

Abstract
Agents that inhibit estrogen production, such as aromatase inhibitors or those that directly block estrogen receptor (ER) activity, such as selective estrogen receptor modulators and selective estrogen receptor degraders, are routinely used in the treatment of ER-positive breast cancers. However, although initial treatment with these agents is often successful, many women eventually relapse with drug-resistant breast cancers. To overcome some of the challenges associated with current endocrine therapies and to combat the development of resistance, there is a need for more durable and more effective ER-targeted therapies. Here we describe and characterize a novel, orally bioavailable small-molecule selective estrogen receptor degrader, RAD1901, and evaluate its therapeutic potential for the treatment of breast cancer. RAD1901 selectively binds to and degrades the ER and is a potent antagonist of ER-positive breast cancer cell proliferation. Importantly, RAD1901 produced a robust and profound inhibition of tumor growth in MCF-7 xenograft models. In an intracranial MCF-7 model, RAD1901-treated animals survived longer than those treated with either control or fulvestrant, suggesting the potential benefit of RAD1901 in the treatment of ER-positive breast cancer that has metastasized to the brain. Finally, RAD1901 preserved ovariectomy-induced bone loss and prevented the uterotropic effects of E2, suggesting that it may act selectively as an agonist in bone but as an antagonist in breast and uterine tissues. RAD1901 is currently under clinical study in postmenopausal women with ER-positive advanced breast cancer.
Keywords: anticancer activity, blood–brain barrier, breast cancer, estrogen receptor inhibition, selective estrogen receptor degraders, selective estrogen receptor modulators, tissue selectivity
Introduction
Estrogen and the estrogen receptor (ER) are known to be prominent drivers of breast tumorigenesis and breast cancer progression 1–5. Agents that inhibit estrogen production, such as aromatase inhibitors, and those that directly block ER activity, such as selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs), are routinely used in the treatment of ER-positive breast cancers. SERMs bind to the ER and can act as receptor antagonists or mixed agonists/antagonists by modulating receptor conformation and regulating coactivator and corepressor recruitment to the receptor 6–8. In contrast, SERDs act by binding to and degrading the ER, thereby providing an alternative mechanism by which estrogen-dependent breast cancers can be targeted. Fulvestrant is currently the only SERD approved for the treatment of ER-positive metastatic breast cancers 9. Despite its clinical efficacy, the utility of fulvestrant has been limited by the amount of drug that can be administered in a single injection and its reduced bioavailability. A recent imaging study, using 18F-estradiol-PET, has suggested that, even at a 500 mg dose level, some patients may not show complete ER inhibition, and insufficient dosing may be a reason for therapy failure 10.
One challenge associated with estrogen-directed therapies is that they may have undesirable effects on uterine, bone, and other tissues. This is because, as a member of the steroid hormone nuclear receptor superfamily 2,11,12, the ER directs transcription of estrogen-responsive genes in a wide variety of tissues and cell types 6,13,14. These effects can be particularly pronounced, as endogenous levels of estrogen and other ovarian hormones diminish during menopause.
Further, although initial treatment with these agents is often successful, many women eventually relapse with drug-resistant breast cancers. Mutations affecting the ER have emerged as a potential mechanism for the development of this resistance 15. To overcome some of the challenges associated with current endocrine therapies and to combat the development of resistance, there is a need for more durable and more effective ER-targeted therapies.
In this report, we describe the identification and characterization of a novel, orally bioavailable, small-molecule SERD developed to specifically bind to the ER.
Methods
Reagents
RAD1901 [Fig. 1; (6R)-6-(2-(N-(4-(2-(ethylamino)ethyl)benzyl)-N-ethylamino)-4-methoxyphenyl)-5,6,7,8-tetrahydronaphthalen-2-ol dihydrochloride] was manufactured by IRIX Pharmaceuticals Inc. (Florence, South Carolina, USA). RAD1901 was stored as a dry powder, formulated for use as a homogenous suspension in 0.5% (w/v) methylcellulose in deionized water, and in animal models, it was administered by oral gavage. Tamoxifen, raloxifene, and estradiol (E2) were obtained from Sigma-Aldrich (St. Louis, Missouri, USA) and administered by subcutaneous injection. Fulvestrant was obtained from Tocris Biosciences (Minneapolis, Minnesota, USA) and administered by subcutaneous injection. Other laboratory reagents were purchased from Sigma-Aldrich unless otherwise specified.
Fig. 1.
Structure of RAD1901.
Animals
The study protocols were reviewed and approved by the Radius Institutional Animal Care and Use Committee. Mice were obtained from Charles River Laboratories (Stone Ridge, New York, USA). Female Sprague–Dawley rat pups (p11) were maintained in a temperature and humidity controlled room with a 12 h light cycle. Animals were allowed to acclimate for 1 week, with ad-libitum access to both food (TD 291615; Teklad, Madison, Wisconsin, USA) and water. Female athymic nude mice [Crl:NU(NCr)-Foxn1nu; Charles River Laboratories] were fed ad-libitum water (reverse osmosis, 1 ppm Cl) and an NIH 31 modified and irradiated lab diet consisting of 18.0% crude protein, 5.0% crude fat, and 5.0% crude fiber. The mice were housed on irradiated bedding in static microisolators on a 12 h light cycle at 20–22°C and 40–60% humidity.
