

Abstract
A new concept in the field of heart failure research points to a role of misfolded proteins, forming pre-amyloid oligomers (PAOs), in cardiac toxicity. This is largely based on few studies reporting the presence of PAOs, similar to those observed in neurodegenerative disease, in experimental models and human heart failure. As the majority of proteinopathies are sporadic in nature, protein post-translational modifications (PTMs) likely play a major role in this growing class of diseases. In fact, PTMs are known regulators of protein folding and of the formation of amyloid species in well-established proteinopathies. Proteomics has been instrumental in identifying both chemical and enzymatic PTMs, with a potential impact on protein mis-/folding. Here we provide the basics on how proteins fold along with a few examples of PTMs known to modulate protein misfolding and aggregation, with particular focus on the heart. Due to its innovative content and the growing awareness of the toxicity of misfolded proteins an “Alzheimer’s theory of heart failure” is timely. Moreover, the continuous innovations in proteomic technologies will help pinpoint PTMs that could contribute to the process. This nuptial between biology and technology could greatly assist in identifying biomarkers with increased specificity as well as more effective therapies.
1. Introduction
According to the American Heart Association there are over 5 million individuals suffering from heart failure (HF) in the US [1] and 60 million worldwide [2]. The World Alzheimer’s Report shows a similar prevalence with more than 5 million cases of Alzheimer’s Disease (AD) in the United States, and 35 million worldwide [3]. Genetic distribution, environmental modifiers and disease pathology also overlap in these diseases.
The majority of both HF [4] and AD cases [5] occur as sporadic events without a defined Mendelian, inherited profile and common genetic variants have been described in both illnesses [4, 6]. However, non-genetic stressors and environmental modifiers play an important role in the onset, development and severity of these diseases [5, 7, 8]. As an example risk factors such as hypercholesterolemia and metabolic syndrome predispose to both AD and HF [9–11].
The accumulation of amyloid deposits causing neuritis, neuronal cell death and cognitive impairment, is the hallmark of AD. Similarly, recent evidence reporting the presence of protein aggregates in human HF suggests that these two diseases may also share a common pathology [4, 12–14]. In recent years, due to the poor correlation between large, insoluble aggregates and the severity of the clinical manifestation in AD patients [15] there has been a shift in focus from the toxicity of large aggregates to that of smaller, potentially more insidious entities [16, 17]. These species, which are also referred to as pre-amyloid oligomers (PAOs), have also been found in the heart of small and large animal models as well as in human HF [4, 8, 13, 18]. Although, most of the mouse models showing cardiac accumulation of PAOs carry genetic mutations, amyloid-like oligomers were observed in a large animal model of dyssynchronous HF, in the absence of genetic mutations [8]. This study points to the role of post-translational modifications of proteins (PTMs), which may lead to their mis-/folding as a unifying mechanism underlying the pathogenesis of at least some forms of HF, and is independent from gene mutations [8].
The technological advances in protein biochemistry collectively referred to as “proteomics”, routinely allow researchers to screen for hundreds to thousands of PTMs and this has largely contributed to the ongoing effort of mapping protein PTMs. A concept which is becoming increasingly clear is that the number and variety of PTMs have been far underestimated and that these could represent the integrators of the dynamic adaptation of the phenotype (both healthy and diseased) to the environment [7]. The importance and contribution of PTMs to protein misfolding is gaining mounting attention also in the field of proteinopathies (reviewed by Nilsson and Abedini [19, 20]). Moreover, the number of studies addressing the role of PTMs in heart disease is also increasing [7]. In this review we will provide examples of the modifications that are known to impact on the formation of amyloid species and we will focus on those proteins and known PTMs that could be relevant for PAOs formation in the failing heart.
2. PTMs and Protein Misfolding
Protein folding is largely determined by a protein’s primary sequence. In fact, folding mainly occurs through a “trial and error” process where thermodynamics and chaperone proteins ultimately lead to the tertiary structure [21, 22]. Errors in this process occur normally and are exacerbated by cellular stressors and/or mutations in the protein sequence. Repair mechanisms are in place as part of the normal process of protein maturation as well as normal protein turnover, together with mechanisms for the removal of damaged proteins so that aminoacids can be recycled into functional ones and potentially toxic entities can be destroyed [23] (reviewed in the heart in [14, 24]).
Failure to correct mistakes in the folding of nascent polypeptides or in mature proteins as well as defects in their turnover (protein degradation) can lead to the accumulation of misfolded proteins. These in turn can generate “seeds” which, through a nucleation process lead to the formation of PAOs. Once oligomers are formed they can grow into fibrils, which are larger entities. The growth of fibrils is usually exponential and leads to the accumulation of large deposits which are visible under the electron microscope and which we refer to here as aggregates [20, 25]. Post-translational modifications are known to play a role at different stages in the formation of amyloid species (Figure 1).
Figure 1. Mechanisms of formation of amyloid species.
