

Abstract
Recent studies indicate that the release of high mobility group box 1 (HMGB1) following nerve injury may play a central role in the pathogenesis of neuropathic pain. HMGB1 is known to influence cellular responses within the nervous system via two distinct receptor families; the Receptor for Advanced Glycation End-products (RAGE) and Toll-like receptors (TLRs). The degree to which HMGB1 activates a receptor is thought to be dependent upon the oxidative state of the ligand, resulting in the functional isoforms of all-thiol HMGB1 (at-HMGB1) acting through RAGE, and disufide HMGB1 (ds-HMGB1) interacting with TLR4. Though it is known that dorsal root ganglia (DRG) sensory neurons exposed to HMGB1 and TLR4 agonists can influence excitation, the degree to which at-HMGB1 signaling through neuronal RAGE contributes to neuropathic pain is unknown. Here we demonstrate that at-HMGB1 activation of nociceptive neurons is dependent on RAGE and not TLR4. To distinguish the possible role of RAGE on neuropathic pain, we characterized the changes in RAGE mRNA expression up to one month after tibial nerve injury (TNI). RAGE mRNA expression in lumbar dorsal root ganglion (DRG) is substantially increased by post-injury day (PID) 28 when compared with sham injured rodents. Protein expression at PID28 confirms this injury-induced event in the DRG. Moreover, a single exposure to monoclonal antibody to RAGE (RAGE Ab) failed to abrogate pain behavior at PID 7, 14 and 21. However, RAGE ab administration produced reversal of mechanical hyperalgesia on PID28. Thus, at-HMGB1 activation through RAGE may be responsible for sensory neuron sensitization and mechanical hyperalgesia associated with chronic neuropathic pain states.
1. Introduction
Recent studies indicate inflammatory mediators released by nerve injury play a central role in the pathogenesis of chronic pain conditions [1]. Although poorly understood, a key feature of these inflammatory events is the presence of Danger Associated Molecular Patterns (DAMPs; alarmins) [2]. A DAMP of particular interest to the injured nervous system is high mobility group protein box-1 (HMGB1; previously known as amphoterin) [3]. HMGB1, a nuclear protein that binds DNA and regulates gene expression is structurally composed of two tandem DNA-binding domains, Box A and B, and a highly acidic C-terminal tail composed of a string of aspartate and glutamate residues [4–7]. Originally described as a membrane-associated protein that regulated neurite outgrowth during development, it is now known that HMGB1 also plays a crucial role in the inflammatory responses associated with tissue injury, reparative responses and disease [8–12] and may contribute significantly to chronic neuropathic pain states [13, 14].
The action of HMGB1 on different cell types is known to differ dramatically based on the oxidation state of the protein. When first released into the extracelluar space, HMGB1 is initially in the all-thiol state (at-HMGB1) and is thought to largely act on a member of the Ig superfamily, the Receptor for Advanced Glycation End-products (RAGE) [15]. There are also reports that at-HMGB1 can form a complex with CXCL12 and act through CXCR4 [16]. Once present in an oxidative environment, cysteines 23 and 46 of HMGB1 Box A form a sulfide bond, effectively producing the disulfide isoform of HMGB1 (ds-HMGB1). ds-HMGB1 appears to primarily act on the receptor toll-like receptor 4 (TLR4) in order to influence the production of inflammatory cytokines [16, 17]. ds-HMGB1 can then be further reduced by sulfonation of cysteine 106 in the Box B domain of the ligand, resulting in an inert form [18].
It has been suggested that release of HMGB1 from injured neurons can contribute to seizure activity associated with epilepsy; however, the receptor responsible for this pathological activity is still disputed. [11, 19]. The actions of HMGB1 have also been implicated in both inflammatory and neuropathic pain conditions, though it is unclear as to whether the TLR4 or RAGE receptor is responsible [13, 14, 20–23]. Endotoxin-mediated TLR4 activation is known to directly increase neuronal excitation states in acutely dissociated nociceptive neurons [24–27] and administration of xenobiotic TLR4 agonists can produce tactile behavioral hypersensitivity in uninjured rodents [27]. Since the effects of HMGB1 could be mediated by either TLR4 and RAGE depending on the oxidation state of the protein, we set out to determine the contribution of at-HMGB1 on neuronal excitation of nociceptive neurons using a small molecule inhibitor of TLR4 [28] and a neutralizing antibody against RAGE, 11E6 [29, 30]. Moreover, as little direct evidence exists to support a direct role for RAGE in pain, additional studies examined tibial nerve injury (TNI)-induced RAGE expression in associated lumbar DRG and the ability of RAGE neutralizing antibody to reverse injury-induced behavioral hypersensitivity in the rat across time. Our data indicates that at-HMGB1 elicits neuronal excitation via RAGE, in acutely dissociated sensory neurons. In addition, the use of the neutralizing RAGE antibody reverses tactile pain hypersensitivity. This evidence, together with the increased expression of RAGE in the sensory ganglia, identifies a new potential therapeutic target which appears to contribute to pathological pain.
2. Methods
2.1 Animals
Pathogen-free, adult female and male Sprague-Dawley (S/D) rats (150–200 g; Harlan Laboratories, Madison, WI) were housed in temperature (23 ± 3°C) and light (12-h light: 12-h dark cycle; lights on at 07:00 h) controlled rooms with standard rodent chow and autoclaved tap water available. Experiments were performed during the light cycle. Animals were randomly assigned to the treatment groups. All animal related experiments were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine. All procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health and the ethical guidelines established by the International Association for the Study of Pain.
2.2 Tibial Nerve Injury
All rodents were anesthetized during the procedure with isoflurane (4% induction, 2% maintenance). To model neuropathic pain, we performed a tibial nerve injury (TNI) [23; 30; 58]. S/D rats 150–200g were anesthetized using isoflurane at 4% induction and 2% maintenance. Under anesthesia, the right sciatic nerve was isolated under aseptic surgical conditions by blunt dissection of the femoral biceps muscle, without damaging the epimycium. The sciatic nerve and its three branches were isolated: the sural, common peroneal and tibial nerves; only the tibial nerve was tightly-ligated with 5-0 silk and transected distal to the ligation. The removal of an additional 2–4mm of distal nerve stump was removed in order to prevent re-innervation by the proximal end of the nerve. The overlying muscle and skin was then sutured in two separate layers. Sham-injured animals were subjected to all preceding procedures with the exception of ligation and transection.