Cell lines
MCF-7 and T47D cells (human mammary metastatic adenocarcinoma) were purchased from American Type Culture Collection (Rockville, Maryland, USA) and were routinely maintained in phenol red-free minimal essential medium (MEM) containing 2 mmol/l l-glutamine and Earle’s BSS, 0.1 mmol/l nonessential amino acids, and 1 mmol/l sodium pyruvate supplemented with 0.01 mg/ml bovine insulin and 10% fetal bovine serum (Invitrogen, Carlsbad, California, USA), at 5% CO2.
Estrogen receptor competitor assay
Competitive ER binding assays were performed using fluorescence polarization (PolarScreen; Invitrogen), as specified by the manufacturer. Briefly, RAD1901 was serially diluted in ES2 screening buffer to concentrations ranging from 10−5 to 10−12 mol/l. Aliquots (25 μl) of each dilution were added to a black 384-well microtiter plate, in triplicate. The ER–Fluormone complex was prepared as directed, with 2 nmol/l Fluormone ES2 and 30 nmol/l ER. A 25 μl aliquot of this preparation was added to each reaction well. The plates were sealed and incubated in the dark at room temperature for 4 h. Polarization values for each well were measured and plotted against the concentration of the test compound. The IC50 was determined from at least three independent experiments, with E2 serving as a positive control.
MCF-7 proliferation assays
For proliferation assays, MCF-7 cells were plated at 10 000 cells/well in 96-well culture plates in MEM with 10% fetal bovine serum, and they were allowed to adhere for 48 h. Thereafter, the cultures were washed with serum-free MEM and incubated in 100 μl serum-free MEM. After 72 h, the medium was removed and replaced with 2% charcoal-stripped FBS–MEM containing 10 pmol/l E2 with either RAD1901 or additional E2 at concentrations ranging from 10−15 to 10−6 mol/l. The medium was removed after 48 h of incubation and the cells were lysed by adding 100 μl of CellTiter Glo (Promega, Madison, Wisconsin, USA), diluted 1 : 1 in water, per well. The plates were gently mixed on a plate shaker for 10 min before the luminescent signal was measured on a luminometer. The EC50 and IC50 of the test compound were defined as the concentrations that resulted in half-maximum shift.
ELISA-based assay of ERα expression in MCF-7 and T47D cells
The levels of ERα expression in the cultured MCF-7 and T47D cells were determined using a commercial ELISA-based assay (DuoSet; R&D Systems, Minneapolis, Minnesota, USA) according to the manufacturer’s instructions. Briefly, MCF-7 or T47D cells were cultured in 10 cm dishes to ~75% confluence in EMEM growth medium supplemented with 10% FBS and 10 µg/ml human insulin (Sigma-Aldrich). Twenty-four hours before treatment, the growth medium was replaced with phenol red-free RPMI-1640 growth medium. A stock solution of 10 mmol/l RAD1901 was prepared in dimethyl sulfoxide (DMSO). Dilutions of RAD1901 were prepared in RPMI growth medium (doses ranging from 10 to 0.5 nmol/l). Controls included 0.1% DMSO alone (vehicle), 1 nmol/l E2, 100 nmol/l fulvestrant, and 1 µmol/l tamoxifen. Plated cells were treated with RAD1901 or controls for 48 h, and then incubated for 15 min with ice-cold lysis buffer [1 mmol/l EDTA, 0.5% Triton X-100, 5 mmol/l NaF, 6 mol/l urea, 1 mmol/l sodium orthovanadate, 2.5 mmol/l sodium pyrophosphate, and 1× HALT protease inhibitor cocktail (R&D Systems)]. Lysates were centrifuged at 2000g for 5 min, and the supernatant was diluted 1 : 1 in lysis buffer. Ninety-six-well plates were coated overnight with capture antibody (1 µg/ml), washed three times in the manufacturer’s wash buffer, blocked with blocking buffer for 2 h, and washed again. The prepared plates were incubated with 100 µl of the prepared cell lysate for 2 h, washed, incubated with biotinylated detection antibody for 2 h, and washed again. After a 20-min incubation with streptavidin–horseradish peroxidase, the plates were washed and incubated with substrate solution for 20 min. The reaction was stopped with stop solution, and the plates were analyzed on a microplate reader (OD450).
Assessment of uterotropic activity
Sprague–Dawley rat pups were weaned at 19 days of age, randomized into groups (n=4), and administered vehicle (aqueous methylcellulose), E2 (0.01 mg/kg), raloxifene (3 mg/kg), tamoxifen (1 mg/kg), RAD1901 alone (0.3–100 mg/kg), or RAD1901 (0.01–10 mg/kg) in combination with E2 (0.01 mg/kg), either by subcutaneous injection or by oral gavage, as appropriate (see reagents above), once daily for three consecutive days. Twenty-four hours after the final dose, all animals were killed by carbon dioxide inhalation. Body weights and wet uterine weights were recorded for each animal. Similar assays were also conducted with RAD1901 (0.03–100 mg/kg) in mice (Charles River Laboratories, Montreal, Quebec, Canada).
Fresh uterine tissue from each rat was fixed in 4% paraformaldehyde, dehydrated with ethanol, and embedded in JB4 plastic resin. Sections were cut at 8 μm and stained with 0.1% toluidine blue O. The thickness of the endometrial epithelium was measured using a Zeiss Axioskop 40 microscope (Carl Zeiss Light Microscopy, Goettingen, Germany) with the Spot Advanced program; the mean of nine measurements per specimen was calculated.
Uterine complement component 3 (C3) gene expression
To determine the relative expression levels of C3 in the treated uterine tissue, RNA was extracted from the remaining tissue using the Micro to Midi Total RNA Purification Kit (Invitrogen) according to the manufacturer’s instructions. RNA was quantified, and equal amounts were reverse-transcribed using the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, California, USA).