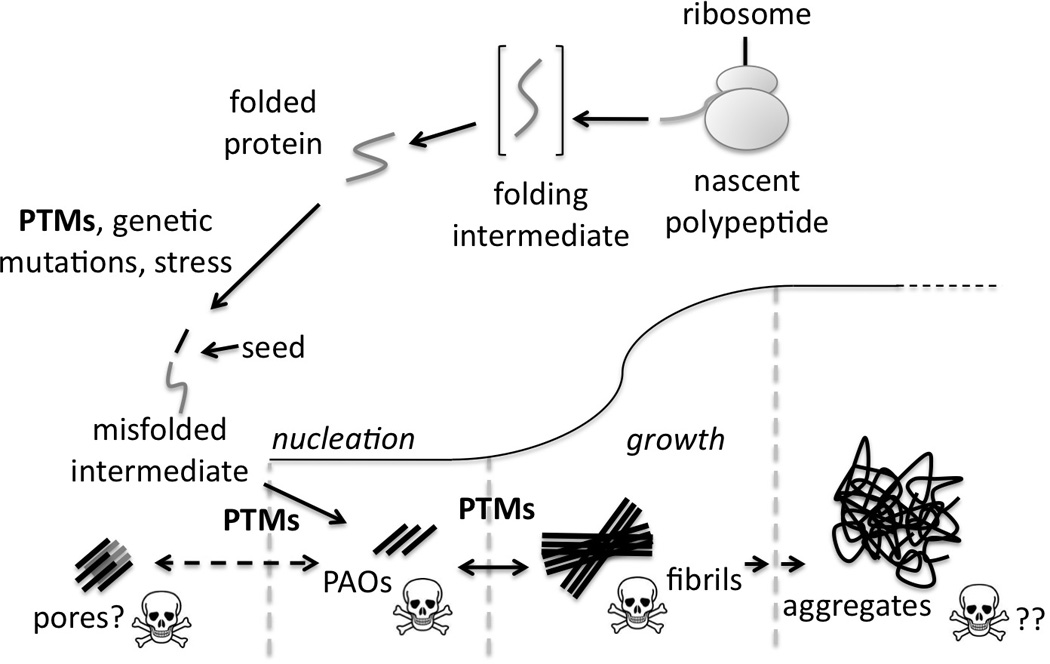
Role of PTMs (chemical and enzymatic), at different stages of amyloid formation. Proteolytic cleavage is a common PTM in many examples of protein misfolding. The resulting “seed” starts a nucleation process which leads to the formation of intermediate pre-amyloid oligomers and fibrils. These grow into large aggregates which are commonly referred to as amyloid. A recent acquisition in the field is that PAOs are the toxic species whereas larger aggregates could represent a by-product of cell detoxification.
Two excellent reviews have been written on the subject by Nilsson and Abedini [19, 20]. In this review we use examples which are relevant for heart failure from these two reviews and the current literature. Post-translational modifications can be catalyzed by enzymes or result from chemical reactions, which take place in the cell under certain conditions (e.g. oxidation).
Enzymatic PTMs are generally controlled, however, under stressed or diseased conditions this regulation can be ineffective, for instance due to the unbalanced activation of their relative upstream signaling pathways [8]. When enzymatic PTMs result in excessive or differential modifications they can impact on the propensity of protein to aggregate [20]. Examples of enzymatic PTMs playing a role in protein misfolding include, but are not limited to proteolytic cleavage, glycosilation, sulfation, phosphorylation and ubiquitination [19, 20, 26, 27].
Proteolytic cleavage is one of the enzymatic PTMs which have been linked extensively to protein misfolding and amyloid deposition in pathological conditions (such as in neurodegenerative diseases and aging [19, 20]). A classic example of the specificity of these events is provided by the amyloid-inducing peptide generated by γ-secretase in AD. A sequence of proteolytic cleavages occurring to the amyloid precursor protein (APP) leads to the formation of Aβ peptides of different length. Notably, the last cleavage is catalyzed by γ-secretase, which can cleave at different sites (38, 40 and 42). Of these, the Aβ42 product showed the highest propensity to form toxic PAOs in vivo [28, 29]. This, along with other examples, points to a role of specific PTMs in triggering amyloid formation [20]. In the case of proteolytic cleavage this is not entirely surprising as evolution has carefully selected protein sequences in order to reduce the chance of misfolding [25]. However, this primary defense from misfolding may lose efficacy when part of the protein sequence is removed.
Pathological but discrete proteolytic processing has been implicated in the formation of the majority of known amyloid oligomers and fibrils found in a wide array of well-established proteinopathies which includes mutant forms of transthyretin T (TTR) in cardiac amylodosis [19]. A few examples of proteins forming amyloid upon proteolytic cleavage from Nilsson’s review are listed in Table 1.
Table 1.
Examples of known cleaved amyloid-forming precursors.
| Protein | Function/Disease |
|---|---|
| Pmel17 | Melanosome biogenesis (physiological) |
| Lithostathine | AD, pancreatic stones |
| Gelsolin (Asp 187 to Asn or Tyr) | Familial Amyloidosis Finnish type (FAF) |
| Cystatin C (Leu 68 to Gln) | Hereditary Cystatin C Amyloid Angiopathy (HCCAA) |
| ApoAIV/TTR | Cardiac Amyloidosis |
| ApoAI | Atherosclerothic plaques, and Familial Amyloid Polyneuropathy (FAP) Type III |
| ApoSAA | AA amyloid fibrils |
| α/β tubulin/BRI | Familial British (FBD) and Danish dementia |
| Β2Microglobulin | Long-term dialysis |
| TTR | Familial Amyloid Polyneuropathy (FAP) and senile systematic amyloidosis (SSA) |
| α-synuclein | Parkinson’s, Dementia with Lewy Bodies (DLB), AD, Multiple System Atrophy (MSA) and Amyotrophic Lateral Sclerosis (ALS) |
| Aβ | AD |
| PrPc | Creutzfeldt-Jakob |
| Huntingtin | Hungtinton’s |
| Glycoprotein B | HSV1/AD |
Adapted with permission from[19]
Notably, in a couple of the examples (Gelsolin and Cystatin C) genetic mutations make the protein more susceptible to proteolytic cleavage and the consequent formation of amyloid. Agnetti et al. recently proposed that proteolytic cleavage of the intermediate filament protein desmin, possibly modulated by phosphorylation, could lead to the formation of amyloid-like oligomers in experimental HF, in the absence of genetic mutations [8]. The role of chemical PTMs in this process needs to be addressed as increased levels of desmin conjugated with advanced glycation end-products (AGEs) have been observed in a mouse model of dilated cardiomyopathy [30].