2.3 Reagents
All reagents were freshly prepared in buffer on day of use. The neutralizing monoclonal RAGE antibody (11E6) was generated against the murine C2-domain of RAGE [29, 30] (Abbvie Laboratories, Deerfield, IL). Administration of 11E6 or control (Ig1 non-neutralizing) antibody was given as intraperitoneal injections. A TLR2 [[31]; CU-CPT22] and a TLR4 [[28]; compound 15] small molecule inhibitor were synthesized as previously described (Kindly provided by Hang Yin, University of Colorado). LPS was purchased by Sigma (St. Louis, MO). Non-oxidizable, chemotaxis-HMGB1 (all-thiol) and cytokine-HMGB1 (disulfide) was purchased from HMGBiotech (Milan, Italy; <1.0 endotoxin per 1 g of the protein by the LAL method), and was reconstituted in sterile 0.1% BSA/PBS.
2.4 Behavioral assessment
All rodents were habituated to testing chambers for at least two days. Rodents were randomly assigned to sham or injured test groups. All baseline testing occurred before and after TNI. The incidence of foot withdrawal in response to mechanical indentation of the plantar surface of each hindpaw was measured with a flat-tipped cylindrical probe (Von Frey filament) measuring 200 μm in diameter [6; 34]. Von Frey filaments capable of exerting forces of 10, 20, 40, 60, 80 and 120 mN with a uniform tip diameter was applied to a designated loci present on the plantar surface of the foot. During each test, the rodent was placed in a transparent plastic cage with a wire grid floor with ~1°–1 cm openings. The cage was elevated so that stimulation was applied to each hind foot from beneath the rodent. The filaments were applied in order of ascending force, and each filament was applied alternately to each foot. The duration of each stimulus was approximately 1 s and the inter-stimulus interval was approximately 10–15 s. The incidence of foot withdrawal was expressed as a percentage of the 6 applications of each stimulus and the percentage of withdrawals was then plotted as a function of force. The von Frey withdrawal threshold was defined as the force that evoked a minimum detectable withdrawal observed on 50% of the tests given at the same force level. For cases in which none of the specific filaments used evoked withdrawals on exactly 50% of the tests, linear interpolation was used to define the threshold. Pre-TNI baseline behavioral assessment was established in all rodents. The rats were tested pre dose and 1 h post dose on days 7, 14, 21 and 28. Optimum 11E6 dosing was established using TNI animals at day 28 using 1, 5, 10, and 15 mg/kg (data not shown). For drug studies, all behavioral assessments were performed by blinded pain assessors.
2.5 RNA isolation and RT-qPCR
Lumbar dorsal root ganglia (DRG; L4-L5) were dissected from adult female Sprague Dawley rats, frozen in liquid nitrogen, and maintained at −80°C until processed for RNA extraction. Total RNA was extracted from the samples using the RNeasy RNA extraction and purification kit (Qiagen). Single stranded cDNA was synthesized using reverse transcriptase (Bioline) with oligo-dT primers. Quantitative PCR was performed as previously described [10]. Briefly, resultant cDNA samples were amplified on an ABI PRISM 7900HTSequence Detection System (Applied Biosystems) using the reporter, SYBR Green. The PCR reaction was as follows: 1x, 50°C, 2 min; 1x, 95°C, 10 min; 45x, 95°C, 15 s, 60°C, 1 min; 1x, 25°C, hold. To check for DNA contamination, PCR was run using an L27 (ribosomal housekeeping gene) primer pair, whose PCR product crosses an intron. The mRNA level for each gene (x) relative to L27 mRNA (internal control) was calculated using the following equation where Ct refers to threshold cycles: mRNA (x%) = 2Ct (L27)-Ct(x) x 100.
2.6 Western blot analysis
Female animals were sacrificed and transcardially-perfused with saline and tissue was removed and frozen immediately with liquid nitrogen and stored at −80°C. The fresh frozen L4-L5 DRG tissue samples, ipsilateral to the injury, were homogenized in RIPA buffer with protease/phosphatase inhibitors and protein concentration was determined using the BCA protein assay (Thermoscientific). Samples (40 μg/lane) were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane. After incubation in 10% non-fat milk blocking solution overnight at 4°C, the membrane was incubated with rabbit anti-RAGE (1:1,000; Sigma–Aldrich) followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch). The membrane was probed again with a monoclonal anti-β actin antibody (1:5,000; Sigma– Aldrich, St. Louis, MO). Immunopositive bands were detected by enhanced chemiluminescence (ECL) and measured by a densitometric analysis (UnscanIt; Silk Scientific Inc., Orem, UT, USA).
2.7 Preparation of acutely dissociated dorsal root ganglion neurons
The L4-L6 DRGs were acutely dissociated using methods described by Ma and LaMotte [32]. Briefly, L4-L6 DRGs were removed from uninjured female animals. The DRGs were treated with collagenase A and collagenase D in HBSS for 20 min (1 mg/ml; Roche Applied Science, Indianapolis, IN), followed by treatment with papain (30 U/ml, Worthington Biochemical, Lakewood, NJ) in HBSS containing 0.5 mM EDTA and cysteine at 35 C. The cells were then dissociated by mechanical trituration in culture media containing 1 mg/ml bovine serum albumin and trypsin inhibitor (Worthington Biochemical, Lakewood, NJ). The culture media was Ham’s F-12 mixture, DMEM, supplemented with 10% fetal bovine serum, penicillin and streptomycin (100 μg/ml and 100 U/ml) and N2 (Life Technologies). The cells were then plated on coverslips coated with poly-L lysine and laminin (BD bioscience) and incubated for 2–3 h before more culture media was added to the wells. The cells were then allowed to sit undisturbed for 12–15 h to adhere at 37° C (with 5% CO2).
Following some in vitro experiments, cells were fixed for 10 minutes using 2% buffered paraformaldehyde, washed with saline and incubated with blocking buffer (3% BSA/3% horse serum/0.4% Triton-X; Fisher Scientific, Pittsburgh PA) for 1 hour, followed by overnight incubation with the goat polyclonal antisera generated against TLR4 L14 extracellular monoclonal antibody (1:200 dilution; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and rabbit anti-RAGE (1:1,000; Sigma–Aldrich) at 4° C. Additional monoclonal antibodies were used against CGRP (1;100; Rockland Inc) and IB4 conjugated directly to fluorescein isothiocyanate (IB4-FITC; Sigma-Aldrich). After primary incubation, secondary antibodies (anti-rabbit, anti-goat or anti-mouse conjugated to CY3 and CY2, made in donkey at 1:800; Jackson ImmunoResearch, West Grove, PA) were used to visualize cells. Coverslips were washed in PBS for 5 min each (×3), stained with Hoechst 33258 nuclear marker (Invitrogen Corporation, Carlsbad CA) and mounted with a PBS/glycerol solution onto glass slides.