Quantitative PCR was performed using the ABI Prism 7300 System (Applied Biosystems). PCR was performed using the TaqMan Universal Master Mix, with probe sets for C3 and for the 18S ribosomal RNA as a reference gene. Thermal cycling conditions comprised an initial denaturation step at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min.
Relative gene expression was determined by normalizing each sample to the endogenous control (18S) and comparing with a calibrator (vehicle). Relative gene expression was determined using the following equation: 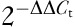 (where Ct=cycle threshold or the cycle number at which the PCR product was first detected, ΔCt=normalized sample value, and ΔΔCt=normalized difference between dosed subjects and the vehicle). Five replicate gene expression determinations were conducted for each dose, within each study.
(where Ct=cycle threshold or the cycle number at which the PCR product was first detected, ΔCt=normalized sample value, and ΔΔCt=normalized difference between dosed subjects and the vehicle). Five replicate gene expression determinations were conducted for each dose, within each study.
Bone loss in ovariectomized rats
As a model of postmenopausal bone loss 16, ovariectomy was performed on anesthetized adult female Sprague–Dawley rats, with sham surgery as a control. Following surgery, ovariectomized rats were treated once daily for 4 weeks with vehicle, E2 (0.01 mg/kg), or RAD1901 (0.1, 0.3, 1, and 3 mg/kg), administered as described above, with 20 animals per group. Animals in the sham surgery group were vehicle treated. All animals were killed by carbon dioxide inhalation 24 h after the final dose. Bone mineral density was assessed at baseline and again after 4 weeks of treatment by PIXImus dual emission X-ray absorptiometry (Lunar Corp/GE, Madison, Wisconsin, USA).
At necropsy, the left femur of each animal was removed, dissected free of soft tissue, and stored in 70% ethanol before analysis. A detailed qualitative and quantitative three-dimensional (3D) evaluation was conducted using a micro-CT40 system (Scanco Systems, Wayne, Pennsylvania, USA). For each specimen, 250 image slices of the distal femur metaphysis were acquired. Morphometric parameters were determined using a direct 3D approach in preselected analysis regions. Parameters determined in the trabecular bone included bone volume density, bone surface density, trabecular number, trabecular thickness, trabecular spacing, connectivity density, and apparent bone density.
MCF-7 xenograft models
Female athymic nude mice [Crl:NU(NCr)-Foxn1nu] were used for tumor xenograft studies. Three days before tumor cell implantation, estrogen pellets (0.36 mg E2, 60-day release; Innovative Research of America, Sarasota, Florida, USA) were implanted subcutaneously between the scapulae of all test animals using a sterilized trochar. MCF-7 human breast adenocarcinoma cells were cultured to mid-log phase in RPMI-1640 medium containing 10% fetal bovine serum, 100 U/ml penicillin G, 100 μg/ml streptomycin sulfate, 2 mmol/l glutamine, 10 mmol/l HEPES, 0.075% sodium bicarbonate, and 25 μg/ml gentamicin. On the day of tumor cell implantation, the cells were trypsinized, pelleted, and resuspended in PBS at a concentration of 5×107 cells/ml. Each test mouse received 1×107 MCF-7 cells implanted subcutaneously in the right flank, and tumor growth was monitored. Volume was calculated using the following formula: tumor volume (mm3)=l×w2/2, where w=width and l=length in mm of an MCF-7 tumor. When necessary, tumor weight was estimated on the basis of the assumption that 1 mm3 of tumor volume was equivalent to 1 mg tumor wet weight.
Fourteen days after tumor cell implantation (designated as day 1 of the study), mice were 9 weeks of age, with body weights ranging from 21.4 to 32.5 g, individual tumor volumes ranging from 75 to 144 mm3, and a group mean tumor volume (MTV) of 108 mm3. The mice were randomized into nine groups of 15 animals each and treated with vehicle, tamoxifen (1 mg/animal every other day), fulvestrant (0.5 mg/animal daily), or RAD1901 (0.3, 1, 3, 10, 30, 60, 90, and 120 mg/kg daily), as described above.
Tumor volumes were evaluated twice per week. The tumor endpoint was defined as an MTV of 1500 mm3 in the control group. Animals were also monitored for partial regression (PR) and complete regression responses. Treatment tolerability was assessed by body weight measurements and frequent observation for clinical signs of treatment-related adverse effects. Animals with weight loss exceeding 30% for one measurement, or exceeding 25% for three measurements, were humanely killed and their deaths were classified as treatment-related deaths. Acceptable toxicity was defined as a group mean body weight loss of less than 20% during the study and not more than one treatment-related death among 10 treated animals, or 10%. At the end of the study, the animals were killed by terminal cardiac puncture under isoflurane anesthesia. RAD1901 concentrations in the plasma and tumor were determined using liquid chromatography–tandem mass spectrometry.
Effect of RAD1901 in an intracranial MCF-7 tumor model
For intracranial tumor models, female athymic nude mice [Crl:NU(NCr)-Foxn1nu] were prepared as above, except that each test mouse received 1×106 MCF-7 cells implanted intracranially. Five days after tumor cell implantation (designated as day 1 of the study), mice were randomized into three groups of 12 animals each and treated with vehicle, fulvestrant (0.5 mg/animal daily), or RAD1901 (120 mg/kg daily), as described above.