Chemical or non-enzymatic PTMs are uncontrolled and have been mainly investigated for their impact on pharmaceutical protein preparations [19]. Among non-catalytic modifications a prominent role in amyloid formation is covered by Met oxidation and the addition of advanced glyaction end products to Lys and Arg residues. A remarkable difference with respect to enzymatic modifications is that chemical ones are likely to depend on a protein’s “age”. For instance, it has been suggested that deamidation of Asp and Gln can act as a molecular clock and that it may be used by the cell as a signal so that “old” proteins can be replaced [19, 31]. Notably, the study by Robinson and Robinson [31] proposing this theory represent one early example of the utility of mass spectrometry (MS) in addressing these aspects.
3. Functional Classes of Amyloid-Forming Proteins
Protein aggregation and the formation of PAOs and fibrils are common to many proteins regardless of the primary sequence, their structure and function. In fact sometime, as in naturally disordered proteins, misfolding is part of a protein’s biological function [25]. Surprisingly, some proteins, which have been implicated in the formation of amyloid in heavily studied proteinopathies, do not have a well-established physiological function. Theses include APP which generates Alzheimer’s Aβs and huntingtin in Huntington’s disease [19, 32].
Of the amyloid-forming proteins with a known physiological function few belong to the cytoskeleton or associate with it. This could be due to the fact that cytoskeletal proteins are more prone to oligomerize because of their function. Additionally, in the heart, they could be more prone to stress-induced misfolding due to their mechanical roles and cell-wide distribution. Lastly, cytoskeletal proteins have been pinpointed as major players in the pathological remodeling seen in heart failure by several authors (reviewed in [33]).
The cytoskeleton of metazoan cells is constituted by three distinct structures: 1) tubulin microtubules which are mainly involved in trafficking; 2) actin microfilaments which are classically involved in cell motility and 3) intermediate filaments which are constituted of different proteins depending on the cell-type and whose function is still object of debate [33, 34]. For each of these classes, which also include the ancillary proteins interacting with these structures, there are examples of differentially modified proteins forming amyloid species and aggregates in both the brain and the heart.
In the brain, the protein tau is a member of microtubule-associated proteins (MAPs) and its hyper-phosphorylated form is a well-established component of neurofibrillary tangles in AD (reviewed in [35]). A role for Tyr nitration was postulated in the formation of tau aggregates whereas a link between deamidation and isomerization of tau and the formation of aggregates has not been established although these PTMs have also been reported [19, 20]. In the heart other MAPs (such as MAPS4) are known to be differentially modified (e.g. phosphorylated) during pressure overload [36].
Gelsolin is an actin-binding protein and its mutant form (Asn187Tyr) has been implicated in the Finnish type of familial amyloidosis (FAF) with a cardiac involvement [37]. As mentioned, the aminoacidic substitution renders the protein more susceptible to proteolytic cleavage whereas the wild-type is resistant to it [19].
Intermediate filament proteins, in particular, desmin and its chaperon α-B-crystallin (CryAB), have been implicated in the formation of amyloid-like oligomers and aggregates in the heart by several studies. Mutations in both desmin and CryAB are known to precipitate a pathological phenotype characterized by skeletal and cardiac muscle involvements (reviewed by [38]). Interestingly, the first study showing PAOs deposition in human heart failure [4, 39] was centered on a murine model of desmin-related myopathy based on a mutation of CryAB (R120G [39]). Intermediate filaments are known to be heavily modified, and these PTMs often impact on their oligomerization (reviewed in [34]). Agnetti et al. reported the induction of modified desmin forms in a well-established in vitro model of cardiac hypertrophy and a concomitant increase of desmin-positive protein aggregates by filter assay, a test commonly used to quantify protein aggregates [40]. More recently the same group reported the increase of phosphorylated and cleaved desmin forms in both experimental and human heart failure. In the experimental (canine) model, desmin-positive amyloid-like oligomers were also increased in the failing phenotype, suggesting a possible relationship between desmin PTMs and aberrant assembly [8].
4. Mechanisms of Amyloid Toxicity
As mentioned amyloid aggregates are formed by misfolded protein units (monomers), which build into progressively larger structures (oligomers) to mature fibrillar aggregates (Figure 1). These can distribute differently in the affected tissues giving rise to variously named aggregates in different diseases, also depending on the location (e.g. intra- vs. extracellular). Examples include Lewy bodies in Parkinson’s disease, neurofibrillary tangles in Alzheimer’s, nuclear and cytoplasmic inclusions in Huntington’s [16]. Protein aggregates and PAOs have also been described in the intracellular and extracellular space in different forms of human HF [4, 12, 13].
As in many interdisciplinary subjects the terminology that is used in established fields (such as neuroscience) may not be readily accessible to researchers in other fields. This is why the term oligomer can generate confusion when used in the context of broad protein biochemistry or proteomics as protein oligomers refer to physiological complexes (quaternary structure). The key message is that the term amyloid describes proteins which aggregate in the form of insoluble fibrils as the result of protein misfolding. Amyloid fibrils are also characterized by periodic structures that exhibits affinity for dyes (such as Congo-red and Thyoflavin [41]) or conformational antibodies [42].
In a simplified view, monomers tend to organize into oligomers which have been also defined as amyloidogenic, amyloid-like or pre-amyloid [8, 43, 44]. According to many, this is the rate-limiting step and it is mainly due to the exposure of hydrophobic domains which are normally inaccessible in the folded protein but become available to interact with the environment (and other proteins), even transiently, when protein are misfolded [16]. The exposed hydrophobic domains act as a “trap” for other proteins during the nucleation phase. The step following oligomers formation is referred to as fibrillization, it is exponential and leads to the formation of fibrils. Like PAOs, these species are also characterized by abundance in β-sheets, which, in the case of fibrils tend to organize perpendicularly to the axis of the fibril [25]. As fibrils grow they tend to reorganize into amorphous large formations, which we refer to here as aggregates. These can be also formed by inherently disordered polypeptides [45].