2.8 Ca2+ imaging
The dissociated DRG cells were loaded with fura-2 AM (3 mM, Invitrogen Corp., Carlsbad, CA USA) for 25 minutes at room temperature in a balanced sterile salt solution (BSS) (NaCl (140 mM), Hepes (10 mM), CaCl2 (2 mM), MgCl2 (1 mM), glucose (10 mM), KCl (5 mM). The cells were rinsed with the BSS and mounted onto a chamber that was placed onto the inverted microscope. Intracellular calcium was measured by digital video microfluorometry with an intensified CCD camera coupled to a microscope and MetaFluor software (Molecular Devices Corp., Downington, PA USA). Cells were illuminated with a 150 W xenon arc lamp, and the excitation wavelengths of the fura-2 (340/380 nm) were selected by a filter changer. Sterile solution was applied to cells prior to HMGB1 application, any cells that responded to buffer alone were not used in neuronal responsive counts. HMGB1 (27 μM) was applied directly into the coverslip bathing solution. HMGB1 was purchased from R&D Systems (Minneapolis, MN, USA; <1.0 endotoxin per 1 g of the protein by the LAL method), and was reconstituted in sterile 0.1% BSA/PBS. 27 μM of HMGB1 was applied for calcium imaging. A period of three minutes was allowed for observation after the treatment was introduced to the bath. After HMGB1 application, LPS (1μg/mL) and capsaicin (3 nM) were added. Calcium imaging traces were analyzed by two independent analyzers and only responses that were in agreement between two individuals were used in the counts.
2.9 Electrophysiology
Sharp-electrode intracellular recordings were obtained from primary afferent neurons 12–18 hours after dissociation. Coverslips were transferred to a recording chamber that was mounted on the stage of an inverted microscope (Nikon Eclipse Ti, Nikon Instruments Inc., Melville, NY). The chamber was perfused with a bath solution containing (mM): NaCl 120, KCl 3, CaCl2 1, MgCl2 1, Hepes 10, Glucose 10, adjusted to pH 7.4 and osmolarity 300 mosM. The recordings were obtained at room temperature. Intracellular recording electrodes were fabricated from borosilicate glass (World Precision Instruments, Sarasota, FL) and pulled on a Flaming/Brown micropipette puller (P-98, Sutter Instruments, Novato, CA). Electrodes were filled with 1.0 M KCl (impedance: 40–80 MΩ) and positioned by a micromanipulator (Newport Corporation, Irvine, CA). −0.1 nA current injection was used to bridge-balance the electrode resistance. Diameter was measured by visual examination prior to electrode impalement using a calibrated eyepiece graticule within the microscope. The size of the soma to be recorded was classified according to its diameter as small (≤30 μm), medium (31–45 μm) and large (≥45 μm). Electrophysiological recordings were performed with continuous current-clamp in bridge mode using an AxoClamp-2B amplifier, stored digitally via Digidata 1322A interface, and analyzed offline with pClamp 9 software (Axon Instruments, Union City, CA). A neuron was accepted for study only when it exhibited a resting membrane potential (RMP) more negative than −45 mV. For each neuron isolated for study, a continuous recording was obtained for 1 minute without the delivery of any external stimulus. Neuronal excitability of small and medium diameter dissociated DRG sensory neurons was measured by injecting 1-s current pulses into the soma every 30 s. Current was adjusted in order to elicit 1–2 action potentials per current injection under baseline conditions. Non-responding cells were discarded. Following 3 control current injections, at-HMGB1 (27 μM) or LPS (1μg/mL) was applied to the coverslip and current injections continued every 30 s. Neuronal excitability was measured as number of action potentials elicited per current pulse before and immediately after addition of HMGB1 (15 and 45 s, respectively). If at-HMGB1 increased neuronal excitability, RAGE 11E6 (50 μM), TLR4 small molecule inhibitor (compound 15, 50 μM), or TLR2 inhibitor (CU-CPT22, 50 μM) was added to the bath to determine if at-HMGB1-elicited neuronal excitation could be reversed. If LPS increased neuronal excitability, RAGE 11E6 was added to the bath to determine if LPS-elicited neuronal excitation could be reversed. Excitation and blocking of excitation was analyzed within the first two minutes of application of RAGE 11E6, TLR2 or TLR4 inhibitors.
2.10 Statistics
GraphPad Software (LaJolla, CA) was used to determine the statistical significance. Results were expressed as mean ± SEM. When only two groups were compared, Student’s unpaired t test was used. Multiple comparisons were evaluated by Dunnett’s multiple comparison test after one-way ANOVA. p< 0.05 was considered to be statistically significant. GraphPad Software was used to determine the statistical significance of differences in calcium response among HMGB1 oxidation state groups using Chi-square test with Yates correction with p<0.05 set as statistical significance.
3. Results
3.1 Neuronal RAGE-immunoreactivity colocalizes with TLR4, the isolectin IB4 and calcitonin-gene related peptide (CGRP) expression
We previously reported that the TLR4 receptor is localized to both peptidergic and non-peptidergic sensory neurons within the dorsal root ganglia [27]. We examined cells in vitro for co-expression of RAGE and TLR4, IB4 and CGRP. Dissociated L4 and L5 DRGs removed from naïve animals and cultured for 16–20 hours, exhibited a near complete colocalization of TLR4 and RAGE in small and medium diameter neurons (Fig. 1B, C) with little to no TLR4 or RAGE immunoreactivity present in non-neuronal cells (Fig. 1A). Further examination revealed that there was some colocalization of the RAGE with the non-peptidergic marker of nociceptive neurons, IB4 (Fig. 1E, F), and the peptidergic nociceptive neuronal marker, CGRP (Fig. 1H, I).
Figure 1. RAGE, TLR4, IB4 and CGRP immunohistochemistry in primary sensory neuron cultures.

Images depicting corresponding images of the cell nucleus label DAPI (A), RAGE (B) and TLR4 (C). Note that the majority of RAGE immunoreactive neurons are also positive for TLR4 (yellow arrows indicates co-expression). Corresponding images of DAPI (D), RAGE (E), and the non-peptidergic marker of nociceptive neurons, IB4 (F). A minority of IB4 cells exhibit RAGE (yellow arrow indicates co-expression). Corresponding images of DAPI (G), RAGE (H), and the peptidergic marker of nociceptive neurons, CGRP (I). Note that there are numerous RAGE immunoreactive neurons which are also positive for CGRP. Scale bar is 50 μm.
