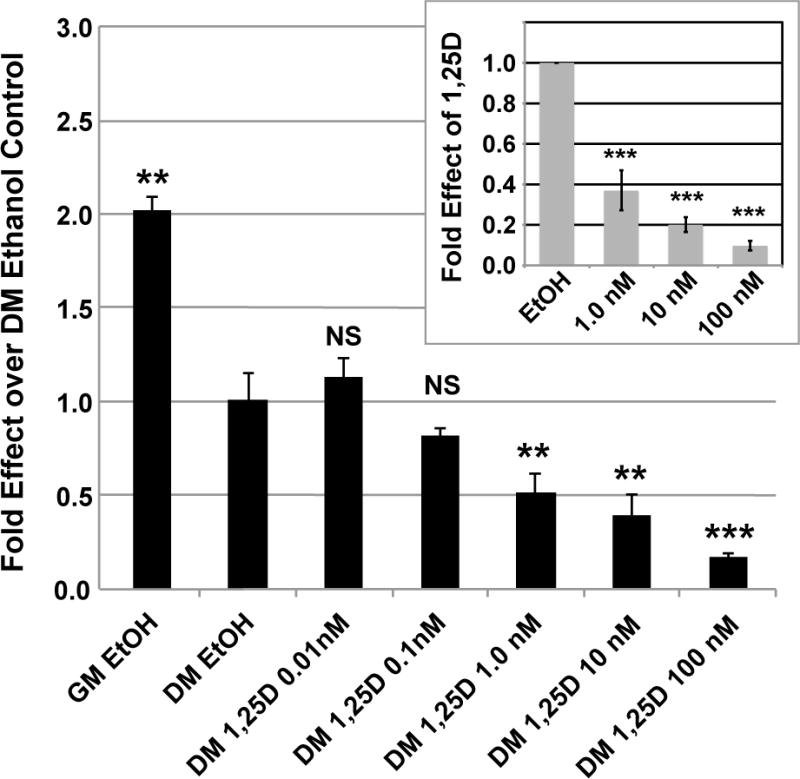
3T3-L1 cells were grown as described in Methods and treated with 1,25D for 24 hours at the indicated concentrations. mRNA levels for FGF23 were determined by real-time PCR. GM is growth medium and DM is differentiation medium. Inset depicts the average of three independent experiments identical to that illustrated in the main body of the figure, except that only doses of 1, 10 and 100 nM 1,25D were tested. Each bar represents the average of three independent experiments performed in triplicate ± standard deviation. All statistics are calculated relative to the DM EtOH set at 1.0. ** p < 0.01, *** p < 0.001
Vitamin D hormone control of other adipocyte-expressed genes
To determine if the repression of FGF23 by 1,25D in adipocytes is unique, the effect of the hormone on other adipocyte-expressed genes was investigated. Initially, for reference, after 3T3-L1 are transferred from growth medium to differentiation medium, leptin is induced 6-fold, leptin receptor is repressed 20-fold, and Cyp24a1 is relatively unchanged (data not shown). As shown in Fig. 3A and Fig. 3C, leptin expression is downregulated by 1,25D in 3T3-L1 differentiated adipocytes. This effect is statistically significant at 100 nM 1,25D, but because of the high variability of the data, it is difficult to define dose responsiveness. Nevertheless, repression of leptin is evident, and is unusually rapid in that maximal repression occurs within only 2 hours of hormone treatment (data not shown). In contrast, leptin receptor mRNA is unaffected by treatment of 3T3-L1 differentiated adipocytes with 1,25D (Fig. 3B). CYP24A1 was employed as a highly-induced positive control for VDR-mediated 1,25D action to ensure that the tested cells expressed VDR appropriately and responded to the 1,25D ligand in the expected fashion. Indeed, Fig. 3D illustrates a dramatic, dose-dependent enhancement of CYP24A1 mRNA concentrations elicited by the vitamin D hormone (nearly 500-fold at the maximal dose of 100 nM), indicating that the differentiated 3T3-L1 mouse adipocyte system represents a valid model for probing VDR-targeted gene expression. Furthermore, as summarized in Fig. 4, among other genes expressed in 3T3-L1 differentiated adipocytes, 1,25D induces C/EBPβ, whereas PPARγ, resistin and aP2 are all regulated biphasically, with induction by 1 nM 1,25D and dose-dependent repression by 10–100 nM hormone. Thus, among the genes regulated by 1,25D in adipocytes, FGF23 stands out as unique with respect to its unequivocal repression by 1,25D in a classic dose-dependent fashion. To assess the physiologic significance of FGF23 repression by 1,25D in adipose, it will ultimately be necessary to monitor FGF23 in intact mouse and human fat tissue in order to investigate possible in vivo target pathways affecting metabolism.
Figure 3. 1,25D represses leptin mRNA while inducing Cyp24a1 in 3T3-L1 adipocytes.
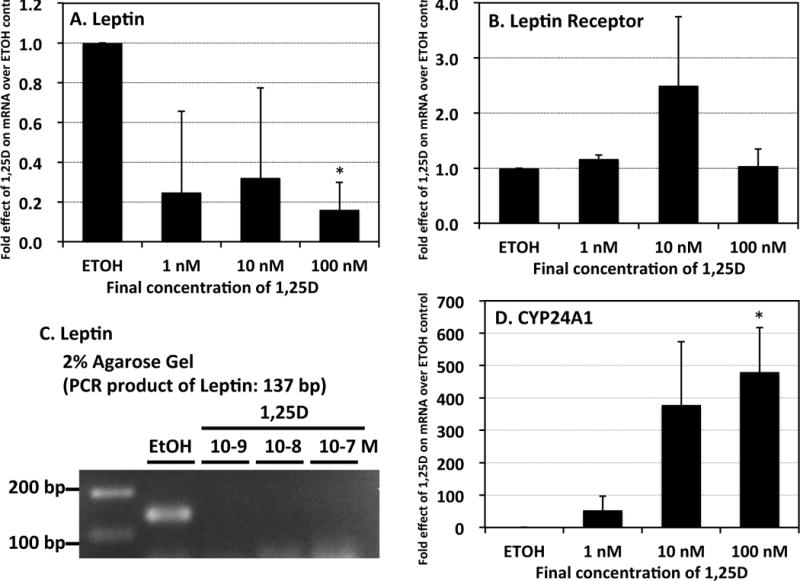
Differentiated 3T3-L1 adipocytes were treated with 1,25D for 24 hours at the indicated concentrations, and mRNA levels for the indicated gene products were determined by real-time PCR (A, B and D) or by gel electrophoresis of PCR products after 40 cycles (panel C). CYP24A1 is a positive control known to be upregulated by 1,25D. Statistical significance is depicted in the body of each panel. Each bar represents the average of three independent experiments performed in triplicate ± standard deviation. *1,25D-treated groups statistically significantly different from ETOH control (p<0.05).