The endpoint was defined as mortality or 3× survival of the control group, whichever came first. Treatment tolerability was assessed by body weight measurements and frequent observation for clinical signs of treatment-related adverse effects. Animals with weight loss exceeding 30% for one measurement, or exceeding 25% for three measurements, were humanely killed and their deaths were classified as treatment-related deaths. Acceptable toxicity was defined as a group mean body weight loss of less than 20% during the study and not more than one treatment-related death among 10 treated animals, or 10%. At the end of the study, the animals were killed by terminal cardiac puncture under isoflurane anesthesia. RAD1901 and fulvestrant concentrations in the plasma and tumor were determined using liquid chromatography-tandem mass spectrometry.
Data analysis
Statistical analysis and graphical presentations were performed using Prism for Windows 6.02 (GraphPad Software Inc., La Jolla, California, USA). Descriptive data are generally expressed as mean±SD, as indicated. Statistical evaluations of the differences between groups were assessed using parametric or nonparametric t-tests as indicated in the text or figures, with a prespecified α of 0.05.
For tumor xenograft models, the treatment outcome was the percent tumor growth inhibition (TGI), defined as the percent difference between baseline and end of study. The data set for TGI analysis included all animals in each group, excluding any that died because of treatment-related or non-treatment-related causes. The threshold for potential therapeutic activity was defined as a treatment effect of at least 60% TGI. Results were analyzed using the Kruskal–Wallis or the Mann–Whitney test, with a prespecified α of 0.05.
Results
RAD1901 exhibits preferential binding affinity for ERα
In competitive receptor binding assays, the IC50 for RAD1901 on ERα was 48 versus 870 nmol/l for ERβ. For the E2 control, the IC50 values for ERα and ERβ were 0.4 and 0.3 nmol/l, respectively.
RAD1901 inhibits expression of ERα in cultured breast tumor cell lines
To further understand the effects of RAD1901 on the ER, ERα expression was evaluated. Treatment of MCF-7 cells with 1 µmol/l tamoxifen had no effect on the expression of ERα, whereas 100 nmol/l fulvestrant completely inhibited ERα expression (Fig. 2a). RAD1901 treatment exhibited dose-dependent inhibition of ERα expression, with a calculated EC50 of 0.6 nmol/l in these experiments. E2 at a concentration of 1 nmol/l also induced complete inhibition of ERα expression, consistent with published findings 17. A similar dose-dependent decrease in ERα expression was also observed after RAD1901 treatment in T47D cells (data not shown).
Fig. 2.
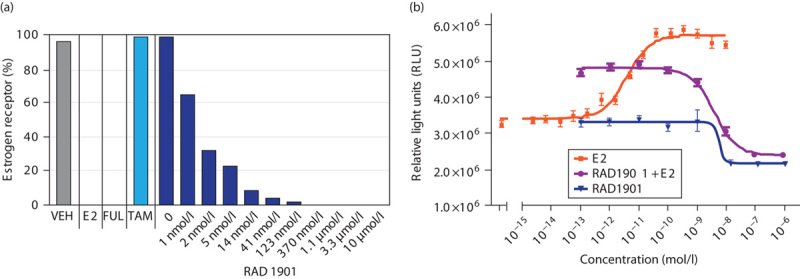
Effects of RAD1901 on proliferation and ERα expression in cultured MCF-7 cells. (a) ERα expression in MCF-7 cells after 48 h in the presence of vehicle (0.1% DMSO), E2 (1 nmol/l), fulvestrant (100 nmol/l), tamoxifen (1 µmol/l), or RAD1901 (10 µmol/l to 0.5 nmol/l). Levels of ERα receptor expression are expressed as percent of vehicle control. (b) For proliferation assays, MCF-7 cells were treated for 48 h in the presence of increasing concentrations of E2, RAD1901, or RAD1901 in the presence of 10 pmol/l E2. DMSO, dimethyl sulfoxide; ERα, estrogen receptor-α; FUL, fulvestrant; TAM, tamoxifen; VEH, vehicle.
RAD1901 inhibits in-vitro proliferation of cultured, E2-stimulated MCF-7 cells in a dose-dependent manner
Treatment of ER-positive MCF-7 cells with E2 resulted in a potent and dose-dependent increase in proliferation, with an EC50 of 4 pmol/l (Fig. 2b). Treatment of cells with RAD1901 in the presence of 10 pmol/l E2 resulted in a dose-dependent decrease in proliferation, with an IC50 value of 4.2 nmol/l. Importantly, RAD1901 treatment (0.1 pmol/l to 1 µmol/l) did not stimulate proliferation in the absence of E2, and resulted in significant inhibition of basal cell proliferation, which was observed at doses of 10 nmol/l and above.
RAD1901 inhibits tumor growth in MCF-7 xenograft models
The antitumor effects of RAD1901 were characterized using an MCF-7 xenograft model in mice supplemented with E2 to stimulate tumor growth. RAD1901 inhibited tumor growth in a dose-dependent manner (Fig. 3). Significant TGI responses were seen at doses of 30 and 60 mg/kg RAD1901, with TGI at day 40 being 66% (P<0.05) and 88% (P<0.001), respectively. These were comparable to the TGI observed for tamoxifen and fulvestrant, which on day 40 were 86 and 88%, respectively (Fig. 3a). RAD1901 was well tolerated, with no adverse effect on body weight.
Fig. 3.