3.2 Differential increases in neuronal [Ca2+]i by oxidation state specific HMGB1
To determine whether neuronal RAGE or TLR4 receptors are capable of modulating depolarization-evoked Ca2+ transients, we used ratiometric Ca2+ imaging to measure at-HMGB1 or ds-HMGB1 responses in neurons isolated from rat lumbar DRG. As many LPS-sensitive sensory neurons are activated by capsaicin [27], we also compared the actions of at-HMGB1 and ds-HMGB1 in LPS-sensitive nociceptive and non-nociceptive sensory neurons. Virtually all responsive neurons were small (≤30 μm) to medium diameter cells (31–45 μm). Exposure to media containing at- or ds-HMGB1 at a 27μM concentration for 3 minutes was used as a maximal stimulus. This treatment produced a total of 14 of 96 cells that were responsive to at-HMGB1, of which only 6 cells responded to both at-HMGB1 and LPS (Table 1). Upwards of 70.1% of at-HMGB1 responsive cells were nociceptive (capsaicin-sensitive) (Table 2). These data indicate that many at-HMGB1 responses in vitro were likely mediated by the RAGE receptor.
Table 1.
Differential increases in neuronal [Ca2+]I by oxidation specific HMGB1
| Percent Total Neurons | Percent Responsive to LPS | |
|---|---|---|
| at-HMGB1 | 14.58% (14/96) | 43% (6/14) |
| ds-HMGB1 | 7.55% (8/106) | 100% (8/8) |
Table 2.
Population of nociceptive neurons via capsaicin sensitivity responsive to HMGB1 isoforms
| N = 217 | Percent Responsive to capsaicin |
|---|---|
| at-HMGB1 | 70.1% |
| ds-HMGB1 | 42% |
When sensory neurons were subjected to ds-HMGB1, considerably fewer cells exhibited calcium transients (7.55%; Table 1). Though there was a decrease in the percentage of total responsive cells, 100% of the disulfide HMGB1-responsive neurons responded to LPS (Table 1) and only 42% of the assayed cells were sensitive to capsaicin (Table 2). Together, these results support the functional expression of both RAGE and TLR4 in nociceptive and non-nociceptive DRG neurons in vitro.
3.2 All-thiol HMGB1-elicited increase in sensory neuron excitation is dependent on RAGE and not TLR4
HMGB1-induced neuronal hyperexcitability has been recently described in rodent models of cerebral ischemia, epilepsy [11, 33] and in DRG sensory neurons in vitro [13]. Though it is now known that oxidation state-dependent HMGB1 interactions signal through specific receptor types (RAGE receptor or TLR4 receptor), it is largely unknown what receptor is responsible for neuronal excitation [16, 17, 34]. To determine the identity of the functional receptor by which oxidation state specific forms of HMGB1 can induce changes in sensory neuron excitability, we examined cellular responses using current clamp in the presence of at-HMGB1. If the cells responded to at-HMGB1, a small molecule inhibitor for TLR4 and TLR2, or the RAGE 11E6 antibody was administered followed by repeated current pulses. Due to the limited numbers of cells that were responsive to ds-HMGB1 (see Table 1), we substituted this form of HMGB1 for the known agonist of TLR4, LPS. Administration of the toll receptor antagonists or RAGE 11E6 alone failed to alter resting membrane potentials (data not shown).
Repeated current pulses in the presence of at-HMGB1 produced a significant increase in the excitability of small diameter sensory neurons when compared to baseline levels (1.4 ± 0.2 action potentials (APs) for control vs. 5.7 ± 0.4 APs for at-HMGB1) (Fig. 2A). Bath exposure to at-HMGB1 in combination with the TLR4 small molecule inhibitor (compound 15) and repeated current pulses failed to alter repeated current pulse induced APs (5.1 ± 0.4 APs; n=5; ANOVA, interaction F(3,14) = 52.60, p<0.0001; Bonferroni’s multiple comparisons, p>0.05 for at-HMGB1 vs. at-HMGB1 + Comp.15) (Fig. 2A, B). Following exposure to at- HMGB1 plus RAGE antibody 11E6, we observed a statistically significant decrease in the number of APs when compared with at-HMGB1 alone (1.2 ± 0.1 APs for control vs. 6.8 ± 1.3 APs for at-HMGB1 vs. 1.6 ± 0.2 APs for at-HMGB1 + RAGE Ab; n=5; ANOVA, interaction F(3,14) = 17.36, p < 0.001; Bonferroni’s multiple comparisons, p<0.001 for at-HMGB1 vs. at-HMGB1 + RAGE antibody, 11E6) (Fig 2. C, D). The consequences of at-HMGB1 neuronal exposure in the presence of TLR2 inhibitor (CU-CPT22) were similar to those observed with Compound 15 (1.1 ± 0.1 APs for control vs. 4.3 ± 0.4 APs for at-HMGB1 vs. 3.7 ± 0.2 APs for at-HMGB1 + CU-CPT22; n=5; ANOVA, interaction F(3,14) = 32.43, p<0.0001; Bonferroni’s multiple comparisons, p>0.05 for at-HMGB1 versus at-HMGB1 + CU-CPT22) (Fig. 2E, F).
Figure 2. RAGE neutralizing antibody, but not the TLR4 small molecule inhibitor, Compound 15, suppresses at-HMGB1-dependent neural excitation in acutely dissociated sensory neurons.

Current clamp recordings were performed on small-to-medium (>30 μm – >40 μm) diameter lumbar 4–5 DRG neurons from naive rats. Firing of 1–2 action potentials (APs) was elicited by a 1 second depolarizing current injection (ranging from 0.1 to 2.0 nA depending on the cell) every 30 seconds. Representative recordings demonstrating that application of at-HMGB1 (27μM) increases the number of elicited action potentials in DRG sensory neurons is not reversed by TLR4 small molecule inhibitor (compound 15) (A) or a TLR2 inhibitor (CU-CPT22) (E). In contrast, exposure to RAGE antibody effectively suppresses at-HMGB1-dependent action potential (C). Group data showing that compound 15 (B) and CU-CPT22 (F) do not reverse at-HMGB1-elicited increase in DRG neuron action potential firing while RAGE Ab does reverse increased excitation (D). Representative recording demonstrating that application of LPS (1 μg/ml) increases the number of elicited action potentials in DRG sensory neurons which is unaffected by RAGE antibody (G). In contrast, the neuronal effects of LPS can be suppressed with compound 15 (I). Group data showing that compound 15 (J), but not RAGE Ab (H), reverses LPS-elicited increase in DRG neuron action potential firing. (Ligand treatment compared to receptor inhibitor; *p < 0.05)
3.3 LPS-elicited increase in sensory neuron excitation is not dependent on RAGE
Given the ability of LPS to elicit an inward current in sensory neurons [25–27], we also tested whether the LPS-induced neuronal responses were dependent on TLR4 or RAGE. Representative recordings and grouped data demonstrate that the excitability of sensory neurons was significantly increased by LPS when compared with control levels (Fig. 2G, I). We then utilized the RAGE 11E6 antibody to discern the degree to which LPS-induced neuron response was dependent on the RAGE receptor. RAGE 11E6 did not alter endotoxin-induced APs (1.2 ± 0.1 APs for control vs. 4.3 ± 0.4 APs for LPS vs. 4.9 ± 0.4 APs for LPS + RAGE 11E6; n=5; ANOVA, interaction F(3,11) = 32.65, p < 0.0001; Bonferroni’s multiple comparisons, p > 0.05 for LPS vs. LPS + RAGE 11E6) (Fig. 2G, H).