Figure 4. 1,25D induces C/EBPβ mRNA and biphasically regulates PPARγ, resistin and aP2 in 3T3-L1 adipocytes.
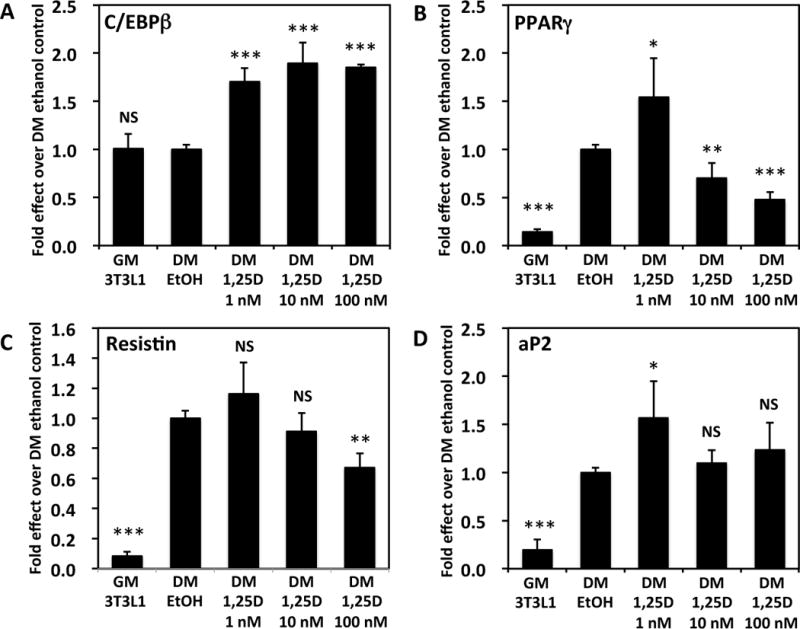
3T3-L1 cells were grown as described in Methods and the legend to Fig. 2, then treated with 1,25D for 24 hours at the indicated concentrations, and mRNA levels determined by real-time PCR for the following genes: (A) C/EBPβ, (B) PPARγ, (C) resistin, and (D) aP2. Each bar represents the average of three independent experiments performed in triplicate ± standard deviation. All statistics are calculated relative to the DM EtOH set at 1.0. * p < 0.05, ** p < 0.01, *** p < 0.001
1,25D induces the expression of FGF23 in UMR-106 osteoblasts; modulation by LPA
To contrast FGF23 regulation in adipocytes vs. in its skeletal endocrine source, UMR-106 osteoblast/osteocyte-like cells were treated with 1,25D, as well as with lysophosphatidic acid (LPA), a known potentiator of 1,25D action to induce osteoblast maturation and alkaline phosphatase expression (Gidley, et al. 2006; Mansell & Blackburn 2013). As illustrated in Fig. 5A, 1,25D dramatically (1920-fold) induces FGF23 in UMR-106 cells, as previously observed in our laboratory and by others (Bergwitz & Juppner 2010; Kolek et al. 2005; Liu et al. 2006). Curiously, LPA and its OMPT agonist blunt the induction of FGF23, whereas the LPA antagonist, Ki16425, slightly amplifies the upregulation of FGF23 by 1,25D (Fig. 5A). A very similar profile of modulation by LPA agonists/antagonists emerges (Fig. 5B) with respect to induction of the positive control gene, CYP24A1, by 1,25D in UMR-106 cells. In contrast, when HEK-293 human embryonic kidney cells are similarly probed for 1,25D control of FGF23 gene expression and its modulation by LPA (Fig. 6A), the results differ markedly from those obtained in osteoblasts. Specifically, FGF23 expression is not affected by 1,25D in HEK-293 cells (Fig. 6A), whereas LPA agonists enhance the action of 1,25D to induce CYP24A1 positive control mRNA (Fig. 6B). The LPA antagonist Ki16425 had no significant effect on 1,25D action in these experiments. Thus, LPA acts more traditionally as a 1,25D-potentiator of CYP24A1 induction in HEK-293 cells as opposed to its reverse effect in UMR-106 osteoblasts. This finding indicates that not only is FGF23 regulation by 1,25D cell-selective in terms of its directionality, but that modulation of this effect by LPA agonists/antagonists differs according to the cell setting.
Figure 5. Induction of FGF23 and Cyp24a1 mRNAs by 1,25D in UMR-106 rat osteoblast/osteocyte-like cells and its modulation by LPA.