A recent acquisition in the field of amyloid-related diseases is that PAOs, rather than large aggregates, may be responsible for cellular toxicity and several studies demonstrate how aggregates may constitute a way for the cell to protect itself from PAOs [17]. The collected evidence points to a mechanism of PAOs’ toxicity involving changes in Ca2+ homeostasis although the exact mechanism leading to Ca2+ perturbation in neurons and cardiomyocytes is still unknown [4, 13, 46]. Intriguingly, it was recently suggested that PAOs could rearrange into pores which may equilibrate with and perturb the activity of biological membranes [47]. However, in addition to this direct toxic effect of PAOs, an aggregate-independent mechanism linked to the loss in the function of the misfolded proteins coexist to define the ultimate disease phenotype [4, 48].
To date, in the field of heart failure, much emphasis has been placed on the mechanisms which prevent the accumulation of misfolded proteins or dispose of them. These include the interaction with molecular chaperones, degradation through the ubiquitin-proteasome system and autophagy [24, 49]. The role of PTMs in these processes is also crucial [23] and the ability of mapping them will likely increase our understanding of which of these pathways are more important for the heart and which can eventually be exploited pharmacologically. Furthermore, it is critical to understand the specific effects of PTMs on the mis-/folding of targeted proteins and how these contribute to the formation of potential toxic species. In fact, chemical modifications of proteins, which may play an important role in the formation of PAOs may not be predictable solely on the basis of biological mechanisms (such as signaling pathways). Therefore, unbiased approaches such as proteomics could be beneficial in tracking down both enzymatic and chemical PTMs.
5. Proteomics to Study Amyloid-Related PTMs
Thanks to the recent developments in mass spectrometry (MS) it has become increasingly clear that the extent of PTMs in biological systems has been largely underestimated in the pre-proteomic era. There are ≈400 PTMs listed in modern protein databases (such as Uniprot) and the quest for their detailed mapping is ongoing [50]. Indeed, a major contribution of proteomics to the development of modern sciences is represented by the revelation of the number and variety of PTMs decorating proteins in living organisms at any given time. As the list of PTMs reported in protein databases increases, more information can be used to dissect the intricate interplay underlying different pathophysiological mechanisms, including amyloid formation or protein toxicity. As such, it is safe to assume that the study of the contribution of PTMs in amyloid formation will greatly benefit from the continuous advances in proteomics.
Despite the emerging consensus on the mechanistic importance of these modifications in amyloid formation, the field of proteomics applied to the study of PTMs on protein mis/folding is still relatively young. Also, the technological advances in the field of proteomics have allowed the discovery of novel PTMs in very well-studied cardiac proteins [51]. Several protocols to study different PTMs have been optimized to date (reviewed in [7]). In fact, not all PTMs can be detected routinely in a mass spectrometer and in the majority of cases tailored approaches have to be implemented.
Three main issues hamper the application of MS to the study of misfolded proteins in the heart. The first is a general limitation with regard to the study of PTMs, which is hindered by their intrinsic liability and the low stoichiometry with respect to other modified forms [7]. As mentioned, one example of a chemical PTM that has been correlated with amyloid formation, for instance during aging, is the addition of advanced glycation end-products (AGEs) to Lys and Arg residues. Under conditions of oxidative stress and increased glucose availability (such as in heart failure), α-dicarbonil compounds (such as glyoxal) can be generated which covalently modify proteins. In humans, increased levels of AGEylated proteins have been reported in diabetic cardiomyopathy (reviewed in [52]). This is due to the oxidative environment and increased abundance of reducing sugars in diabetes. In fact, glycation, different from the enzymatically catalyzed glycosilation, is the product of a chemical reaction between reducing sugars and the amino groups present in Arg and Lys side chains as well as the N-terminus of proteins. The result of this reaction is a Schiff base, which after Amadori and several other chemical rearrangements lead to the formation of AGEylated proteins [20]. There is a number of reducing sugars which can generate AGEs which complicates the spectrum of peptide masses that can be detected in a mass spectrometer [53]. Interestingly, increased levels of circulating N(ε)-(carboxymethyl)lysine (CML) and pentosidine proteins (two common AGEs modifiers) were able to predict hospitalization and CML was independently associated with a higher risk of mortality in a large cohort of heart failure patients [54]. Using different instruments and set-up it is possible to accurately measure the presence of these adducts by MS, in vitro [53]. Due to the variety of AGEs moieties, a considerable optimization may be necessary, especially for complex sample mixtures. For this and other PTMs, strategies to enrich for the modified peptides, for instance by affinity capture, could be developed to address poor stoichiometry and chemical liability. Another PTM, which is relevant in protein misfolding is ubiquitination. This PTM is also traceable by MS due to a signature di-Gly di-peptide inducing an increase of 114.043 Da in the modified peptide [55]. The same signature is originated by neddylation, which may require further downstream validation [56]. These are only two examples of what is already feasible with respect to PTMs that have been linked to protein misfolding. Lastly, the recent reports of the regulatory phosphorylation of ubiquitin mediated by PINK1 in depolarized mitochondria may suggest an added level of complexity due to the interaction of layers of different PTMs [57].
