

Summary
It is commonly assumed that there is a single canonical DNA damage response (DDR) that protects cells from various types of double-strand breaks and that its activation occurs via recognition of DNA damage by the DDR machinery. Recent work suggests that both assumptions may be oversimplifications. Here, we discuss several variations of the DDR in which the pathway is activated by diverse cellular events or generates distinct signaling outcomes. The existence of multiple non-canonical DDRs provides insights into how DNA damage is sensed and suggests a highly modular organization of the DDR.
Introduction
The response to double-strand breaks (DSBs), termed the DNA damage response (DDR), is a fundamental cellular process. It protects the genome by swiftly responding to and repairing potentially lethal double-strand breaks (DSBs) that could trigger genome instability or tumorigenesis (Goldstein and Kastan, 2015). The DDR is traditionally divided into three phases: detection of damage, signal transduction, and downstream actions (Fig. 1A). The response begins when DNA damage is detected, typically via the Mre11-Rad50-Nbs1 (MRN) sensor complex, activating the apical kinases Ataxia-Telangiectasia Mutated kinase (ATM), the DNA-dependent Protein Kinase (DNA-PK) and/or ATR (ATM and Rad3-related). The signal is then transduced through phosphorylation by ATM of the core histone variant H2AX (called γ-H2AX), creating a platform for the MDC1 mediator protein. MDC1, in turn, recruits a myriad of DDR factors, including ubiquitin ligase complexes and chromatin remodelers, but also iterates the MRN-ATM activation loop, propagating the γ-H2AX mark over megabase domains, thus amplifying the DDR signal. Other histone modifications such as ubiquitination catalyzed by the RNF8/RNF168 ubiquitin ligases, modulate the recruitment of repair factors, including 53BP1. The downstream effects of DDR signaling are finally mediated by soluble effector kinases, particularly CHK1 and CHK2, distributing the phosphorylation cascade away from the DSB site to proteins like p53 that execute cell cycle arrest, transcription of DNA damage responsive genes, and other repair pathways to promote the cell’s survival or, alternatively, trigger downstream pathways such as apoptosis and senescence to promote organismal health at the expense of the damaged cell.
Figure 1.
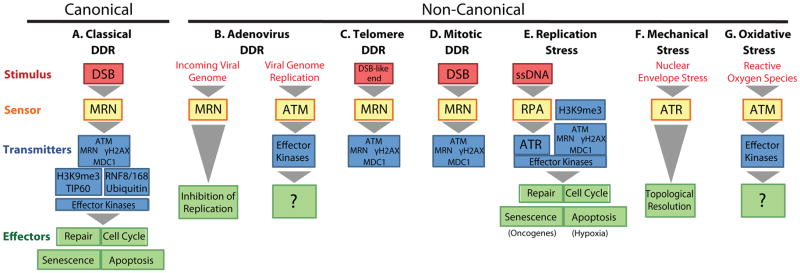
Modular structure of canonical and non-canonical DDRs. Stimuli are in red: damage stimuli, boxes; non-damage stimuli, text. Sensors (yellow) lead to activation of various transmission modules (blue boxes) and downstream effectors (green), each of which can be disengaged for different DDR outcomes.
Given its inherent complexity, it has been helpful to think of the DDR as a singular response starting with DNA damage and ending in cellular responses such as cell cycle arrest or apoptosis. However, its multi-phase molecular nature and the requirement for the DDR to detect and respond to a wide range of stimuli in varying cellular circumstances raises the question of whether multiple, non-canonical DDRs exist. Here we briefly discuss select examples from several biological systems that demonstrate alternative triggers, modes of transmission and cellular outcome of the DDR signaling. These findings highlight the notion that the response to DNA damage may differ considerably depending on cellular context and that not all DDRs are created equal (Fig. 1).
Insights from a virus: Non-canonical DDRs in viral infection
Viruses have evolved a staggering number of different ways to hijack the cellular machinery for their propagation, including the DDR (Luftig, 2014). An elegant example of how analysis of viral responses provides insight into cellular function is work by Shah and O’Shea, in this issue of Cell, in which they dissect how adenovirus evades and exploits the canonical DDR.