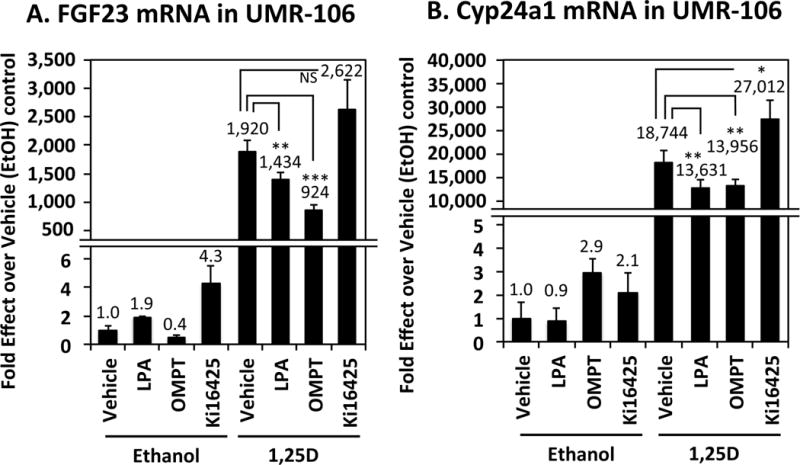
UMR-106 cells were treated for 24 hours with 20 μM lysophosphatidic acid (LPA), 10 μM OMPT (an LPA agonist), 10 μM Ki16425 (an LPA antagonist), 10 nM 1,25D, or combinations thereof as indicated. mRNA levels were determined by real-time PCR for the following genes: (A) FGF23 and (B) Cyp24a1. Each bar represents the average of three independent experiments performed in triplicate ± standard deviation. All statistics are calculated relative to the EtOH control, set at 1.0. * p < 0.05, ** p < 0.01, *** p < 0.001
Figure 6. Control of FGF23 and CYP24A1 mRNAs by 1,25D in HEK-293 embryonic human kidney cells and its modulation by LPA agonists/antagonists.
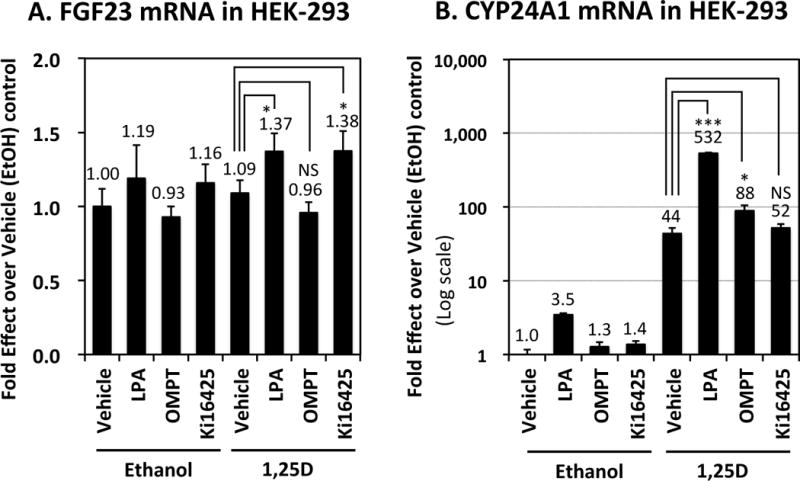
HEK-293 cells were treated for 24 hours with 20 μM lysophosphatidic acid (LPA), 10 μM OMPT (an LPA agonist), 10 μM Ki16425 (an LPA antagonist), 10 nM 1,25D, or combinations thereof as indicated. mRNA levels were determined by real-time PCR for the following genes: (A) FGF23 and (B) Cyp24a1. Each bar represents the average of three independent experiments performed in triplicate ± standard deviation. All statistics are calculated relative to the EtOH control, set at 1.0. * p < 0.05, *** p < 0.001
Dissection of the mouse FGF23 promoter by informatics, truncation and mutagenesis
We have re-examined the mouse FGF23 proximal promoter (DNA sequence listed in Fig. 7) for transactivator binding sites that might mediate control of FGF23 mRNA expression. As depicted in Fig. 7, we observed a collection of conserved, consensus cis-elements between −110 and −347 bp in relation to the start of transcription. Most prominent in this collection are three ETS1, two Nurr1 (bold type), two GATA, and single AP1 and LEF-1 sites. It is noteworthy that AP1 and LEF-1 cis elements are often associated with VDREs in 1,25D-regulated genes (Luderer, et al. 2011; Ozono, et al. 1990). Meir et al. (Meir, et al. 2014), recently provided evidence that PTH induces FGF23 in osteoblasts via activation of Nurr1, a transcription factor involved in the development of midbrain dopaminergic neurons (Sakurada, et al. 1999). Strikingly, one of the conserved Nurr1 sites in the FGF23 promoter (Meir et al. 2014), exactly overlaps the 3′ putative half element in the proposed new VDRE (Fig. 7). Equally important, ETS1, which was implicated as a partner with VDR in controlling FGF23 in a previous study (Saini et al. 2013), possesses a cis-docking site in the mouse FGF23 promoter that is only six bp 5′ of a newly discovered candidate VDRE/Nurr1 (AGTGGGgacAGGTCA) site at −334 bp, highlighted in light purple in Fig. 7. This novel VDRE is distinct from the VDRE reported by Liu et al. (Liu et al. 2006) at −1124 bp that is also highlighted (dark gray) in Fig. 7. We hypothesized that the −110 to −346 bp region of the FGF23 promoter constitutes a proximal cis regulatory module anchored by the adjacent ETS1 and VDRE sites. To test this hypothesis, we dissected the mouse FGF23 promoter kindly supplied by Drs. M. Ito and K. Miyamoto of the Tokushima University (Ito et al. 2005). By employing progressively truncated promoter-reporter constructs, we obtained evidence that a VDRE is located somewhere between −200 and −400 bp in the mouse FGF23 promoter (Fig. 8A and B). The activity of the FGF23 promoter fragments was unaffected by phosphate concentration (Fig. 8A vs 8B), suggesting that phosphate-responsive elements must reside outside of the 1.0 kb promoter. To determine if the VDRE localized to the −200 to −400 interval corresponds to the newly revealed candidate VDRE shown in Fig. 7, this VDRE/Nurr1 and/or its adjacent ETS1 site were inactivated by point mutation within the context of a −0.6 kb promoter fragment-luciferase construct, and the mutated plasmids evaluated for 1,25D responsiveness. As illustrated in Fig. 8C, the −0.6 kb promoter fragment-luciferase construct yields a 4-fold response to 1,25D. This effect is significantly diminished to 2.2 to 2.4-fold by mutation of either the VDRE/Nurr1 or ETS1 site, or both simultaneously, with all responses by the mutated promoters not significantly different from the minimal promoter (−0.06 kb) control construct. The observation that inactivation by site-directed mutagenesis of either the VDRE/Nurr1 or ETS1 elements abolishes transcriptional stimulation by 1,25D (Fig. 8C) provides evidence for a novel, composite ETS1-VDRE/Nurr1 cis-element that may be central to the CRM in the mouse FGF23 promoter that mediates at least part of the response of this gene to induction by 1,25D. Finally, we probed the responsiveness of the FGF23 promoter to calcium, as David et al. (David et al. 2013) recently called attention to calcium as a stimulator of FGF23 secretion by osteoblasts. Indeed, as illustrated in Fig. 8D, high calcium significantly (1.7 to 2.0-fold) induces the −1.0 kb mouse FGF23 promoter-reporter construct, when it is transfected into MC3T3-E1 osteoblast-like cells. Therefore, as documented by data in Figs. 8C and D, we conclude that 1,25D and calcium comprise FGF23 inducers that function by stimulating the proximal promoter of the mouse gene and likely other species, as the CRM components of this promoter are largely conserved across mammalian species.