A recent example of mass spectrometry applied to the study of PAOs’ PTMs addressed the role of protein cross-linking which can be catalyzed by transglutamminases. In this study Tay et al. used a targeted approach combined with in vitro experiments to predict the mass of several peptide fragments originated by cross-linked peptides [29]. The study shows that targeted approaches can be used to monitor specific PTMs which could be challenging to measure and monitor with proteome-wide approaches and underscore the importance of targeted approaches in the future of proteomics [58]. Alternative, emerging strategies which include top-down, catalyzed fragmentation, deuterium-exchange and others have also been used to infer about both PTMs and structural changes in amyloid-forming peptides [59, 60]. These approaches are likely to facilitate the discovery of novel functions for PTMs on protein mis-/folding.
The second issue with studying PTMs in misfolded protein is the access of proteolytic enzymes commonly used in bottom-up approaches [7] to the backbone of proteins which may be tightly packed into amyloid. Intriguingly, it has been suggested that there is a gradient of PTMs, which builds up along with the organization of misfolded protein from oligomers to higher structural states (such as fibrils and aggregates) [19]. This could represent an advantage as this gradient is likely related to the temporal scale of the aggregation process. By focusing on the “layers” of modifications that are differentially represented from the surface towards the core of one specific amyloid form, in theory, one could infer about the temporal and perhaps causal role of different PTMs in the different stages of the process. In fact, it is reasonable to assume that PTMs which trigger aberrant oligomerization are buried in the core whereas those that are mainly present in the outer layers could represent the result of non-specific chemical reactions which occur over time. As misfolded proteins can overcome the capacity of clearance mechanisms, such as the proteasome [61], accumulated aggregates can last a relatively long time and therefore can accumulate chemical PTMs on the surface. Finally, when it comes to certain chemical modifications (such as Met oxidation, Asn and Gln deamination, etc.), it may be hard to distinguish artifactual protein modifications that occur during sample collection, handling and preparation for MS analysis from those present in vivo. This is also a general challenge in proteomic study, especially involving fresh, human biological samples (such as heart tissue and plasma). For clinical and translational studies it is therefore essential that the correct protocols, which preserve the “native” state of the proteome are implemented [7].
6. Targeting PAOs as a Therapeutic Approach
With the recognition that protein misfolding participates to the pathogenesis of a large number of diseases affecting various organs, a substantial effort has been invested in identifying therapeutic solutions to target the misfolding of proteins and their toxic effect. An updated description of the therapeutic strategies targeting protein misfolding can be found in previous comprehensive reviews [16, 62].
Due to the increasing awareness of the role played by PTMs in affecting protein folding their role as potential therapeutic targets for protein misfolding has also gained attention. In some cases PTMs push the equilibrium of fibrillization forward, whereas in others they tend to stabilize PAOs. This fact has to be considered carefully as reducing the formation of relative inert aggregates in favor of an increased accumulation of PAOs would be detrimental [16, 62].
Examples of drugs targeting amyloidogenic PTMs include some non-steroidal anti-inflammatory drugs (NSAIDs) and lithium, which were found to inhibit the proteolysis of APP induced by γ–secretase activity. Other PTMs currently therapeutically targeted are phosphorylation (such as tau phosphorylation via inhibition of tau kinases) and oxidation [16, 62]. The tau kinase Glycogen Synthase Kinase 3 (GSK3) and its inhibitors (such as lithium) are also attracting attention in HF due to the well-established roles of this enzyme in hypertrophic remodeling and, more recently, in targeting potentially amyloidogenic cardiac proteins [8]. Antioxidants have been studied in proteinopathies in that one of the proposed mechanisms of amyloid toxicity is the generation of reactive oxygen species. Protein oxidation tends to promote protein aggregation, generating a vicious cycle. Thus, several antioxidants are currently being investigated for their potential in preventing or alleviating proteotoxicity (reviewed in [63]). The development of new compounds targeting PTMs much depends on the progress in mapping new PTMs and establishing their roles in PAOs formation. Thus the increasing knowledge around the role of PTMs in the process will likely have a critical impact on the development of new targeted therapies.
In addition to PTMs, direct targeting of the stabilization of a protein’s native conformation has been pursued. An example for such approach is provided by the TTR stabilizer Tafamidis in the therapy of transthyretin (TTR) amyloidosis [64]. Other examples include NSAIDs (Diflunisal), glucocorticoid steroids (dexamethasone) and chemotherapy agents (Cycloposphamide; Doxycliclin) which are also being tested for the treatment of TTR amyloidosis [65–69].
Other therapeutic opportunities include the upregulation of chaperone proteins, the natural first defense against protein misfolding or antibodies and peptides which can prevent PAOs formation [62]. Last but not least, one of the few therapeutic approaches which has shown to be valuable for both HF and other proteinopathies is physical exercise, likely acting at different levels of the cascade leading to PAOs toxicity [14, 44, 70–73].
7. Concluding Remarks
It is noteworthy that the majority of the most diffused chronic degenerative diseases affecting the brain, the heart and other organs, are emerging as diseases related to protein misfolding. Interestingly, these are largely sporadic in nature and therefore, a causal link to gene mutations may not always be found. In these cases, environmental events may impact on the phenotype at the post-translational level and unmask otherwise silent illnesses, or even induce them. Although syndromes defined by gene mutations can provide key information on the effects of protein mutations on the phenotype, especially when they occur at sites that are post-translationally modifiable, the nurture (vs. nature) aspects of these chronic conditions and possibly many other diseases, may offer a more accessible therapeutic approach. This is a crucial reason why considerable efforts be devoted to the detailed mapping of PTMs: so that their functional roles can be established and that this information can be exploited to generate more specific diagnostic and therapeutic approaches. We are confident that proteomics will greatly assist with this task. The functional validation of the role of new PTMs may be challenging as this is largely achieved by single site mutagenesis to mimic modifications. To mimic phosphorylation and protein cleavage by manipulating the coding DNA in transgenic organisms (e.g. by mutation to Asp, or truncation) is rather straightforward. However, it may not be as simple to mimic the remaining several hundreds of known PTMs, and those that have not yet been uncovered.