It has long been known that human Adenovirus 5 (Ad5), a linear dsDNA virus, escapes the DNA damage surveillance machinery by selectively targeting the MRN sensor complex for degradation and sequestration (Stracker et al., 2002). However, the purpose of MRN inactivation has remained unclear and was paradoxical, since it did not prevent the activation of a global DDR during the course of infection. Shah et al. now delineate a model where cells first enlist an MRN- and ATM-dependent local DDR to prevent early viral replication while still allowing normal cellular replication so as not to endanger proliferation and the purging of viral genomes from the nucleus during cell division. In infections where viral replication is successful in escaping the first line of defense, a later MRN-independent global DDR signaling pathway is activated (Shah and O’Shea 2015). These findings are of significance in that they define two distinct DDR mechanisms which both differ considerably from the classical DDR. The first non-canonical DDR represents an interrupted DDR in which upstream portions of the canonical pathway are activated, but the signal is not transmitted downstream; the second involves an MRN-independent, and thus non-canonical, means to activate signaling (Fig. 1B). The presence, and particularly the complex interplay, of these non-canonical DDRs, which both use some, but not all parts of the canonical pathway, highlight that the DDR can be activated in multiple ways to adapt to various cellular situations and needs.
Emerging non-canonical DDRs
The observations of these virally-induced non-canonical DDRs are the proverbial tip of the iceberg, as several variations on the DDR have been described. These DDRs are non-canonical both in that their stimuli are diverse and in that they only use portions of the canonical DDR pathway.
DDR activation in telomere maintenance
Telomeres, via a specialized protein complex called shelterin, serve to protect chromosomes ends from detection as DNA damage and fusion. The discovery of accumulation of DDR proteins at telomeres thus seemed paradoxical. Closer inspection, however, revealed that telomeres are partly deprotected after replication and recruit DDR proteins in G2 phase of the cell cycle to reconstruct the end protection complex (Verdun et al., 2005). This telomere DDR is non-canonical in that it is interrupted and only activates MRN and ATM but none of the diffusible DDR signaling proteins, such as CHK2 and p53, and thus does not trigger downstream cell cycle inhibition (Fig 1C). The interrupted nature of the telomere maintenance DDR is likely mediated by the residual activity of the shelterin protein TRF2, which disengages the DDR pathway from the RNF8/RNF168/53BP1 module by recruiting deubiquitylating enzymes (Okamoto et al., 2013). This provides a potential mechanism for utilizing upstream DDR enzymes for their DNA binding activities without unnecessary or deleterious downstream effects such as telomere fusion. Interestingly, the telomere-maintenance DDR bears considerable resemblance to the MRN antiviral pathway of Ad5 in that it is spatially localized and interrupted. This similarity may also extend to function in the establishment of DNA structures to inhibit enzymatic activities, such as DNA repair/end fusion in telomeres and DNA replication in virally infected cells.
Chromatin-mediated DDR
Although DDR activation at telomeres and viral genomes does not use DNA damage as a trigger, DNA ends are involved. DDR signaling can also occur in the complete absence of DNA ends or breaks (Bakkenist and Kastan 2003; Soutoglou and Misteli 2008). For example, non-damaging treatments induce inactive ATM dimers to dissociate and form kinase-active monomers (Bakkenist and Kastan, 2003). The upstream activation feedback loop of the canonical DDR – the γ-H2AX/MDC1/MRN/ATM module - can be also activated independently of a damage stimulus by the experimental binding of upstream DDR proteins to chromatin, effectively nucleating the signaling complex and initiating the phosphorylation cascade (Soutoglou & Misteli, 2008). This suggests that free DNA ends are not an intrinsically required part of the DDR machinery and that the main amplification module of the DDR can be disconnected from the rest of the pathway (Fig. 1A). A physiological equivalent of this type of non-canonical DDR is seen in Human T-cell Leukemia Virus (HTLV), which tethers MDC1 to chromatin for sequestering DDR proteins away from the viral DNA (Luftig, 2014).