A previously reported small molecule inhibitor of TLR4, Compound 15, was then used as a chemical probe to further investigate the molecular mechanism of LPS-induced neuron response [27, 28]. Similar to our previous observations, Compound 15 completely blocked the increased excitability of LPS (1.2 ± 0.1 APs for control vs. 4.9 ± 0.8 APs for LPS vs. 1.7 ± 0.2 APs for LPS + Comp. 15; n=5; ANOVA, interaction F(3,14) = 29.95, p < 0.0001; Bonferroni’s multiple comparisons, p < 0.001 for LPS vs. LPS + Comp. 15) (Fig. 2I, J). In contrast, Compound 15 demonstrated no effect upon blocking the increased neuronal hyperexcitability induced by HMGB1 treatment (Fig. 2A, B).
3.4 RAGE expression in the Lumbar DRG following Tibial Nerve Injury
Previous findings suggest that HMGB1 may be important for neuropathic pain behavior in rodents [14] and that HMGB1 can be released for extended periods of time following peripheral nerve injury (Feldman et al., 2012). Based on the kinetic breakdown of at-HMGB1 in different oxidative environments, it is likely that the neuronal RAGE receptor may be associated with nociceptive behavior in the rodent. In order to better understand the potential influence of RAGE in rodent neuropathic pain models, we studied changes in expression of RAGE and other genes known to be modified following peripheral nerve injury including the α2β1 auxiliary subunit of the voltage-dependent calcium channel (CACNA2D1) and the voltage-gated sodium channel isoform 1.8 (NaV1.8) SCN10A. We initially quantified the mRNA level of individual genes in the DRG using quantitative real time PCR. Although CACNA2D1 mRNA appeared to increase from naïve levels to post-injury day 14 and 28, the data was not significantly different (Fig. 3A; n=4, ANOVA, F= 2.51; p> 0.05). SCN10A mRNA level comparing naive control tissue with post-injury day 14 and 28 also lacked a statistically different level of significance (Fig. 3B; n=4, ANOVA, F= 0.62; p> 0.05). RAGE expression was strongly upregulated by PID 28 when compared to both PID 14 and naïve control tissue (Fig. 3C; n=4, ANOVA, F= 8.41; p< 0.05). Given the pronounced effect of TNI on RAGE mRNA expression within the L4-L6 DRGs at PID28, we next compared the TNI-induced changes on RAGE protein expression levels in L4-L6 DRGs. Lumbar DRG derived from TNI rodents exhibited a 3-fold increase in RAGE protein levels at PID28 (Fig. 4; n=6, ANOVA, F= 12.93; p< 0.05).
Figure 3. Tibal Nerve Injury (TNI) alters the expression of neuronal transcripts in dorsal root ganglion (DRG) derived from TNI rats.
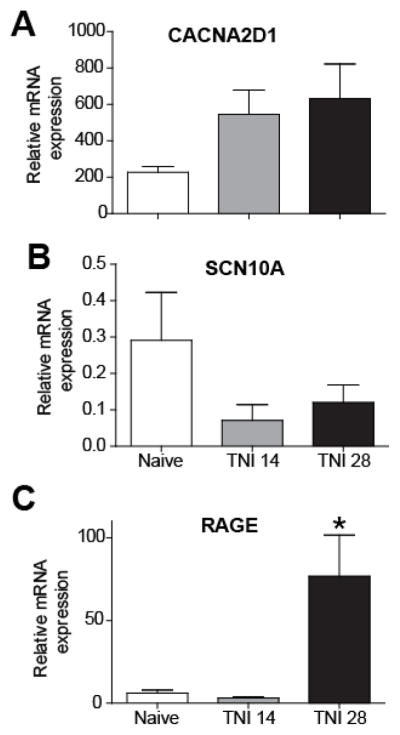
(A–D) RT-PCR analysis showing the mRNA expression profile of CaV alpha2delta1 (A) NaV1.8 (B) and RAGE (*p< 0.05) (C) at different time points following TNI; post injury day (PID) 14 and PID 28 (n=3). RT-PCR data were analyzed using the Ct method and mRNA expression levels are expressed relative to L27-ribosomal housekeeping gene.
Figure 4. RAGE protein expression following TNI at post injury day 28.

Immunoblot of RAGE in L4/5 DRGs from naïve, sham injured and TNI ipsilateral to the injury at [PID] 28 (n=3, sham versus injury; *p< 0.05). Actin was used as a loading control to which samples were normalized.
3.5 Effects of RAGE 11E6 on pain hypersensitivity in the TNI model of neuropathic pain
To determine the degree to which neutralization of RAGE contributes to chronic pain hypersensitivity induced by nerve injury, we performed nociceptive behavioral tests in the presence of a RAGE neutralizing antibody. TNI significantly reduced the pressure and tactile withdrawal thresholds of the hind paw on the ipsilateral side (Fig. 5; n=3, 70.54 ± 1.37 mN; p>0.05) and a single systemic injection of RAGE 11E6 (10 mg/kg) had no effect on uninjured rodents (data not shown). Systemic administration of RAGE 11E6 failed to affect tactile hyperalgesia when administered at 7, 14, or 21 days. However, tactile withdrawal thresholds of the hind paw ipsilateral to TNI at PID28 were transiently returned to baseline levels by 4 hours (Fig. 5; n=6–8, 58.33 ± 2.21 mN, ANOVA; p< 0.01). The finding that RAGE 11E6 did not elevate tactile hypersensitive levels above the pre-injury state shows that the animals ability to detect noxious tactile stimulus was not altered by the use of the antibody. Similar observations of RAGE 11E6 behavioral reversal in age-matched male rodents subjected to TNI was also observed (data not shown; n=6–8 per post injury time point).
Figure 5. Decreased tactile hyperalgesia following intraperitoneal injection of a neutralizing RAGE antibody in tibial nerve-injured rats at Day 28.