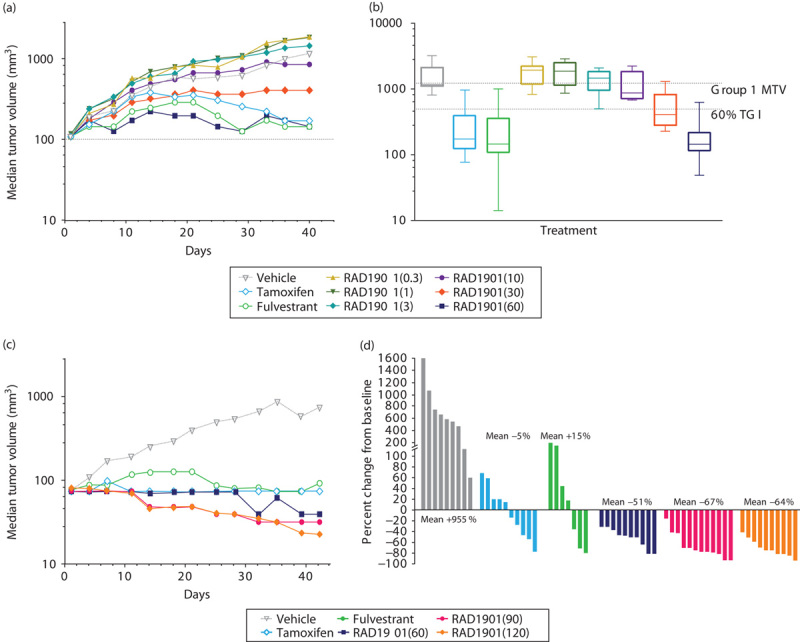
Dose-dependent inhibition of tumor growth by RAD1901 in an MCF-7 mouse xenograft model. Female nude mice were implanted with 1×107 MCF-7 cells subcutaneously in the right flank. Fourteen days after implantation, tumor volumes were recorded and treatment was initiated with vehicle, tamoxifen (1 mg/animal every other day), fulvestrant (0.5 mg/animal daily), or RAD1901 (0.3, 1, 3, 10, 30, 60, 90, and 120 mg/kg daily). Tumor volume was evaluated twice per week until the study endpoint. (a) Mean tumor volumes over time for daily doses of 0.3, 1, 3, 10, 30, 60 mg/kg RAD1901, tamoxifen, and fulvestrant. (b) Box and whisker plots show the day 40 tumor volume by group. The box represents the 25th through 75th percentiles of observations, the line represents the mean of the observations, and the whiskers represent the extreme observations. (c) Mean tumor volumes over time for daily doses of 60, 90, and 120 mg/kg RAD1901, tamoxifen, and fulvestrant. (d) Tumor volumes from individual animals at day 42, with the mean percent change detailed for each group.
To explore whether higher doses of RAD1901 induced a more robust effect in this model, doses of RAD1901 up to 120 mg/kg were assessed (Fig. 3b and c). As previously observed, RAD1901 induced significant tumor inhibition at day 42, with a TGI of 94% in the 60 mg/kg group (with 2/10 PRs), 97% in the 90 mg/kg group (with 8/10 PRs), and 96% in the 120 mg/kg group (with 7/10 PRs). Although both tamoxifen and fulvestrant decreased tumor growth, the inhibition was not as profound as that observed with RAD1901. Tamoxifen treatment yielded a TGI of 90% (with 2/10 PRs) and fulvestrant treatment resulted in 87% TGI (with 1/10 PR). RAD1901 inhibition in the 90 and 120 mg/kg groups was significantly greater relative to inhibition by both tamoxifen (P<0.05) and fulvestrant (P<0.05). RAD1901 was well tolerated at these higher dose levels.
At the end of study, the concentration of RAD1901 in plasma was 344±117 ng/ml and in the tumor was 11118±3801 ng/ml for the 60 mg/kg dose level. A similar tumor-to-plasma ratio was also observed at lower dose levels, at which tumor concentrations were ∼20–30-fold higher than in plasma.
RAD1901 promotes survival in a mouse xenograft model of brain metastasis
The potential ability of RAD1901 to cross the blood–brain barrier and inhibit tumor growth was evaluated using an MCF-7 intracranial tumor xenograft model. An intracranial model was selected to directly assess the effects of RAD1901 on inhibiting an established brain tumor and survival was the primary endpoint to evaluate efficacy. Kaplan–Meier survival analysis demonstrated that RAD1901 significantly prolonged survival compared with fulvestrant (P<0.0001; Fig. 4). No animal in the control or fulvestrant group survived beyond day 20 and day 34, respectively, whereas 41% (5/12) of the RAD1901-treated animals survived until the end of the study at day 54.
Fig. 4.
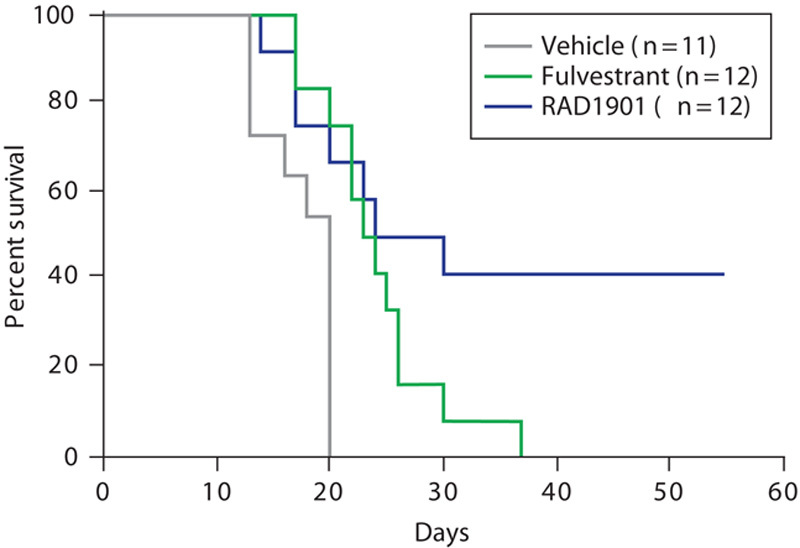
Effect of RAD1901 on mouse survival in an intracranial MCF-7 tumor model. Female nude mice were implanted with 1×106 MCF-7 cells intracranially. Five days after tumor cell implantation (designated as day 1 of the study), mice were randomized into three groups of 12 animals each and treated with vehicle, fulvestrant (0.5 mg/animal daily), or RAD1901 (120 mg/kg daily).