Several studies have suggested that chromatin alterations may activate the DDR. Modulation of higher-order chromatin structure by exposure to hypotonic conditions or inhibition of histone deacetylation results in activation of ATM signaling (Bakkenist and Kastan, 2003). Changes in chromatin structure around DSBs during the canonical DDR include transient formation of repressive chromatin domains marked by tri-methylated lysine 9 of histone H3 (H3K9me3) that stimulate the binding and phosphorylation of the acetyltransferase TIP60, which activates ATM (Ayrapetov et al., 2014). Mimicking this chromatin context for DDR activation with defined H3K9me3 domains alone is sufficient for the activation of upstream portions of the DDR signaling pathway (Fig. 1A; Burgess et al., 2014). However, similar to telomere DDR, this DDR signaling remains localized and does not activate functional downstream modules, establishing another restricted non-canonical DDR.
Mitotic DDR
An interrupted DDR is also seen in mitosis. Upon DNA damage of condensed chromosomes, the upstream signaling loop, including formation of the γ-H2AX-MDC1 platform, is initiated (Giunta et al., 2010). However, the signaling is restrained by the mitotic kinase-mediated inactivation of downstream signaling through CHK2 and the phosphorylation-mediated inhibition of the RNF8/RNF168/53BP1 branch of the DDR until cells progress through mitosis (Lee et al., 2014; Orthwein et al., 2014). The interruption of mitotic DDR has important consequences since telomeres are prone to uncapping and fusions during mitosis (Lee et al., 2014; Orthwein et al., 2014). This provides another layer of protection to telomere instability using non-canonical DDR pathways. Interestingly, it has been noticed that some apparently undamaged mitotic cells also exhibit upstream ATM signaling and form γ-H2AX and MDC1 foci on condensed mitotic chromosomes (Burgess et al., 2014, Giunta et al., 2009). While it can be not be excluded that these foci are due to undetectable damage on mitotic chromosomes, they may reflect upstream DDR activation triggered by the highly compacted chromatin of mitotic chromosomes which may be pre-emptively scanned to mark damage for immediate repair upon mitotic exit (Fig. 1D).
Replication stress-induced DDR
Replication stress is an activator of both canonical and non-canonical DDRs. Hypoxic conditions to induce replication stress lead to standard activation of ATR by ssDNA in stalled replication forks, but also trigger rapid activation of ATM prior to formation of DSBs (Olcina et al, 2013). Consistent with the absence of DSBs, ATM activation is MRN-independent and, despite robust γ-H2AX/MDC1 phosphorylation, RNF8 and downstream repair factors are not recruited, yet downstream signaling to p53 is initiated. The activation of ATM was enhanced by hypoxia-induced H3K9me3, suggesting, again, that chromatin structure may promote DDR signaling (Fig. 1E). Interestingly, signaling during chronic hypoxia preferentially prompts apoptosis, but the outcome from oncogene-induced replication stress, whether from cellular or viral oncogenes, is senescence. This could potentially highlight differential downstream consequences of non-canonical DDR activation, though the effects of other hypoxia-induced signaling pathways can not be excluded.
Mechanical stress-induced DDR
Mechanical stress on the nuclear envelope has recently been found to activate ATR. In this scenario, ATR acts as a mechanosensor for chromatin topological structures that transmit a mechanical stimulus to the nuclear envelope through their attachment to the nuclear lamina (Kumar et al., 2014). The activation of the ATR signaling cascade upon mechanical stress does not result in the canonical activation of downstream checkpoints or apoptosis, and appears to be localized to the nuclear envelope (Fig. 1F). Rather, this response helps resolve the torsional stress to protect the perinuclear chromatin and avoid chromosomal aberrations such as DSBs and collapsed forks, representing another example of uncoupling of upstream portions of the DDR from downstream events.