Acknowledgments
The authors are grateful for the editorial assistance to Kate E. Fiske, BS. This work is supported by the American Heart Association (SDG 12SDG9210000), the NIH Proteomic innovation contract P01HL081427 and PPG on cardiac resynchronization therapy PO1-HL077180, Fondazione del Monte, and RFO University of Bologna (Bologna, Italy) to GA; the NIH exploratory/developmental Research grant award R21HL102716, the Research Project Grant R01HL098468, and the American Heart Association Innovative Research Grant 14IRG18980028 to FdM.
List of abbreviations
- AD
Alzheimer’s disease
- AGE
advanced glycation end products
- APP
amyloid precursor protein
- Aβ
amyloid beta
- CryAB
α-B-crystallin
- FAF
familial amyloidosis Finnish type
- GSK3
Glycogen Synthase Kinase 3
- HF
heart failure
- MAPs
microtubule associate proteins
- NSAIDs
Non-steroidal anti-inflammatory drugs
- PAO
pre-amyloid oligomers
- PINK1
PTEN-induced putative kinase 1
- PTMs
post-translational modifications
- TTR
transthyretin
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, et al. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMurray JJ, Petrie MC, Murdoch DR, Davie AP. Clinical epidemiology of heart failure: public and private health burden. European heart journal. 1998;19(Suppl P):P9–P16. [PubMed] [Google Scholar]
- 3.Prince M, Prina M, Guerchet M. World alzheimer report. London: Alzheimer’s Disease International (ADI); 2013. [Google Scholar]
- 4.Gianni D, Li A, Tesco G, McKay KM, et al. Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy. Circulation. 2010;121:1216–1226. doi: 10.1161/CIRCULATIONAHA.109.879510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campion D, Dumanchin C, Hannequin D, Dubois B, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999;65:664–670. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li D, Parks SB, Kushner JD, Nauman D, et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 2006;79:1030–1039. doi: 10.1086/509900. doi:S0002-9297(07)63465-X [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agnetti G, Husberg C, Van Eyk JE. Divide and conquer: the application of organelle proteomics to heart failure. Circ. Res. 2011;108:512–526. doi: 10.1161/CIRCRESAHA.110.226910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agnetti G, Halperin VLA, Kirk J, Chakir K, et al. Desmin modifications associate with amyloid-like oligomers deposition in heart failure. Cardiovasc. Res. 2014 doi: 10.1093/cvr/cvu003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pappolla MA, Bryant-Thomas TK, Herbert D, Pacheco J, et al. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology. 2003;61:199–205. doi: 10.1212/01.wnl.0000070182.02537.84. [DOI] [PubMed] [Google Scholar]
- 10.Razay G, Vreugdenhil A, Wilcock G. The metabolic syndrome and Alzheimer disease. Arch Neurol. 2007;64:93–96. doi: 10.1001/archneur.64.1.93. [DOI] [PubMed] [Google Scholar]
- 11.Neubauer S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 12.Sanbe A, Osinska H, Saffitz JE, Glabe CG, et al. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A. 2004;101:10132–10136. doi: 10.1073/pnas.0401900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Despa S, Margulies KB, Chen L, Knowlton AA, et al. Hyperamylinemia contributes to cardiac dysfunction in obesity and diabetes: a study in humans and rats. Circ. Res. 2012;110:598–608. doi: 10.1161/CIRCRESAHA.111.258285. 10.1161/CIRCRESAHA.111.258285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willis MS, Patterson C. Proteotoxicity and cardiac dysfunction--Alzheimer's disease of the heart? N. Engl. J. Med. 2013;368:455–464. doi: 10.1056/NEJMra1106180. 10.1056/NEJMra1106180. [DOI] [PubMed] [Google Scholar]
- 15.Katzman R, Terry R, DeTeresa R, Brown T, et al. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Annals of neurology. 1988;23:138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- 16.Rochet JC. Novel therapeutic strategies for the treatment of protein-misfolding diseases. Expert Rev. Mol. Med. 2007;9:1–34. doi: 10.1017/S1462399407000385. [DOI] [PubMed] [Google Scholar]
- 17.Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010;11:777–788. doi: 10.1038/nrm2993. 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- 18.Lefebvre T, Guinez C, Dehennaut V, Beseme-Dekeyser O, et al. Does O-GlcNAc play a role in neurodegenerative diseases? Expert Rev. Proteomics. 2005;2:265–275. doi: 10.1586/14789450.2.2.265. [DOI] [PubMed] [Google Scholar]
- 19.Nilsson MR. In: Amyloid Proteins. the Beta Sheet Conformation and Disease. Sipe JD, editor. Weinheim: WILEY-VCH Verlag GmbH & Co. KGaA; 2005. p. 81. [Google Scholar]
- 20.Abedini A, Gupta R, Marek P, Meng F, et al. In: Protein Misfolding Diseases: Current and Emerging Principles and Therapies. Ramirez-Alvarado M, Kelly JW, Dobson CM, editors. John Wiley and Sons, Inc; 2010. p. 131. [Google Scholar]
- 21.Gorza L, del Monte F. Protein unfolding in cardiomyopathies. Heart Fail Clin. 2005;1:237–250. doi: 10.1016/j.hfc.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 22.Mannini B, Cascella R, Zampagni M, van Waarde-Verhagen M, et al. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. U. S. A. 2012;109:12479–12484. doi: 10.1073/pnas.1117799109. 10.1073/pnas.1117799109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandri M, Robbins J. Proteotoxicity: An underappreciated pathology in cardiac disease. J. Mol. Cell. Cardiol. 2013 doi: 10.1016/j.yjmcc.2013.12.015. 10.1016/j.yjmcc.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Robbins J. Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell. Cardiol. 2013 doi: 10.1016/j.yjmcc.2013.11.006. 10.1016/j.yjmcc.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 26.Walter J, Haass C. Posttranslational modifications of amyloid precursor protein : ectodomain phosphorylation and sulfation. Methods in molecular medicine. 2000;32:149–168. doi: 10.1385/1-59259-195-7:149. [DOI] [PubMed] [Google Scholar]