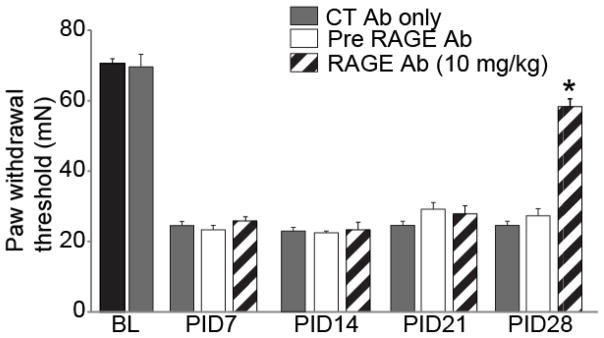
Paw withdrawal threshold (PWT) in rodents subjected to TNI (n = 6–8, white bars) were significantly reduced when compared with pre-TNI thresholds (n = 6–8, black bar) for at least 28 days. Administration of control, non-neutralizing antibody did not alter PWT at baseline or after TNI at 7, 14, 21 or 28 days (n = 6–8, CT Ab; gray bars). A humanized monoclonal antibody to RAGE (RAGE Ab; 10 mg/kg body weight; n = 6–8 each) was administered intraperitoneally and ipsilateral PWT was assessed using the von Frey filament test. Behavior was tested at 1 h post injection and again at 4 hours (data not shown). A single injection of RAGE Ab did not produce a change in PWT at TNI PID 7, 14, and 21 (striped bar) that differed from pre-RAGE Ab (white bars). At TNI PID 28 a single injection of RAGE Ab successfully reversed TNI decreases in PWT when compared with pre-RAGE Ab treatment (*p< 0.05).
4. Discussion
Our study provides evidence that the ligand at-HMGB1 acting through RAGE is a critical regulator of nociceptive signaling and sensitization in vitro. Moreover, as ongoing pain following peripheral nerve injury is thought to be maintained in part by activity in sensitized primary afferent neurons and could be influenced by an injury-induced upregulation of de novo receptors [35], latent upregulation of RAGE in the DRG corresponds with the ability of a monoclonal antibody against RAGE to modulate neuropathic pain-related behavior in the rat.
4.1 HMGB1 receptor signaling in the nervous system
There is growing evidence that the presence of cytosolic HMGB1 in the nervous system following insult contributes to neuropathologic neuronal excitability, including epilepsy, neuropathic pain and migraine [11, 13, 36]. A number of pain studies have demonstrated that the perisciatic or intrathecal administration of HMGB1 can produce rapid and transient mechanical and thermal hyperalgesia in rodents [14, 20, 21]. However, given the possibility that either of the two isoforms of HMGB1 may be present after injury to the nervous system [37], there is little knowledge regarding which of the cogent HMGB1 receptors, RAGE and TLRs, contribute to changes in neuropathological conditions. Although sensory neurons exhibit both functional TLR4 and RAGE receptors, our studies show that RAGE 11E6 completely inhibits at-HMGB1-dependent neuronal excitability in vitro. In contrast, there is no evidence to suggest that at-HMGB1 activates neuronal TLR4, considering a small molecule inhibitor of TLR4 failed to reverse at- HGMB1 elicited neuronal excitability in vitro.
4.2 RAGE signaling in the nervous system
HMGB1 acting through RAGE was initially implicated in neurite outgrowth in embryonic neurons and migration of numerous immune and non-immune cells types during states of inflammation [38–41]. The relevance of HMGB1/RAGE to inflammation is relatively well described for the pathogenesis of diabetes, cardiovascular disease and various cancers [42]. Whether HMGB1 activation of neuronal RAGE and the resultant increase in neuronal excitability during or following nerve injury-induced inflammation, represents a neuropathological consequence, is generally unknown. Initial reports in mice suggest that pharmacological and genetic blockade of RAGE impairs both innate immune response and nerve regeneration following sciatic nerve crush [43, 44]. Though the degree to which this injury altered tactile hyperalgesic behavior was not tested, the potential effects may not have manifested measurable changes given that both the nerve conduction velocities and gait analysis were tested for only three weeks post-nerve injury while the behavioral effects of increased RAGE expression in the rat DRG following peripheral nerve injury could only be adequately assayed at four weeks. A mechanism that might account for this latent change in RAGE could be due to a HMGB1 feed forward mechanism that exists in some immune cells and could serve to increase the levels of RAGE ligands and subsequently increased RAGE expression [45].
4.3 Oxidation State dependent HMGB1 signaling in the nervous system
Functional delays in the expression of RAGE in the affected DRG and the latent ability of RAGE neutralization to reverse tactile behavior in the rodent may also be dependent on changes in the state of HMGB1. It is well known that HMGB1 may form a complex with other molecules to enhance proinflammatory responses, including LPS, IL-1, bacterial DNA, CXCL12, and viral RNA [46–48]. The state-dependent characteristics that contribute to protein/receptor interaction appear to depend on the molecular properties of three cysteine residues; the redox state of cysteine (C) 106, and a disulfide bond between C23 and C45 [49, 50]. For example, during active inflammation the predominant form of HMGB1 is C106 thiol (all-thiol) and disulfide bond C23–45 (disulfide-HMGB1); however, when inflammation begins to subside, HMGB1 terminally oxidizes its cysteine residues and greatly diminishes biological activity (oxidized-HMGB1) [51–53]. Moreover, an oxidizing environment following inflammation or injury may promote HMGB1 cytokine activity [54]. These changes in HMGB1 conformation over time combined with the feed forward expression mechanisms that exist may account for the delayed influence observed in this neuropathic pain model.
Several published works have shown the importance of HMGB1 in the propagation of pain pathology, and when combined with the duality of function of the HMGB1 molecule, the change of the expression of the RAGE receptor as time progresses in vivo, as well as the efficacy of the antibody against the RAGE receptor with pain reversal, the conclusion can be drawn that RAGE and its signaling pathway are integral to pain pathology at later stages. Results of further study regarding the RAGE signaling axis may uncover targets for novel therapeutics and possibly have direct impact upon current clinical practice, through the discovery of potential biomarkers for pathologies of chronic inflammation, and through better understanding of the mechanisms of inflammation as they relate to disease. RAGE could possibly be a lynchpin to understanding the complete pathway leading to the pathogenesis of neuropathic pain.
Acknowledgments
This work was supported by the Indiana Spinal Cord & Brain Injury Research Grant (TL1-TR001107; FAW); NIH (DK100905 to FAW); the National Scientist Development from the American Heart Association (SDG5280023 to RK); the Neurofibromatosis New Investigator Award from the DOD/CDMRP (NF1000099 to RK). Additional support for YMA as an Indiana CTSI Predoctoral trainee was provided by UL1 TR001108, NIH/NCATS (A. Shekhar, PI).
Footnotes
The authors declare no conflicts of interest.