Figure 7.
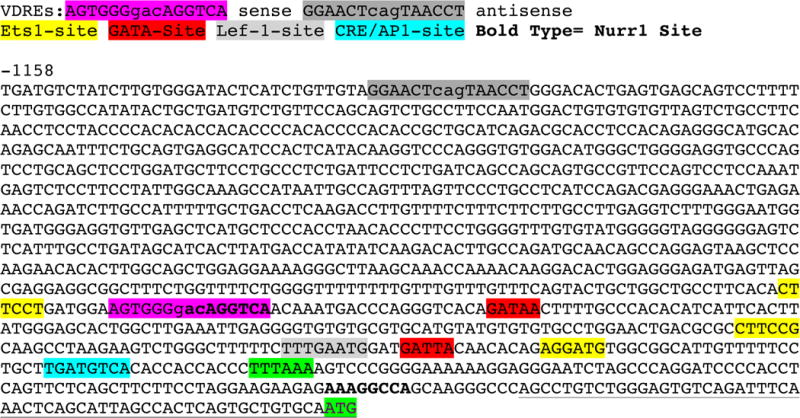
Nucleotide sequence of the mouse FGF23 promoter. The color-coded legend for cis elements is depicted above the sequence. Nurr1 sites are depicted in bold type. The transcription start site begins at the 5′ end of the underlined sequence. The TATA-box and the ATG start codon for translation are highlighted in light green.
Figure 8.
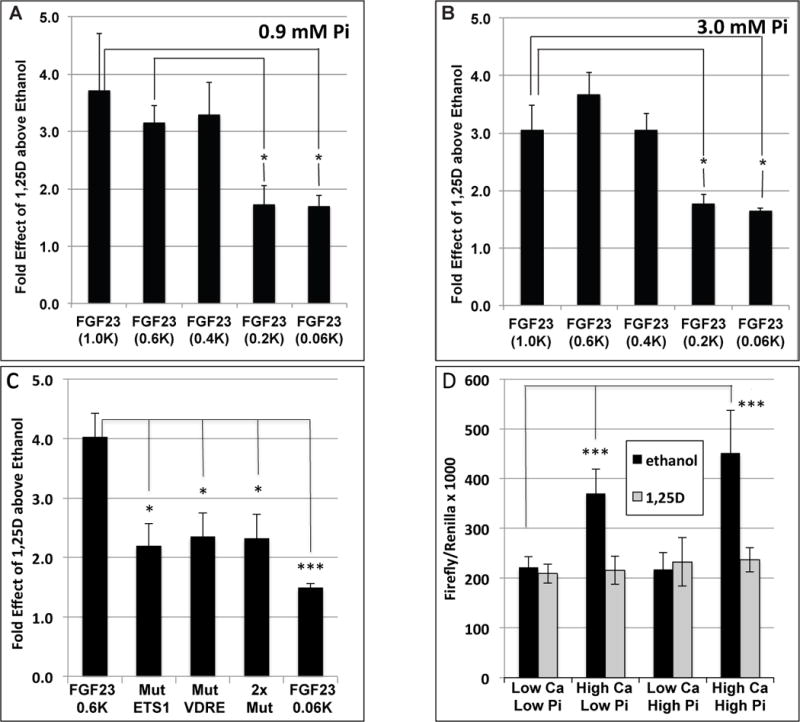
Dissection of the mouse FGF23 proximal promoter. (A) A promoter fragment containing 1.0 kb of promoter sequence (1.0K) was linked to a luciferase reporter and transfected into K562 cells. Transcription of the reporter gene was stimulated 3 to 4-fold by 1,25D in the presence of 0.9 mM (low) phosphate. This transcriptional effect is significantly reduced to less than 2-fold when progressively 5′-truncated promoter fragments (e.g. 0.4K or 0.2K) are tested. (B) Profile of transcriptional stimulation by 1,25D of FGF23 promoter fragments, transfected into K562 cells, is essentially unaffected by 3.0 mM (high) phosphate concentration when compared to the 0.9 mM phosphate results. (C) When transfected into K562 cells, a −0.6 kb promoter fragment-luciferase construct of the mouse FGF23 promoter yields a 4-fold response to 1,25D. This effect is significantly diminished to 2.2 to 2.4-fold by mutation of either the VDRE or ETS1 site, or both simultaneously, with all responses not significantly different from the minimal promoter (−0.06 kb) control construct. (D) High (6.0 mM) calcium, but not high (3.0 mM) phosphate, significantly (1.7 to 2.0-fold) induces the −1.0 kb mouse FGF23 promoter-reporter construct, when it is transfected into MC3T3-E1 osteoblast-like cells.