Oxidative stress-induced DDR
Oxidative stress activates ATM via direct oxidation (Guo et al., 2010). This non-canonical activation of ATM occurs in the absence of DSBs and does not require MRN nor does it lead to formation of significant γ-H2AX domains. Although the main amplification module of the canonical DDR is not activated during oxidative stress, soluble factors such as CHK2 and p53 are phosphorylated by oxidation-activated ATM (Fig. 1G). This mechanism may be clinically relevant for Epstein-Barr virus (EBV). In many EBV malignancies the only viral protein expressed is the episome-maintaining EBNA-1 protein, which has been shown to upregulate oxidase activity, leading to activation of the DDR and promoting EBV transformation of B-lymphocytes (Gruhne et al., 2009). Eventually, the increased reactive oxygen species in the cell leads to damage and canonical DDR, but whether the initial ATM activation in this case occurs by direct oxidation has not been determined. Use of a direct activation mechanism by the virus may serve to limit amplification of the response and in this way protect the virus from downstream DDR events such as apoptosis until downstream tumor suppression pathways are genetically inactivated by genome instability.
Sensing mechanisms of the DNA damage response
The fact that DDRs can be activated by multiple means suggests that the sensing machinery, like the DDR itself, is not a single one-size-fits-all entity, but that there are multiple sensors involved in the various DDR pathways.
The best-characterized DNA damage sensor is the MRN complex. It is a dimer of the Mre11, Rad50, Nbs1 trimer, which in theory can assemble into up to 216 distinct states, notwithstanding post-translational modifications (Williams et al., 2010). Therefore, MRN has considerable intrinsic flexibility to adopt the role of multiple sensors. In the telomeric DDR, where the stimulus is much like a DSB, MRN is likely sensing the ends in the conformation used for the canonical response to DSBs, which is that of a symmetrical dimer of two MRN complexes, exposing the ATM binding domain.
MRN also binds replication forks by adopting an asymmetrical conformation, which blocks ATM binding. However, MRN activity is not required for the activation of ATM or ATR during replication stress and MRN instead appears to be involved in resolution of intermediates in replication stress rather than damage sensing (Bruhn et al., 2014). Similarly, although MRN is recruited to a number of non-canonical DDR stimuli, such as tethering-induced chromatin domains and mitotic chromatin (Burgess et al., 2014; Giunta et al., 2010), it is likely that its role in these cases is not as a sensor but as an upstream DDR factor contributing to the amplification of the signal.
The notion that chromatin structure is a DDR trigger is intriguing, yet a sensor has not been identified. A candidate for a chromatin-structure sensor in the DDR may be the histone acetyltransferase TIP60, which binds to H3K9me3, a known DDR stimulus, and subsequently activates ATM (Sun et al., 2009). Since H3K9me3 and TIP60 are also known to be involved in the canonical DDR, it is tempting to speculate that chromatin structure may also contribute to canonical DDR transmission (Ayrapetov et al., 2014). In support, it has been recognized that upon formation of DSBs, heterochromatin forms transiently in the vicinity of breaks and as such may augment DDR signaling as indicated by its stimulation of the recruitment of downstream factors such as BRCA1 (Khurana et al., 2014). An attractive feature of chromatin structure as a DDR trigger is its potential to rapidly amplify the signal due to the many binding sites generated in the heterochromatinized domain spreading along the flanking regions of the damaged site. Further elucidation of the role of chromatin structure as a DDR trigger and the nature of sensors is required.
Examination of non-canonical DDRs also indicates that DDR kinases are sensors themselves, thus bypassing the upstream damage-sensing module of the canonical DDR. Activation of ATR by mechanical stimuli occurs in a highly localized fashion in domains at the nuclear envelope that contain activated ATR in the absence of RPA or ATRIP, suggesting local mechanosensing by the kinase itself (Kumar et al., 2014). Precisely how ATR becomes activated by a mechanical stimulus is an outstanding question, but its structure indicates that it could occur through forces acting on its highly elastic N-terminal domain, transmitting mechanical information to the C-terminal kinase domain (Perry and Kleckner, 2003). This mechanism could also be at play in replication stress, since S-phase chromatin dynamics produce mechanical forces that are transmitted to the nuclear envelope by lamin-associated chromatin domains. It is also possible that ATR contributes to sensing of the chromatin structure-based DDR mechanisms, although the function of ATR as a sensor, thus far, appears to be localized to the nuclear envelope, while H3K9me3 domains also activate DDR in the nuclear interior (Burgess et al., 2014). A sensing function of DDR kinases is finally also suggested by the direct activation of ATM by oxidation, which is required for a rapid response to oxidative stress, preceding DNA damage and perhaps potentiating the DDR. It will be interesting to see if this direct ATM activation through oxidation also occurs in other non-canonical DDR mechanisms, particularly in the virus world. The multiple non-nuclear targets of ATM and ATR identified in phospho-proteomic screens could serve as a guide for elucidating the non-canonical signaling pathways of these kinases (Bensimon et al., 2011).