Dr. Khanna is presently located at the Department of Pharmacology, College of Medicine, The University of Arizona, Tucson, AZ.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Yohance M. Allette, Medical Science Training Program, Department of Anatomy, Indiana University School of Medicine, Indianapolis, IN.
Michael R. Due, Department of Anesthesia, Indiana University School of Medicine, Indianapolis, IN.
Sarah M. Wilson, Medical Neuroscience Graduate Program, Paul and Carole Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, IN
Polina Feldman, Medical Neuroscience Graduate Program, Paul and Carole Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, IN.
Matthew S. Ripsch, Department of Anesthesia, Indiana University School of Medicine, Indianapolis, IN
Rajesh Khanna, Program in Medical Neurosciences, Paul and Carole Stark Neurosciences Research Institute; Departments of Pharmacology and Toxicology and Biochemistry and Molecular Biology.
Fletcher A. White, Program in Medical Neurosciences, Paul and Carole Stark Neurosciences Research Institute; Department of Anesthesia, Indiana University School of Medicine, Indianapolis, IN
References
- 1.Calvo M, Dawes JM, Bennett DL. The role of the immune system in the generation of neuropathic pain. Lancet Neurol. 2012;11:629–42. doi: 10.1016/S1474-4422(12)70134-5. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 3.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bianchi ME, Falciola L, Ferrari S, Lilley DM. The DNA binding site of HMG1 protein is composed of two similar segments (HMG boxes), both of which have counterparts in other eukaryotic regulatory proteins. EMBO J. 1992;11:1055–63. doi: 10.1002/j.1460-2075.1992.tb05144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Czura CJ, Wang H, Tracey KJ. Dual roles for HMGB1: DNA binding and cytokine. J Endotoxin Res. 2001;7:315–21. doi: 10.1177/09680519010070041401. [DOI] [PubMed] [Google Scholar]
- 6.Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P. HMGB1: guiding immunity from within. Trends Immunol. 2005;26:381–7. doi: 10.1016/j.it.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Giese K, Cox J, Grosschedl R. The HMG domain of lymphoid enhancer factor 1 bends DNA and facilitates assembly of functional nucleoprotein structures. Cell. 1992;69:185–95. doi: 10.1016/0092-8674(92)90129-z. [DOI] [PubMed] [Google Scholar]
- 8.Parkkinen J, Raulo E, Merenmies J, Nolo R, Kajander EO, Baumann M, et al. Amphoterin, the 30-kDa protein in a family of HMG1-type polypeptides. Enhanced expression in transformed cells, leading edge localization, and interactions with plasminogen activation. J Biol Chem. 1993;268:19726–38. [PubMed] [Google Scholar]
- 9.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–61. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 10.Ulloa L, Tracey KJ. The “cytokine profile”: a code for sepsis. Trends Mol Med. 2005;11:56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–9. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Takahashi HK, Liu K, Wake H, Liu R, Maruo T, et al. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke. 2011;42:1420–8. doi: 10.1161/STROKEAHA.110.598334. [DOI] [PubMed] [Google Scholar]
- 13.Feldman P, Due MR, Ripsch MS, Khanna R, White FA. The persistent release of HMGB1 contributes to tactile hyperalgesia in a rodent model of neuropathic pain. J Neuroinflammation. 2012;9:180. doi: 10.1186/1742-2094-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibasaki M, Sasaki M, Miura M, Mizukoshi K, Ueno H, Hashimoto S, et al. Induction of high mobility group box-1 in dorsal root ganglion contributes to pain hypersensitivity after peripheral nerve injury. Pain. 2010;149:514–21. doi: 10.1016/j.pain.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 15.Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, Rauvala H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002;62:4805–11. [PubMed] [Google Scholar]
- 16.Venereau E, Schiraldi M, Uguccioni M, Bianchi ME. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol Immunol. 2012 doi: 10.1016/j.molimm.2012.10.037. [DOI] [PubMed] [Google Scholar]
- 17.Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1) Mol Med. 2012;18:250–9. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iori V, Maroso M, Rizzi M, Iyer AM, Vertemara R, Carli M, et al. Receptor for Advanced Glycation Endproducts is upregulated in temporal lobe epilepsy and contributes to experimental seizures. Neurobiol Dis. 2013;58:102–14. doi: 10.1016/j.nbd.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Chacur M, Milligan ED, Gazda LS, Armstrong C, Wang H, Tracey KJ, et al. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain. 2001;94:231–44. doi: 10.1016/S0304-3959(01)00354-2. [DOI] [PubMed] [Google Scholar]
- 21.O’Connor KA, Hansen MK, Rachal Pugh C, Deak MM, Biedenkapp JC, Milligan ED, et al. Further characterization of high mobility group box 1 (HMGB1) as a proinflammatory cytokine: central nervous system effects. Cytokine. 2003;24:254–65. doi: 10.1016/j.cyto.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Otoshi K, Kikuchi S, Kato K, Sekiguchi M, Konno S. Anti-HMGB1 neutralization antibody improves pain-related behavior induced by application of autologous nucleus pulposus onto nerve roots in rats. Spine (Phila Pa 1976) 2011;36:E692–8. doi: 10.1097/BRS.0b013e3181ecd675. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura Y, Morioka N, Abe H, Zhang FF, Hisaoka-Nakashima K, Liu K, et al. Neuropathic pain in rats with a partial sciatic nerve ligation is alleviated by intravenous injection of monoclonal antibody to high mobility group box-1. PLoS One. 2013;8:e73640. doi: 10.1371/journal.pone.0073640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hua XY, Chen P, Fox A, Myers RR. Involvement of cytokines in lipopolysaccharide-induced facilitation of CGRP release from capsaicin-sensitive nerves in the trachea: studies with interleukin-1beta and tumor necrosis factor-alpha. J Neurosci. 1996;16:4742–8. doi: 10.1523/JNEUROSCI.16-15-04742.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS Sensitizes TRPV1 via Activation of TLR4 in Trigeminal Sensory Neurons. J Dent Res. 2011 Jun;90(6):759–64. doi: 10.1177/0022034511400225. [DOI] [PubMed] [Google Scholar]
- 26.Ochoa-Cortes F, Ramos-Lomas T, Miranda-Morales M, Spreadbury I, Ibeakanma C, Barajas-Lopez C, et al. Bacterial cell products signal to mouse colonic nociceptive dorsal root ganglia neurons. Am J Physiol Gastrointest Liver Physiol. 2010;299:G723–32. doi: 10.1152/ajpgi.00494.2009. [DOI] [PubMed] [Google Scholar]