DISCUSSION
Like PTH, FGF23 inhibits renal Npt2a and Npt2c to elicit phosphaturia (Shimada, et al. 2004a). In contrast to PTH that is downregulated by 1,25D in parathyroid glands, FGF23 is upregulated by 1,25D in osteocytes (Bergwitz & Juppner 2010; Kolek et al. 2005; Liu et al. 2006), which are a major source of endocrine FGF23 production by bone. Hyperphosphatemia enhances osteocytic FGF23 production independently of 1,25D, rendering FGF23 the ideal phosphaturic counter-1,25D hormone because it inhibits renal phosphate reabsorption, and 1,25D biosynthesis via repression of CYP27B1, while enhancing 1,25D degradation by inducing CYP24A1 in all tissues (Shimada et al. 2004a). In this fashion, FGF23 allows osteocytes to communicate with the kidney to govern circulating 1,25D as well as phosphate levels, thereby preventing excess 1,25D function and hyperphosphatemia, either of which lead to ectopic calcification.
In the past year, new actions of FGF23 have been revealed via generation of FGF23-null mice. For example, FGF23 deficiency leads to mixed hearing loss and middle ear malformation in mice (Lysaght, et al. 2014). FGF23 also regulates renal sodium handling and blood pressure (Andrukhova, et al. 2014a), as well as cardiac structure and function (Agarwal, et al. 2014). Additionally, FGF23 is a negative regulator of prenatal and postnatal erythropoiesis (Coe, et al. 2014), and is a stimulator of renal calcium reabsorption through the TRPV5 channel (Andrukhova, et al. 2014b). Many, but not all, of these novel actions of FGF23 involve the kidney and the vascular system. The function of FGF23 in adipose is unknown, but in the present communication we report a surprising repression of FGF23 expression by 1,25D in adipocytes, contrasting with all previous reports of FGF23 induction by 1,25D in other cell types. The mechanism of this repression is also unknown, and is apparently unique for 1,25D control of adipose-expressed genes, differing in direction and 1,25D dose response when compared with regulation of genes such as C/EBPβ, PPARγ, resistin, aP2, the leptin receptor, and Cyp24a1. The only similarity between the effect of 1,25D on FGF23 and another gene is that of leptin, a gene product that itself induces FGF23. Thus, the influence of 1,25D to repress adipose FGF23 may be related to curbing some deleterious off-target effect of FGF23 in the adipocyte, such as adipose growth (Oldknow, et al. 2015) or malignancy (Jacobs, et al. 2011). With respect to obesity, FGF23 has been implicated in the regulation of energy metabolism, and mouse models with elevated FGF23 display increased adiposity (Oldknow et al. 2015), suggesting that 1,25D attenuation of FGF23 may curtail obesity. We (Kolek et al. 2005) originally reported that, in osteocyte-like UMR-106 cells, FGF23 mRNA levels are dramatically upregulated by 1,25D. We later determined that this FGF23 induction is potentiated by leptin, and inhibited by IL-6 (Saini et al. 2013), further linking FGF23 homeostasis with adipose hormones and cytokines, possibly implicating FGF23 repression by 1,25D in the modulation of energy metabolism and the prevention of malignancy.
In the current study, we re-examined the mouse FGF23 proximal promoter for transactivator binding sites that might mediate control of FGF23 mRNA expression. Notably, ETS1, which was implicated as a partner with VDR in controlling FGF23 in a previous study (Saini et al. 2013), possesses a cis-docking site in the mouse FGF23 promoter that is only six bp 5′ of a newly discovered candidate VDRE/Nurr1 site (AGTGGGgacAGGTCA), highlighted in light purple in Fig. 7. By employing progressively truncated promoter-reporter constructs, we deduced that a 1,25D-responsive region must be present between −200 and −400 bp in the mouse FGF23 promoter (Fig. 8A and B). When two elements within this interval, namely the candidate VDRE/Nurr1 site at −334 bp and/or its adjacent ETS1 site at −346 bp, were inactivated by point mutation, transcriptional activation was diminished to a level not significantly different from that of the minimal promoter (−0.06 kb) control construct (Fig. 8C). Therefore, it was concluded that the −200 to −399 bp region of the mouse FGF23 promoter contains a cis regulatory module (CRM) anchored by the adjacent ETS1 and VDRE/Nurr1 sites.
With respect to 1,25D induction of murine FGF23, there are other considerations for the −399 bp region of the FGF23 promoter (Fig. 7). In addition to the ETS1-VDRE/Nurr1 composite element, there exist two additional ETS1 sites that are conserved across species, as well as two conserved GATA cis-elements of unknown significance (Barthel et al. 2007). The ETS1 protein is known to complex with VDR (Dittmer 2003), and to confer ligand- and AF-2-independent transcriptional activation properties on nuclear receptors such as VDR (Tolon, et al. 2000). The multiple conserved ETS1 cis-elements in the FGF23 gene region (Saini et al. 2013) therefore render this transcription factor a likely candidate for the recruitment of VDR for the purpose of regulating FGF23 gene expression. Finally, the ETS1 protein is induced by 1,25D to amplify the response (Saini et al. 2013), and this process would be inhibited by cycloheximide, perhaps accounting for the reported cycloheximide sensitivity of the induction of FGF23 by 1,25D in UMR-106 osteocyte-like cells.
Equally interesting is the existence of a Lef-1 site in the FGF23 promoter (Fig. 7), a cis-element that is often associated with VDREs in 1,25D-regulated genes (Luderer et al. 2011). This Lef-1 site is yet another target for 1,25D, possibly secondary to the repression of sclerostin (St John et al. 2014) a Wnt signaling inhibitor that is crucial in bone remodeling. Moreover, PTH increases FGF23 mRNA levels, and this effect is mimicked by the PKA activator, forskolin. PTH also decreases SOST mRNA encoding sclerostin, which is a PTH receptor (PTH1R) target; this action of PTH was abrogated by added sclerostin. Therefore, PTH increases FGF23 through the PKA and Wnt pathways (Lavi-Moshayoff, et al. 2010). Thus, by analogy with PTH action on FGF23 expression, we suspect that the response of FGF23 expression to 1,25D/VDR may not be a direct one, primarily because VDR has never been observed to associate with putative VDREs in the FGF23 gene region as monitored by ChIP-seq analysis (St John et al. 2014).