- 27.Georgopoulou N, McLaughlin M, McFarlane I, Breen KC. The role of post-translational modification in beta-amyloid precursor protein processing. Biochemical Society symposium. 2001:23–36. doi: 10.1042/bss0670023. [DOI] [PubMed] [Google Scholar]
- 28.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. doi:nrm2101 [pii]. [DOI] [PubMed] [Google Scholar]
- 29.Tay WM, Bryant JG, Martin PK, Nix AJ, et al. A mass spectrometric approach for characterization of amyloid-beta aggregates and identification of their post-translational modifications. Biochemistry. 2012;51:3759–3766. doi: 10.1021/bi300316d. 10.1021/bi300316d. [DOI] [PubMed] [Google Scholar]
- 30.Diguet N, Mallat Y, Ladouce R, Clodic G, et al. Muscle creatine kinase deficiency triggers both actin depolymerization and desmin disorganization by advanced glycation end products in dilated cardiomyopathy. J. Biol. Chem. 2011;286:35007–35019. doi: 10.1074/jbc.M111.252395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson NE, Robinson AB. Molecular clocks. Proc. Natl. Acad. Sci. U. S. A. 2001;98:944–949. doi: 10.1073/pnas.98.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller UC, Zheng H. Physiological functions of APP family proteins. Cold Spring Harbor perspectives in medicine. 2012;2:a006288. doi: 10.1101/cshperspect.a006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hein S, Kostin S, Heling A, Maeno Y, Schaper J. The role of the cytoskeleton in heart failure. Cardiovasc. Res. 2000;45:273–278. doi: 10.1016/s0008-6363(99)00268-0. [DOI] [PubMed] [Google Scholar]
- 34.Snider NT, Omary MB. Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2014;15:163–177. doi: 10.1038/nrm3753. 10.1038/nrm3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baird FJ, Bennett CL. Microtubule defects & Neurodegeneration. J. Genet. Syndr. Gene Ther. 2013;4:203. doi: 10.4172/2157-7412.1000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chinnakkannu P, Samanna V, Cheng G, Ablonczy Z, et al. Site-specific microtubule-associated protein 4 dephosphorylation causes microtubule network densification in pressure overload cardiac hypertrophy. J. Biol. Chem. 2010;285:21837–21848. doi: 10.1074/jbc.M110.120709. 10.1074/jbc.M110.120709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maury CP. Gelsolin-related amyloidosis. Identification of the amyloid protein in Finnish hereditary amyloidosis as a fragment of variant gelsolin. J. Clin. Invest. 1991;87:1195–1199. doi: 10.1172/JCI115118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clemen CS, Herrmann H, Strelkov SV, Schroder R. Desminopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:47–75. doi: 10.1007/s00401-012-1057-6. 10.1007/s00401-012-1057-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanbe A, Osinska H, Saffitz JE, Glabe CG, et al. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10132–10136. doi: 10.1073/pnas.0401900101. doi:10.1073/pnas.0401900101 [doi]; 0401900101 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agnetti G, Bezstarosti K, Dekkers DH, Verhoeven AJ, et al. Proteomic profiling of endothelin-1-stimulated hypertrophic cardiomyocytes reveals the increase of four different desmin species and alpha-B-crystallin. Biochim. Biophys. Acta. 2008;1784:1068–1076. doi: 10.1016/j.bbapap.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis. 2010;17:101–104. doi: 10.3109/13506129.2010.526812. [DOI] [PubMed] [Google Scholar]
- 42.Glabe CG. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberts BE, Duennwald ML, Wang H, Chung C, et al. A synergistic small-molecule combination directly eradicates diverse prion strain structures. Nat. Chem. Biol. 2009;5:936–946. doi: 10.1038/nchembio.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maloyan A, Gulick J, Glabe CG, Kayed R, Robbins J. Exercise reverses preamyloid oligomer and prolongs survival in {alpha}B-crystallin-based desmin-related cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5995–6000. doi: 10.1073/pnas.0609202104. doi:0609202104 [pii]; 10.1073/pnas.0609202104 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abedini A, Schmidt AM. Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 2013;587:1119–1127. doi: 10.1016/j.febslet.2013.01.017. 10.1016/j.febslet.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285:12463–12468. doi: 10.1074/jbc.R109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerf E, Sarroukh R, Tamamizu-Kato S, Breydo L, et al. Antiparallel beta-sheet: a signature structure of the oligomeric amyloid beta-peptide. Biochem. J. 2009;421:415–423. doi: 10.1042/BJ20090379. 10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]
