

Abstract
The human herpesvirus-6 (HHV-6) infection induces cell-cycle arrest. In this study, we found that the HHV-6-encoded U14 protein induced cell-cycle arrest at G2/M phase via an association with the cellular protein EDD, a mediator of DNA-damage signal transduction. In the early phase of HHV-6 infection, U14 colocalized with EDD dots in the nucleus, and similar colocalization was also observed in cells transfected with a U14 expression vector. When the carboxyl-terminal region of U14 was deleted, no association of U14 and EDD was observed, and the percentage of cells in G2/M decreased relative to that in cells expressing wild-type U14, indicating that the C-terminal region of U14 and the U14–EDD association are critical for the cell-cycle arrest induced by U14. These results indicate that U14 is a G2/M checkpoint regulator encoded by HHV-6.
Introduction
Human herpesvirus-6 (HHV-6), a member of the beta herpesvirus family, is classified into two viruses, HHV-6A and HHV-6B [1]. HHV-6B causes exanthem subitum, and persists as a lifelong latent infection [2,3], whereas the pathogenesis of HHV-6A remains unknown. Viral reactivation later in life can lead to severe and even fatal diseases in immune-compromised individuals [4,5]. The genome size of HHV-6 is approximately 160 kbps and encodes about 100 ORFs [6,7,8]. Some of these ORFs are unique to HHV-6, and the functions of some ORFs remain unclear.
Previously, we found that the HHV-6-encoded U14 protein is expressed at an early phase of infection, and is distributed in a dot-like pattern in the nucleus. During the late phase of infection, its localization changes to the cytoplasm mainly, and the protein is incorporated into virions, possibly functioning as a tegument protein [9]. We wished to understand why U14 localizes as nuclear dots in the early phase. Based on this localization and the timing of expression, we speculated that U14 functions in viral replication. Therefore, we tried to identify cellular molecules that associate with U14. We also found that U14 interacts with cellular p53, and that p53 is incorporated into virions along with U14 [9].
In this study, we identified a HECT E3 ubiquitin ligase, E3 identified by differential display (EDD), which associates with U14. Previous work showed that EDD is involved in the regulation of cell proliferation and tumorigenesis [10,11]. Furthermore, EDD plays a role in DNA-damage signaling: EDD binds the DNA-dependent protein kinase-interacting protein CIB1, modulates the activity of CHK2, and is an established mediator of G1/S, intra-S, and G2/M phase checkpoints [11,12,13]. EDD inhibits ataxia telangiectasia mutated (ATM)-mediated phosphorylation of p53, and also plays a role in ensuring smooth G1/S progression [14].
To promote their own replication, viruses control cell-cycle progression [15,16,17,18,19]. HHV-6 infection also induces cell-cycle arrest. For example, infection of T cells with HHV-6 is associated with cell-cycle arrest at the G1/S or G2/M phase [20,21,22,23,24,25], and HHV-6B infection of Molt3 cells causes cell-cycle arrest at the G1 phase concomitant with accumulated and phosphorylated p53 [25]. Inhibition of cell proliferation by viruses can occur through both p53-dependent and-independent pathways [25,26,27]. HHV-6B induces p53 Ser392 phosphorylation by an atypical pathway independent of CK2 and p38 kinases [28]. The cell-cycle arrest induced by HHV-6 in the G2/M phase is accompanied by inhibition of Cdc2-cyclin B1 kinase activity and a significant increase in phosphorylation of Cdc2 at the Tyr15 inhibitory site [23]. HHV-6 alters the E2F1/Rb pathway and E2F1 localization and causes cell-cycle arrest in infected cells [24]. This also causes an elevation in levels of the transcription factor E2F1, which promotes expression of genes important for viral DNA synthesis, such as U27 and U79 [24,29]. During HHV-6B infection, the majority of cellular p53 is inactivated and stabilized in the cytoplasm, most likely due to a reduction in p53 degradation [30]. Recent work showed that HHV-6 DR6 protein has the ability to inhibit the G2/M transition independently of p53 [31]. In addition, HHV-6B U19 inhibits p53-dependent cell death [32].