Furthermore, control of FGF23 transcription by PTH has recently been discovered to be dependent on the induction of Nurr1 by PTH/PKA signaling (Meir et al. 2014). Conserved Nurr1 elements have been reported (Meir et al. 2014) in the FGF23 proximal promoter and immediately preceding the start site for transcription as highlighted in bold type in Fig. 7. Although there is a conserved CREB/AP-1 site just 11 bp upstream of the TATA-box in the rodent FGF23 genes (see Fig. 7), Meir and colleagues (Meir et al. 2014) demonstrate that the effect of PTH via PKA/cAMP is apparently through induction of Nurr1, although it remains possible that the Nurr1 site just 3′ of the TATA-box mediates cooperation between Nurr1 and CREB to activate transcription. However, Meir and colleagues did not point mutate either of the Nurr1 sites in their study to demonstrate conclusively that one or both Nurr1 elements participated in FGF23 regulation by PTH (Lanske & Razzaque 2014). In the present experiments, our mutagenesis of the candidate VDRE at −334 bp in the mouse FGF23 promoter also would inactivate the Nurr1 site. This maneuver eliminated the response of the promoter to 1,25D (Fig. 7C), but one explanation of this result is that 1,25D/VDR is functioning through Nurr1 as a secondary transcriptional activation factor as does PTH. We favor the mechanism that 1,25D induces FGF23 via primary activation of Nurr1 expression rather than direct binding of VDR to the FGF23 promoter. Nurr1 then would bind to its cognate element adjacent to the ETS1 element and likely cooperate with ETS1 to induce FGF23. Evidence for this cooperation includes elimination of the 1,25D effect on FGF23 induction by point inactivation of the ETS1 site in question (Fig. 7C). Nurr1 has, in fact, been shown to interact with ETS1 in controlling the MMP-1 promoter in cartilage, but in this case the relationship between the two transcription factors is an antagonistic one that does not require a Nurr1 consensus element (Mix, et al. 2007). We do not know if the more proximal Nurr1 site 3′ of the TATA-box is relevant to the (secondary) action of 1,25D, but point out that when cells are treated with 1,25D, there persists a significant 1.5 to 2.0-fold increase in reporter expression in −0.06 kb truncated promoter-reporter constructs (Fig. 7A, B, C) that retain the proximal Nurr1 site. This residual effect may indicate that the proximal Nurr1 site is indeed active. But, more importantly, this new model of 1,25D action is consistent with evidence currently published on 1,25D induction of FGF23 as follows: 1) the secondary mechanism accounts for the cycloheximide sensitivity of the process (Haussler et al. 2010), 2) invoking Nurr1 is in concert with negative ChIP-seq results for 1,25D-dependent association of VDR-RXR with putative VDREs in the FGF23 gene region in osteocytes (St John et al. 2014), 3) the secondary mechanism invoking Nurr1 is entirely consistent with a diminution of the 1,25D-effect on the promoter when a Nurr1 site is altered by mutagenesis, and 4) Nurr1 expression is reduced in rat brain by developmental vitamin D-deficiency (Kesby, et al. 2013). Moreover, the requirement for a cAMP increase to induce Nurr1 and, secondarily, FGF23 in the case of PTH induction of FGF23, is likely pertinent to the control of FGF23 by 1,25D. Notably, we have demonstrated in the present results (Fig. 5A) that LPA, a known activator of Gi to suppress cAMP (Mansell & Blackburn 2013), attenuates the ability of 1,25D to induce FGF23 in UMR-106 cells. Thus, we conclude that Nurr1, in combination with upstream PKA signaling, may be the new key to understanding FGF23 regulation by the calcemic hormones PTH and 1,25D. Indeed, one of the presentations of McCune Albright Syndrome, in which there is constitutive activation of Gs, is hypophosphatemia and FGF23 excess (Dumitrescu & Collins 2008). Vitamin D has been associated with the development of the dopaminergic nervous system (Harms, et al. 2011), a phenomenon dependent on Nurr1 (Sakurada et al. 1999). Therefore, another connection between Nurr1 and 1,25D action could reside in the central nervous system. Thus, a more complete characterization of the relationship between 1,25D and FGF23 could potentially open new vistas far beyond the treatment of bone mineral disorders to include not only energy metabolism and malignancy, but also the prevention of neuropsychiatric disease.
Acknowledgments
The authors are grateful to Drs. M. Ito and K. Miyamoto of the Tokushima University for the kind gift of mouse FGF23 promoter-reporter constructs.
FUNDING: This study was supported by National Institutes of Health grants NIH DK033351 to M.R.H and CA140285 to P.W.J.