The concentration of RAD1901 in plasma was 738±471 ng/ml and in the intracranial tumor was 462±105 ng/ml, supporting the hypothesis that RAD1901 is able to effectively cross the blood–brain barrier. In contrast, concentrations of fulvestrant were substantially lower in the plasma (21±10 ng/ml) and in the intracranial tumor (8.3±0.8 ng/g).
RAD1901 antagonizes E2 stimulation of uterine tissue
The uterotropic effects of RAD1901 were investigated by assessing changes in uterine weight, histology, and C3 gene expression in immature rats. Results from a representative study are shown in Fig. 5. Treatment with E2 (0.01 mg/kg), raloxifene (3 mg/kg), or tamoxifen (1 mg/kg) resulted in significant increases in uterine wet weight compared with treatment vehicle alone, whereas RAD1901 treatment at a range of doses between 0.3 and 100 mg/kg did not significantly affect uterine wet weight (Fig. 5a). Further, when administered in combination with E2 (0.01 mg/kg), RAD1901 antagonized E2-mediated uterine stimulation in a dose-dependent manner, exhibiting significant inhibition of uterotropic activity at doses of 0.1 mg/kg and greater and complete inhibition at 3 mg/kg. The EC50 for RAD1901 was ~0.3 mg/kg. Similar results were obtained in mice; RAD1901 doses of 0.03–100 mg/kg also had no effect on uterine wet weight or epithelial thickness (data not shown).
Fig. 5.
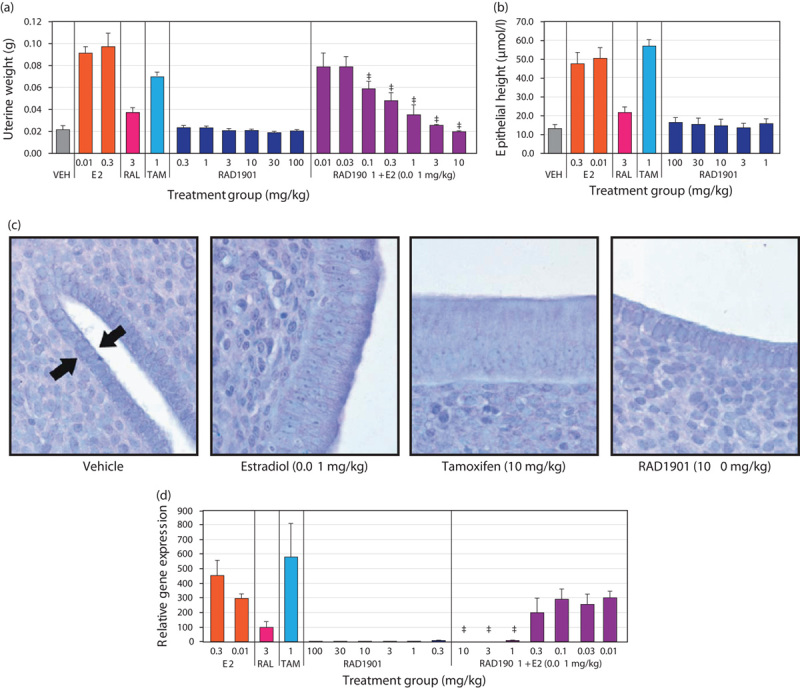
Effect of RAD1901 on uterine tissue. Newly weaned female Sprague–Dawley rats were treated with vehicle, E2 (0.01 mg/kg), raloxifene (3 mg/kg), tamoxifen (1 mg/kg), RAD1901 alone (0.3 to 100 mg/kg), or RAD1901 (0.01 to 10 mg/kg) in combination with E2 (0.01 mg/kg) once daily for three consecutive days. (a) Twenty-four hours after the final dose, rats were killed and uterine wet weights were recorded in grams. (b) Epithelial height was quantified in tissue sections of the uterus. (c) Representative sections of Toluidine Blue O-stained uterine tissue at ×400 magnification. Arrows indicate the uterine epithelium. (d) Total RNA was extracted from uterine tissue and analyzed by quantitative RT-PCR for the level of complement C3 expression relative to the 18S ribosomal RNA housekeeping gene. Data are expressed as mean±SD. *P<0.05 versus vehicle. ‡P<0.05 versus E2. E2, estradiol, RAL, raloxifene; TAM, tamoxifen; VEH, vehicle.
Treatment-dependent changes in uterine tissue were further investigated by quantitative microscopic histological analysis. There was a statistically significant increase in the endometrial epithelial thickness after treatment with E2 at both 0.01 and 0.3 mg/kg (Fig. 5b). A significant increase in epithelial thickness was also observed after treatment with tamoxifen (1 mg/kg) or raloxifene (3 mg/kg). In contrast, RAD1901 treatment did not increase endometrial epithelial thickness up to the highest evaluated dose of 100 mg/kg. Representative images of the endometrial epithelium are shown in Fig. 5c.