Outlook
The diversity of sensors, distinct signaling pathways, and alternate outcomes of signaling point to a high degree of diversity in the DDR pathway. A striking feature that emerges when considering the various DDRs is their modular nature in which upstream sensing, mid-level transmission and downstream effector modules can be functionally separated from each other and be used in combinatorial fashion (Fig. 1). An important aspect of future inquiry into the DDR will be the full elucidation of the series of molecular clutches that have evolved to uncouple DDR modules from each other to generate the diverse set of DDRs required to faithfully maintain genome integrity in a vast spectrum of biological systems and circumstances.
Acknowledgments
We apologize to the large number of authors whose work we could not cite due to space constraints. Thanks to Marc Raley for help with the figure. Work in the author’s laboratory is supported by the Intramural Research Program of the National Institutes of Health (NIH), NCI, Center for Cancer Research.
References
- Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci U S A. 2014;111:9169–9174. doi: 10.1073/pnas.1403565111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Bensimon A, Aebersold R, Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 2011;585:1625–1639. doi: 10.1016/j.febslet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Bruhn C, Zhou ZW, Ai H, Wang ZQ. The essential function of the MRN complex in the resolution of endogenous replication intermediates. Cell Rep. 2014;6:182–195. doi: 10.1016/j.celrep.2013.12.018. [DOI] [PubMed] [Google Scholar]
- Burgess RC, Burman B, Kruhlak MJ, Misteli T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014;9:1703–1717. doi: 10.1016/j.celrep.2014.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double-strand breaks during mitosis. J Cell Biol. 2010;190:197–207. doi: 10.1083/jcb.200911156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–143. doi: 10.1146/annurev-med-081313-121208. [DOI] [PubMed] [Google Scholar]
- Gruhne B, et al. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci U S A. 2009;106:2313–2318. doi: 10.1073/pnas.0810619106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- Khurana S, et al. A Macrohistone Variant Links Dynamic Chromatin Compaction to BRCA1-Dependent Genome Maintenance. Cell Rep. 2014;8:1049–1062. doi: 10.1016/j.celrep.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, et al. ATR Mediates a Checkpoint at the Nuclear Envelope in Response to Mechanical Stress. Cell. 2014;158:633–646. doi: 10.1016/j.cell.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, et al. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol Cell. 2014;54:512–525. doi: 10.1016/j.molcel.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luftig MA. Viruses and the DNA Damage Response: Activation and Antagonism. Annual Review of Virology. 2014;1:605–625. doi: 10.1146/annurev-virology-031413-085548. [DOI] [PubMed] [Google Scholar]
- Olcina MM, et al. Replication stress and chromatin context link ATM activation to a role in DNA replication. Mol Cell. 2013;52:758–766. doi: 10.1016/j.molcel.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, et al. A two-step mechanism for TRF2-mediated chromosome-end protection. Nature. 2013;494:502–505. doi: 10.1038/nature11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A, et al. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science. 2014;344:189–193. doi: 10.1126/science.1248024. [DOI] [PubMed] [Google Scholar]
- Perry J, Kleckner N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell. 2003;112:151–155. doi: 10.1016/s0092-8674(03)00033-3. [DOI] [PubMed] [Google Scholar]
- Shah, O’Shea The MRE11 complex and ATM activate distinct signaling responses to defend against DNA viruses versus DNA breaks. Cell. 2015 in press. [Google Scholar]
- Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science. 2008;320:1507–1510. doi: 10.1126/science.1159051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- Sun Y, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009;11:1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdun RE, Crabbe L, Haggblom C, Karlseder J. Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell. 2005;20:551–561. doi: 10.1016/j.molcel.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Williams GJ, Lees-Miller SP, Tainer JA. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair. 2010;9:1299–1306. doi: 10.1016/j.dnarep.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]