- 48.Wolfe MS. When loss is gain: reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:136–140. doi: 10.1038/sj.embor.7400896. doi:7400896 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Willis MS, Bevilacqua A, Pulinilkunnil T, Kienesberger P, et al. The role of ubiquitin ligases in cardiac disease. J. Mol. Cell. Cardiol. 2013 doi: 10.1016/j.yjmcc.2013.11.008. 10.1016/j.yjmcc.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hart GW, Ball LE. Post-translational modifications: a major focus for the future of proteomics. Mol. Cell. Proteomics. 2013;12:3443. doi: 10.1074/mcp.E113.036491. 10.1074/mcp.E113.036491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang P, Kirk JA, Ji W, dos Remedios CG, et al. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation. 2012;126:1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388. 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bodiga VL, Eda SR, Bodiga S. Advanced glycation end products: role in pathology of diabetic cardiomyopathy. Heart Fail. Rev. 2014;19:49–63. doi: 10.1007/s10741-013-9374-y. 10.1007/s10741-013-9374-y. [DOI] [PubMed] [Google Scholar]
- 53.Lopez-Clavijo AF, Duque-Daza CA, O'Connor PB. Tandem mass spectrometry for the study of glyoxal-derived advanced glycation end-products (AGEs) in peptides. Rapid Commun. Mass Spectrom. 2014;28:25–32. doi: 10.1002/rcm.6753. 10.1002/rcm.6753. [DOI] [PubMed] [Google Scholar]
- 54.Willemsen S, Hartog JW, van Veldhuisen DJ, van der Meer P, et al. The role of advanced glycation end-products and their receptor on outcome in heart failure patients with preserved and reduced ejection fraction. Am. Heart J. 2012;164:742–749. e3. doi: 10.1016/j.ahj.2012.07.027. 10.1016/j.ahj.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 55.Sylvestersen KB, Young C, Nielsen ML. Advances in characterizing ubiquitylation sites by mass spectrometry. Curr. Opin. Chem. Biol. 2013;17:49–58. doi: 10.1016/j.cbpa.2012.12.009. 10.1016/j.cbpa.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 56.Jones J, Wu K, Yang Y, Guerrero C, et al. A targeted proteomic analysis of the ubiquitin-like modifier nedd8 and associated proteins. J. Proteome Res. 2008;7:1274–1287. doi: 10.1021/pr700749v. [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koyano F, Okatsu K, Kosako H, Tamura Y, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014 doi: 10.1038/nature13392. [doi]. [DOI] [PubMed] [Google Scholar]
- 58.Marx V. Targeted proteomics. Nat. Methods. 2013;10:19–22. doi: 10.1038/nmeth.2285. [DOI] [PubMed] [Google Scholar]
- 59.Brinkmalm G, Portelius E, Ohrfelt A, Mattsson N, et al. An online nano-LC-ESI-FTICR-MS method for comprehensive characterization of endogenous fragments from amyloid beta and amyloid precursor protein in human and cat cerebrospinal fluid. J. Mass Spectrom. 2012;47:591–603. doi: 10.1002/jms.2987. [doi]. [DOI] [PubMed] [Google Scholar]
- 60.Pan J, Han J, Borchers CH, Konermann L. Structure and dynamics of small soluble Abeta(1–40) oligomers studied by top-down hydrogen exchange mass spectrometry. Biochemistry. 2012;51:3694–3703. doi: 10.1021/bi3002049. [doi]. [DOI] [PubMed] [Google Scholar]
- 61.Patterson C. Search and destroy: the role of protein quality control in maintaining cardiac function. J. Mol. Cell. Cardiol. 2006;40:438–441. doi: 10.1016/j.yjmcc.2006.01.003. doi:S0022-2828(06)00004-6 [pii]; 10.1016/j.yjmcc.2006.01.003 [doi]. [DOI] [PubMed] [Google Scholar]
- 62.Hard T, Lendel C. Inhibition of amyloid formation. J. Mol. Biol. 2012;421:441–465. doi: 10.1016/j.jmb.2011.12.062. [doi]. [DOI] [PubMed] [Google Scholar]
- 63.Ho L, Pasinetti GM. Polyphenolic compounds for treating neurodegenerative disorders involving protein misfolding. Expert review of proteomics. 2010;7:579–589. doi: 10.1586/epr.10.69. [DOI] [PubMed] [Google Scholar]
- 64.Bulawa CE, Connelly S, Devit M, Wang L, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Castano A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congestive heart failure. 2012;18:315–319. doi: 10.1111/j.1751-7133.2012.00303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berk JL, Suhr OB, Obici L, Sekijima Y, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA : the journal of the American Medical Association. 2013;310:2658–2667. doi: 10.1001/jama.2013.283815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kastritis E, Wechalekar AD, Dimopoulos MA, Merlini G, et al. Bortezomib with or without dexamethasone in primary systemic (light chain) amyloidosis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:1031–1037. doi: 10.1200/JCO.2009.23.8220. [DOI] [PubMed] [Google Scholar]
- 68.Venner CP, Lane T, Foard D, Rannigan L, et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood. 2012;119:4387–4390. doi: 10.1182/blood-2011-10-388462. [DOI] [PubMed] [Google Scholar]
- 69.Mikhael JR, Schuster SR, Jimenez-Zepeda VH, Bello N, et al. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood. 2012;119:4391–4394. doi: 10.1182/blood-2011-11-390930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Waard MC, van der Velden J, Bito V, Ozdemir S, et al. Early Exercise Training Normalizes Myofilament Function and Attenuates Left Ventricular Pump Dysfunction in Mice With a Large Myocardial Infarction. Circ. Res. 2007 doi: 10.1161/01.RES.0000262655.16373.37. doi:01.RES.0000262655.16373.37 [pii]; 10.1161/01.RES.0000262655.16373.37 [doi]. [DOI] [PubMed] [Google Scholar]
- 71.Um HS, Kang EB, Leem YH, Cho IH, et al. Exercise training acts as a therapeutic strategy for reduction of the pathogenic phenotypes for Alzheimer's disease in an NSE/APPsw-transgenic model. Int. J. Mol. Med. 2008;22:529–539. [PubMed] [Google Scholar]
- 72.Downing J, Balady GJ. The role of exercise training in heart failure. J. Am. Coll. Cardiol. 2011;58:561–569. doi: 10.1016/j.jacc.2011.04.020. 10.1016/j.jacc.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 73.Bhuiyan MS, Pattison JS, Osinska H, James J, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877. [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]