Consistent with the changes in both uterine weight and endometrial epithelial thickness, E2, tamoxifen, and raloxifene all significantly increased the expression of the estrogen-regulated complement gene, C3 (Fig. 5d). In contrast, RAD1901 did not increase C3 gene expression at any of the doses tested (0.3–100 mg/kg). Furthermore, RAD1901 at 1, 3, and 10 mg/kg significantly suppressed E2-stimulated C3 gene expression.
Treatment with RAD1901 protects against bone loss in ovariectomized rats
The bone-specific effects of RAD1901 were examined in ovariectomized rats. Following ovariectomy, untreated (vehicle control) rats experienced a decrease in bone mineral density both in the entire femur and in the lumbar spine compared with baseline (Table 1). Treatment with E2 was associated with prevention of bone loss in both the femur and the spine. Similarly, treatment with RAD1901 resulted in a dose-dependent and statistically significant suppression of ovariectomy-induced bone loss (data shown for the 3 mg/kg treatment group). At doses of 0.1–3 mg/kg, the bone mineral density in RAD1901-treated rats was unchanged, with no statistically significant difference compared with the E2-treated group.
Table 1.
Effect of RAD1901 on BMD in ovariectomized ratsa

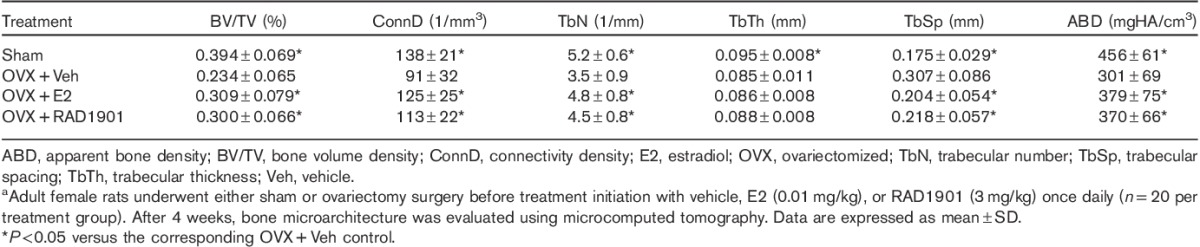
Micro-CT analysis of the distal femur (Table 2) demonstrated that ovariectomy induced significant changes in a number of key microarchitectural parameters when compared with sham surgery. These changes were consistent with a decrease in bone mass and included decreased bone volume, reduced trabecular number, thickness, and density, and increased trabecular separation. Consistent with the preservation of bone mineral density observed after treatment with RAD1901, significant preservation of trabecular architecture was observed in key microstructural parameters (Table 2).
Table 2.
Effect of RAD1901 on femur microarchitecture in ovariectomized ratsa
Discussion
A selective approach to targeting ER activity is a current focus of therapeutic development in breast cancer. We have identified a novel SERD with properties that distinguish it from fulvestrant 18, as well as from the SERMs tamoxifen and raloxifene 19.
RAD1901 had a high affinity for ERα, which resulted in rapid and complete degradation of the ER, which was comparable to that of fulvestrant. RAD1901 was a potent antagonist of breast cancer cells, inhibiting both basal and E2-stimulated MCF-7 cell proliferation.
In xenograft models, RAD1901 demonstrated dose-dependent antitumor activity and induced a more profound antitumor effect when compared with tamoxifen and fulvestrant, without significant adverse effects or toxicity. Although both tamoxifen and fulvestrant were effective at preventing tumor growth in these models, RAD1901 treatment was associated with robust and marked tumor regression and an apparent early onset of effect. Of note, the plasma levels of RAD1901 (at 60–120 mg/kg) are comparable to the human plasma concentrations, where complete ER engagement was observed as measured by 18F-estradiol-PET 20). Similarly, the fulvestrant concentrations observed in our models are comparable to the human exposure observed with the 500 mg dose 21. Taken together, these results suggest that RAD1901 may have an enhanced clinical efficacy profile.
Our characterization of RAD1901 demonstrated improved tissue selectivity when compared with tamoxifen and raloxifene. Importantly, RAD1901 did not stimulate the uterus, as evaluated by changes in uterine wet weight, endometrial epithelial thickness, and C3 gene expression. In addition, RAD1901 antagonized the uterotropic effects of E2, even at doses that yielded effective tumor growth inhibition in the xenograft models.
Use of ER-targeted therapies to treat breast cancer can be associated with a clinically relevant loss of bone mass 22. Ovariectomy-induced bone loss in rats represents a widely used animal model that mimics the development of estrogen deficiency-induced osteopenia in humans and has shown significant value in aiding the identification of bone-sparing or bone-protective agents. Estrogen deficiency induced by ovariectomy in rats produces a high bone turnover state where bone resorption outpaces new bone formation, resulting in a net loss of bone mass 16. In this model, RAD1901 treatment completely prevented ovariectomy-induced bone loss and preserved bone microarchitecture at dose levels as low as 0.1 mg/kg. Together, these results suggest that RAD1901 may act selectively as an agonist in bone but as an antagonist in breast and uterine tissues.