Footnotes
AUTHOR CONTRIBUTIONS
Conception and design: Ichiro Kaneko, Rimpi K. Saini, G. Kerr Whitfield, Peter W. Jurutka, and Mark R. Haussler
Development of methodology: Peter W. Jurutka, G. Kerr Whitfield, Ichiro Kaneko, Rimpi K. Saini, and Kristin P. Griffin
Acquisition of data: Rimpi K. Saini, G. Kerr Whitfield, Kristin P. Griffin, and Ichiro Kaneko
Writing, review, and/or revision of the manuscript: Mark R. Haussler, G. Kerr Whitfield, Peter W. Jurutka, Ichiro Kaneko, and Rimpi K. Saini
Administrative, technical, or material support: G. Kerr Whitfield, Peter W. Jurutka, and Mark R. Haussler
Study supervision: Peter W. Jurutka, G. Kerr Whitfield, and Mark R. Haussler
DECLARATION OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Agarwal I, Ide N, Ix JH, Kestenbaum B, Lanske B, Schiller NB, Whooley MA, Mukamal KJ. Fibroblast growth factor-23 and cardiac structure and function. Journal of the American Heart Association. 2014;3:e000584. doi: 10.1161/JAHA.113.000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrukhova O, Slavic S, Smorodchenko A, Zeitz U, Shalhoub V, Lanske B, Pohl EE, Erben RG. FGF23 regulates renal sodium handling and blood pressure. EMBO Molecular Medicine. 2014a;6:744–759. doi: 10.1002/emmm.201303716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrukhova O, Smorodchenko A, Egerbacher M, Streicher C, Zeitz U, Goetz R, Shalhoub V, Mohammadi M, Pohl EE, Lanske B, et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO Journal. 2014b;33:229–246. doi: 10.1002/embj.201284188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel TK, Mathern DR, Whitfield GK, Haussler CA, Hopper HAI, Hsieh JC, Slater SA, Hsieh G, Kaczmarska M, Jurutka PW, et al. 1,25-Dihydroxyvitamin D3/VDR-mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. Journal of Steroid Biochemistry and Molecular Biology. 2007;103:381–388. doi: 10.1016/j.jsbmb.2006.12.054. [DOI] [PubMed] [Google Scholar]
- Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J. The parathyroid is a target organ for FGF23 in rats. Journal of Clinical Investigation. 2007;117:4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergwitz C, Juppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annual Review of Medicine. 2010;61:91–104. doi: 10.1146/annurev.med.051308.111339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe LM, Madathil SV, Casu C, Lanske B, Rivella S, Sitara D. FGF-23 is a negative regulator of prenatal and postnatal erythropoiesis. Journal of Biological Chemistry. 2014;289:9795–9810. doi: 10.1074/jbc.M113.527150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David V, Dai B, Martin A, Huang J, Han X, Quarles LD. Calcium regulates FGF-23 expression in bone. Endocrinology. 2013;154:4469–4482. doi: 10.1210/en.2013-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer J. The biology of the Ets1 proto-oncogene. Molecular Cancer. 2003;2:29. doi: 10.1186/1476-4598-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet Journal of Rare Diseases. 2008;3:12. doi: 10.1186/1750-1172-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidley J, Openshaw S, Pring ET, Sale S, Mansell JP. Lysophosphatidic acid cooperates with 1alpha,25(OH)2D3 in stimulating human MG63 osteoblast maturation. Prostaglandins and Other Lipid Mediators. 2006;80:46–61. doi: 10.1016/j.prostaglandins.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Harms LR, Burne TH, Eyles DW, McGrath JJ. Vitamin D and the brain. Best Practice & Research Clinical Endocrinology & Metabolism. 2011;25:657–669. doi: 10.1016/j.beem.2011.05.009. [DOI] [PubMed] [Google Scholar]
- Haussler MR, Haussler CA, Whitfield GK, Hsieh JC, Thompson PD, Barthel TK, Bartik L, Egan JB, Wu Y, Kubicek JL, et al. The nuclear vitamin D receptor controls the expression of genes encoding factors which feed the “Fountain of Youth” to mediate healthful aging. Journal of Steroid Biochemistry and Molecular Biology. 2010;121:88–97. doi: 10.1016/j.jsbmb.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussler MR, Whitfield GK, Kaneko I, Forster R, Saini R, Hsieh JC, Haussler CA, Jurutka PW. The role of vitamin D in the FGF23, klotho, and phosphate bone-kidney endocrine axis. Reviews In Endocrine & Metabolic Disorders. 2012;13:57–69. doi: 10.1007/s11154-011-9199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussler MR, Whitfield GK, Kaneko I, Haussler CA, Hsieh D, Hsieh JC, Jurutka PW. Molecular mechanisms of vitamin D action. Calcified Tissue International. 2013;92:77–98. doi: 10.1007/s00223-012-9619-0. [DOI] [PubMed] [Google Scholar]
- Ito M, Sakai Y, Furumoto M, Segawa H, Haito S, Yamanaka S, Nakamura R, Kuwahata M, Miyamoto K. Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. American Journal Of Physiology Endocrinology And Metabolism. 2005;288:E1101–1109. doi: 10.1152/ajpendo.00502.2004. [DOI] [PubMed] [Google Scholar]
- Jacobs E, Martinez ME, Buckmeier J, Lance P, May M, Jurutka P. Circulating fibroblast growth factor-23 is associated with increased risk for metachronous colorectal adenoma. J Carcinog. 2011;10:3. doi: 10.4103/1477-3163.76723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurutka PW, Hsieh JC, Remus LS, Whitfield GK, Thompson PD, Haussler CA, Blanco JC, Ozato K, Haussler MR. Mutations in the 1,25-dihydroxyvitamin D3 receptor identifying C-terminal amino acids required for transcriptional activation that are functionally dissociated from hormone binding, heterodimeric DNA binding, and interaction with basal transcription factor IIB, in vitro. Journal of Biological Chemistry. 1997;272:14592–14599. doi: 10.1074/jbc.272.23.14592. [DOI] [PubMed] [Google Scholar]
- Keisala T, Minasyan A, Lou YR, Zou J, Kalueff AV, Pyykko I, Tuohimaa P. Premature aging in vitamin D receptor mutant mice. Journal of Steroid Biochemistry and Molecular Biology. 2009;115:91–97. doi: 10.1016/j.jsbmb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Kesby JP, Cui X, Burne TH, Eyles DW. Altered dopamine ontogeny in the developmentally vitamin D deficient rat and its relevance to schizophrenia. Frontiers In Cellular Neuroscience. 2013;7:111. doi: 10.3389/fncel.2013.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolek OI, Hines ER, Jones MD, Lesueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK. 1{alpha},25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2005;289:G1036–G1042. doi: 10.1152/ajpgi.00243.2005. [DOI] [PubMed] [Google Scholar]