- 27.Due MR, Piekarz AD, Wilson N, Feldman P, Ripsch MS, Chavez S, et al. Neuroexcitatory effects of morphine-3-glucuronide are dependent on Toll-like receptor 4 signaling. J Neuroinflammation. 2012;9:200. doi: 10.1186/1742-2094-9-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bevan DE, Martinko AJ, Loram LC, Stahl JA, Taylor FR, Joshee S, et al. Selection, Preparation, and Evaluation of Small-Molecule Inhibitors of Toll-Like Receptor 4. ACS Med Chem Lett. 2010;1:194–8. doi: 10.1021/ml100041f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo J, Hsieh C, Wu Z, DiGiammarino EL, Luo F, Fox GB, et al. Office USPaT, editor. Antibodies to receptor of advanced glycation end products (RAGE) and uses thereof. United States: Abbott Laboratories; Abbott Park, IL: Abbott GMbH & Co, KG, Wiesbaden-Delkenheim, DE; 2012. [Google Scholar]
- 30.Strakhova MI, Desiree-Brderson J. Office USPaT, editor. Antibodies to receptor of advanced glycation end products (RAGE) and uses thereof. United States: Abbvie Inc, North Chicago, IL; 2013. [Google Scholar]
- 31.Cheng K, Wang X, Zhang S, Yin H. Discovery of Small-Molecule Inhibitors of the TLR1/TLR2 Complex. Angew Chem Int Ed Engl. 2012 doi: 10.1002/anie.201204910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma C, LaMotte RH. Enhanced excitability of dissociated primary sensory neurons after chronic compression of the dorsal root ganglion in the rat. Pain. 2005;113:106–12. doi: 10.1016/j.pain.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Qiu J, Xu J, Zheng Y, Wei Y, Zhu X, Lo EH, et al. High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke. 2010;41:2077–82. doi: 10.1161/STROKEAHA.110.590463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–7. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White FA, Bhangoo SK, Miller RJ. Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov. 2005;4:834–44. doi: 10.1038/nrd1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Kocak E, Sen ZD, et al. Spreading depression triggers headache by activating neuronal Panx1 channels. Science. 2013;339:1092–5. doi: 10.1126/science.1231897. [DOI] [PubMed] [Google Scholar]
- 37.Zandarashvili L, Sahu D, Lee K, Lee YS, Singh P, Rajarathnam K, et al. Real-time kinetics of high-mobility group box 1 (HMGB1) oxidation in extracellular fluids studied by in situ protein NMR spectroscopy. J Biol Chem. 2013;288:11621–7. doi: 10.1074/jbc.M113.449942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rauvala H, Pihlaskari R. Isolation and some characteristics of an adhesive factor of brain that enhances neurite outgrowth in central neurons. J Biol Chem. 1987;262:16625–35. [PubMed] [Google Scholar]
- 39.Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, et al. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104:1174–82. doi: 10.1182/blood-2003-10-3536. [DOI] [PubMed] [Google Scholar]
- 40.Merenmies J, Pihlaskari R, Laitinen J, Wartiovaara J, Rauvala H. 30-kDa heparin-binding protein of brain (amphoterin) involved in neurite outgrowth. Amino acid sequence and localization in the filopodia of the advancing plasma membrane. J Biol Chem. 1991;266:16722–9. [PubMed] [Google Scholar]
- 41.Fages C, Nolo R, Huttunen HJ, Eskelinen E, Rauvala H. Regulation of cell migration by amphoterin. J Cell Sci. 2000;113 ( Pt 4):611–20. doi: 10.1242/jcs.113.4.611. [DOI] [PubMed] [Google Scholar]
- 42.Rouhiainen A, Kuja-Panula J, Tumova S, Rauvala H. RAGE-Mediated Cell Signaling. Methods Mol Biol. 2013;963:239–63. doi: 10.1007/978-1-62703-230-8_15. [DOI] [PubMed] [Google Scholar]
- 43.Rong LL, Trojaborg W, Qu W, Kostov K, Yan SD, Gooch C, et al. Antagonism of RAGE suppresses peripheral nerve regeneration. FASEB J. 2004;18:1812–7. doi: 10.1096/fj.04-1899com. [DOI] [PubMed] [Google Scholar]
- 44.Rong LL, Yan SF, Wendt T, Hans D, Pachydaki S, Bucciarelli LG, et al. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 2004;18:1818–25. doi: 10.1096/fj.04-1900com. [DOI] [PubMed] [Google Scholar]
- 45.Akirav EM, Preston-Hurlburt P, Garyu J, Henegariu O, Clynes R, Schmidt AM, et al. RAGE expression in human T cells: a link between environmental factors and adaptive immune responses. PLoS One. 2012;7:e34698. doi: 10.1371/journal.pone.0034698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sha Y, Zmijewski J, Xu Z, Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol. 2008;180:2531–7. doi: 10.4049/jimmunol.180.4.2531. [DOI] [PubMed] [Google Scholar]
- 47.Campana L, Bosurgi L, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 for stromal cell-derived factor-1/CXCL12-dependent migration of macrophages and dendritic cells. J Leukoc Biol. 2009;86:609–15. doi: 10.1189/jlb.0908576. [DOI] [PubMed] [Google Scholar]
- 48.Hreggvidsdottir HS, Lundberg AM, Aveberger AC, Klevenvall L, Andersson U, Harris HE. High mobility group box protein 1 (HMGB1)-partner molecule complexes enhance cytokine production by signaling through the partner molecule receptor. Mol Med. 2012;18:224–30. doi: 10.2119/molmed.2011.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312:3526–38. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 50.Sahu D, Debnath P, Takayama Y, Iwahara J. Redox properties of the A-domain of the HMGB1 protein. FEBS Lett. 2008;582:3973–8. doi: 10.1016/j.febslet.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Antoine DJ, Williams DP, Kipar A, Laverty H, Park BK. Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity. Mol Med. 2010;16:479–90. doi: 10.2119/molmed.2010.00126. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Vezzoli M, Castellani P, Campana L, Corna G, Bosurgi L, Manfredi AA, et al. Redox remodeling: a candidate regulator of HMGB1 function in injured skeletal muscle. Ann N Y Acad Sci. 2010;1209:83–90. doi: 10.1111/j.1749-6632.2010.05748.x. [DOI] [PubMed] [Google Scholar]
- 53.Yang QW, Lu FL, Zhou Y, Wang L, Zhong Q, Lin S, et al. HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. J Cereb Blood Flow Metab. 2011;31:593–605. doi: 10.1038/jcbfm.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vezzoli M, Castellani P, Corna G, Castiglioni A, Bosurgi L, Monno A, et al. High-mobility group box 1 release and redox regulation accompany regeneration and remodeling of skeletal muscle. Antioxid Redox Signal. 2011;15:2161–74. doi: 10.1089/ars.2010.3341. [DOI] [PubMed] [Google Scholar]