Current endocrine agents have limited ability to cross the blood–brain barrier; therefore, there are few treatment options for patients with ER-positive breast cancer brain metastases, and new targeted therapies are needed to treat this highly unmet medical need. RAD1901 was shown to have promising efficacy against MCF-7 tumors that had been implanted intracranially. In this orthotropic model, E2-dependent breast cancer cells (MCF-7) implanted into the brain led to rapid tumor growth and high mortality in untreated animals. In our study, treatment with fulvestrant, an agent known to be ineffective at crossing the blood–brain barrier, had a limited effect in reducing mortality. In contrast, RAD1901 was associated with prolonged survival, consistent with the significantly higher tumor concentrations observed with RAD1901 compared with fulvestrant. Given the encouraging efficacy data on an established tumor in the intracranial model, additional studies to evaluate the prevention of brain metastasis are ongoing.
With the evolving knowledge of resistance mechanisms to endocrine therapies, it will be important to further understand the efficacy of RAD1901 on both wild-type and mutant ERs in ongoing studies. In addition, evaluating the molecular profile of RAD1901 in breast cancer cells that are both sensitive and resistant to current endocrine therapies will further guide clinical development. RAD1901 is currently under clinical study in women with ER-positive advanced breast cancer (ClinicalTrials.gov #:NCT02338349).
In summary, RAD1901 is a tissue-selective SERD with good oral bioavailability that produces profound tumor regression in ER-positive breast tumor xenograft models. RAD1901 is therefore an attractive therapeutic candidate for treating estrogen-dependent breast cancers.
Acknowledgements
The authors thank Phillips Gilmore, Oncology Communications Inc., and Jeanne McAdara-Berkowitz, PhD, for professional assistance with manuscript preparation, which was funded by Radius Health Inc.
Conflicts of interest
F.G. and G.H. are employees of Radius Health Inc. For the remaining authors there are no conflicts of interest.
References
- 1.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med 2006; 354:270–282. [DOI] [PubMed] [Google Scholar]
- 2.Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem 2006; 6:181–202. [PubMed] [Google Scholar]
- 3.Stein RA, McDonnell DP. Estrogen-related receptor alpha as a therapeutic target in cancer. Endocr Relat Cancer 2006; 13 (Suppl 1):S25–S32. [DOI] [PubMed] [Google Scholar]
- 4.Yue W, Yager JD, Wang JP, Jupe ER, Santen RJ. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids 2013; 78:161–170. [DOI] [PubMed] [Google Scholar]
- 5.Stingl J. Estrogen and progesterone in normal mammary gland development and in cancer. Horm Cancer 2011; 2:85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinkerton JV, Thomas S. Use of SERMs for treatment in postmenopausal women. J Steroid Biochem Mol Biol 2014; 142:142–154. [DOI] [PubMed] [Google Scholar]
- 7.Swaby RF, Sharma CG, Jordan VC. SERMs for the treatment and prevention of breast cancer. Rev Endocr Metab Disord 2007; 8:229–239. [DOI] [PubMed] [Google Scholar]
- 8.Jordan VC. Chemoprevention of breast cancer with selective oestrogen-receptor modulators. Nat Rev Cancer 2007; 7:46–53. [DOI] [PubMed] [Google Scholar]
- 9.Di Leo A, Jerusalem G, Petruzelka L, Torres R, Bondarenko IN, Khasanov R, et al. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J Clin Oncol 2010; 28:4594–4600. [DOI] [PubMed] [Google Scholar]
- 10.Van Kruchten M, de Vries EG, Glaudemans AW, van Lanschot MC, van Faassen M, Kema IP, et al. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov 2015; 5:72–81. [DOI] [PubMed] [Google Scholar]
- 11.Evans RM. The steroid and thyroid hormone receptor superfamily. Science 1988; 240:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh RR, Kumar R. Steroid hormone receptor signaling in tumorigenesis. J Cell Biochem 2005; 96:490–505. [DOI] [PubMed] [Google Scholar]
- 13.McDonnell DP, Norris JD. Connections and regulation of the human estrogen receptor. Science 2002; 296:1642–1644. [DOI] [PubMed] [Google Scholar]
- 14.Burns KA, Korach KS. Estrogen receptors and human disease: an update. Arch Toxicol 2012; 86:1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herynk MH, Fuqua SA. Estrogen receptors in resistance to hormone therapy. Adv Exp Med Biol 2007; 608:130–143. [DOI] [PubMed] [Google Scholar]
- 16.Kalu DN. The ovariectomized rat model of postmenopausal bone loss. Bone Miner 1991; 15:175–191. [DOI] [PubMed] [Google Scholar]
- 17.Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, Julien J, et al. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem 2015; 58:4888–4904. [DOI] [PubMed] [Google Scholar]
- 18.Wardell SE, Marks JR, McDonnell DP. The turnover of estrogen receptor α by the selective estrogen receptor degrader (SERD) fulvestrant is a saturable process that is not required for antagonist efficacy. Biochem Pharmacol 2011; 82:122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maximov PY, Lee TM, Jordan VC. The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr Clin Pharmacol 2013; 8:135–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hattersley G, David F, Harris A, Clarkin M, Banks K, Williams G, et al. RAD1901, a novel tissue-selective estrogen receptor degrader (SERD) demonstrates estrogen receptor engagement in a phase 1 clinical study. Paper presented at: San Antonio Breast Cancer Meeting; 9–13 December 2014; San Antonio, Texas, USA.
- 21.Fulvestrant Prescribing Information. AstraZeneca website. Available at: http://www1.astrazeneca-us.com/pi/faslodex.pdf. [Accessed 6 May 2015].
- 22.Coleman RE. Hormone- and chemotherapy-induced bone loss in breast cancer. Oncology (Williston Park) 2004; 18 (5 Suppl 3):16–20. [PubMed] [Google Scholar]