- Lanske B, Razzaque MS. Molecular interactions of FGF23 and PTH in phosphate regulation. Kidney International. 2014;86:1072–1074. doi: 10.1038/ki.2014.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. American Journal Of Physiology Renal Physiology. 2010;299:F882–889. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. Journal of the American Society of Nephrology. 2006;17:1305–1315. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
- Luderer HF, Gori F, Demay MB. Lymphoid enhancer-binding factor-1 (LEF1) interacts with the DNA-binding domain of the vitamin D receptor. Journal of Biological Chemistry. 2011;286:18444–18451. doi: 10.1074/jbc.M110.188219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysaght AC, Yuan Q, Fan Y, Kalwani N, Caruso P, Cunnane M, Lanske B, Stankovic KM. FGF23 deficiency leads to mixed hearing loss and middle ear malformation in mice. PLoS One. 2014;9:e107681. doi: 10.1371/journal.pone.0107681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansell JP, Blackburn J. Lysophosphatidic acid, human osteoblast formation, maturation and the role of 1alpha,25-Dihydroxyvitamin D3 (calcitriol) Biochimica et Biophysica Acta. 20131831:105–108. doi: 10.1016/j.bbalip.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Masuda S, Byford V, Arabian A, Sakai Y, Demay MB, St-Arnaud R, Jones G. Altered pharmacokinetics of 1alpha,25-dihydroxyvitamin D3 and 25-hydroxyvitamin D3 in the blood and tissues of the 25-hydroxyvitamin D-24-hydroxylase (Cyp24a1) null mouse. Endocrinology. 2005;146:825–834. doi: 10.1210/en.2004-1116. [DOI] [PubMed] [Google Scholar]
- Meir T, Durlacher K, Pan Z, Amir G, Richards WG, Silver J, Naveh-Many T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney International. 2014;86:1106–1115. doi: 10.1038/ki.2014.215. [DOI] [PubMed] [Google Scholar]
- Meyer MB, Benkusky NA, Pike JW. Selective Distal Enhancer Control of the Mmp13 Gene Identified through Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) Genomic Deletions. Journal of Biological Chemistry. 2015;290:11093–11107. doi: 10.1074/jbc.M115.648394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MB, Goetsch PD, Pike JW. A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1alpha,25-dihydroxyvitamin D3. Journal of Biological Chemistry. 2010a;285:15599–15610. doi: 10.1074/jbc.M110.119958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MB, Goetsch PD, Pike JW. Genome-wide analysis of the VDR/RXR cistrome in osteoblast cells provides new mechanistic insight into the actions of the vitamin D hormone. Journal of Steroid Biochemistry and Molecular Biology. 2010b;121:136–141. doi: 10.1016/j.jsbmb.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mix KS, Attur MG, Al-Mussawir H, Abramson SB, Brinckerhoff CE, Murphy EP. Transcriptional repression of matrix metalloproteinase gene expression by the orphan nuclear receptor NURR1 in cartilage. Journal of Biological Chemistry. 2007;282:9492–9504. doi: 10.1074/jbc.M608327200. [DOI] [PubMed] [Google Scholar]
- Miyagawa K, Yamazaki M, Kawai M, Nishino J, Koshimizu T, Ohata Y, Tachikawa K, Mikuni-Takagaki Y, Kogo M, Ozono K, et al. Dysregulated gene expression in the primary osteoblasts and osteocytes isolated from hypophosphatemic Hyp mice. PLoS One. 2014;9:e93840. doi: 10.1371/journal.pone.0093840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldknow KJ, MacRae VE, Farquharson C. Endocrine role of bone: recent and emerging perspectives beyond osteocalcin. Journal of Endocrinology. 2015;225:R1–R19. doi: 10.1530/JOE-14-0584. [DOI] [PubMed] [Google Scholar]
- Ozono K, Liao J, Kerner SA, Scott RA, Pike JW. The vitamin D-responsive element in the human osteocalcin gene: association with a nuclear proto-oncogene enhancer. Journal of Biological Chemistry. 1990;265:21881–21888. [PubMed] [Google Scholar]
- Pike JW, Meyer MB, Martowicz ML, Bishop KA, Lee SM, Nerenz RD, Goetsch PD. Emerging regulatory paradigms for control of gene expression by 1,25-dihydroxyvitamin D(3) Journal of Steroid Biochemistry and Molecular Biology. 2010;121:130–135. doi: 10.1016/j.jsbmb.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarles LD. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nature Reviews Endocrinology. 2012;8:276–286. doi: 10.1038/nrendo.2011.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini RK, Kaneko I, Jurutka PW, Forster R, Hsieh A, Hsieh JC, Haussler MR, Whitfield GK. 1,25-dihydroxyvitamin D(3) regulation of fibroblast growth factor-23 expression in bone cells: evidence for primary and secondary mechanisms modulated by leptin and interleukin-6. Calcified Tissue International. 2013;92:339–353. doi: 10.1007/s00223-012-9683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurada K, Ohshima-Sakurada M, Palmer TD, Gage FH. Nurr1, an orphan nuclear receptor, is a transcriptional activator of endogenous tyrosine hydroxylase in neural progenitor cells derived from the adult brain. Development. 1999;126:4017–4026. doi: 10.1242/dev.126.18.4017. [DOI] [PubMed] [Google Scholar]
- Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. Journal of Bone and Mineral Research. 2004a;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. Journal of Clinical Investigation. 2004b;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John HC, Bishop KA, Meyer MB, Benkusky NA, Leng N, Kendziorski C, Bonewald LF, Pike JW. The osteoblast to osteocyte transition: epigenetic changes and response to the vitamin D3 hormone. Molecular Endocrinology. 2014;28:1150–1165. doi: 10.1210/me.2014-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolon RM, Castillo AI, Jimenez-Lara AM, Aranda A. Association with Ets-1 causes ligand- and AF2-independent activation of nuclear receptors. Molecular and Cellular Biology. 2000;20:8793–8802. doi: 10.1128/mcb.20.23.8793-8802.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. [Google Scholar]
- Yu X, Sabbagh Y, Davis SI, Demay MB, White KE. Genetic dissection of phosphate- and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone. 2005;36:971–977. doi: 10.1016/j.bone.2005.03.002. [DOI] [PubMed] [Google Scholar]