As described above, several genes encoded by HHV-6 have been shown to contribute to the cell-cycle arrest. Here, we found that U14 protein whose function had been unknown in early phase of HHV-6-infection, induces cell-cycle arrest at G2/M phase via an association with EDD for which the C-terminus of U14 is required. Thus, U14 is a G2/M checkpoint regulator encoded by HHV-6.
Materials and Methods
Cells and viruses
Molt3 cells were cultured in RPMI-1640 medium supplemented with 8% fetal bovine serum (FBS). 293T, 293 and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 8% FBS. Umbilical cord blood mononuclear cells (CBMCs) were cultured as described previously [33]. CBMCs were provided by H. Yamada (Kobe University Graduate School of Medicine, Kobe, Japan) and purchased from the Cell Bank of the RIKEN BioResource Center, Tsukuba, Japan. Regarding CBMC usages, the study was approved by the ethics committee of Kobe University Graduate School of Medicine and National Institute of Biomedical Innovation.
The HHV-6A strain U1102 [34] were used for this study, and HHV-6 cell-free virus was prepared as described previously [35]. CBMCs were infected with HHV-6A, cultured in medium with phosphonoformic acid (PFA; 100 μg/ml), which inhibits viral DNA synthesis, and harvested at 24 hpi.
Antibodies
Monoclonal antibody (Mab) BU14, which recognizes the C-terminal region of HHV-6 U14, was described previously [9]. Mab AgQ1–119, for HHV-6A gQ1 was described previously [36] Anti-HA antibody (clone HA-7; Sigma), rabbit polyclonal antibody against EDD (BETHYLor Abcam), and anti–α-tubulin antibody (Sigma) were purchased from the indicated suppliers. Alexa Fluor 488- or 594-conjugated F(ab’)2 fragment of donkey anti-rabbit or-mouse immunoglobulin G (IgG) (Invitrogen) was used as a secondary antibody.
Immunofluorescence assay
Indirect immunofluorescence assay (IFA) was performed as described previously [33,37,38]. Specific IFA signals were detected using a confocal laser-scanning microscope (Olympus FluoView FV1000; Olympus).
Western blotting
Western blotting was performed as described previously [37].
Immunoprecipitation and LC/MS analysis
To investigate protein–protein associations, 293T cells transfected with U14 expressing plasmid were lysed in TNE buffer (10 mM Tris-HCl [pH 7.8], 0.15 M NaCl, 1 mM EDTA, and 1% NP-40 [Nacalai Tesque]). For immunoprecipitations, antibodies were bound to protein G–Sepharose (GE Healthcare) and then cross-linked with protein G using dimethyl pimelimidate (DMP; Thermo Scientific) [39]. Whole-cell extracts were then incubated with the appropriate protein G–Sepharose-bound antibody. Bound proteins were eluted with 0.1 M glycin (PH 2.5) and neutralized by using 1 M Tris-HCl (PH 9.0). The eluates were further prepared for LC/MS analysis as described elsewhere [40,41]. LC/MS analysis was performed using a Qstar-XL mass spectrometer (Applied Biosystems).
Plasmid construction
N-terminally HA-tagged HHV-6A U14 and its deletion-mutant expression plasmids were constructed using plasmid pCAGGS. pCAGGS was kindly provided by Jun-ichi Miyazaki (Osaka University, Japan) [42]. DNA fragments were amplified by PCR from cDNA from CBMCs infected with the HHV-6A strain U1102, and then inserted in frame into pCAGGS via the NotI and EcoRI sites.
Plasmid transfection
293T cells were transfected with expression plasmids using Lipofectamine 2000 (Invitrogen) or the calcium phosphate method, as described previously [43].
Construction of replication-incompetent recombinant adenovirus vectors
U14 and its mutant genes were excised from each pCAGGS-MCS-expression plasmid using the KpnI and NotI sites, and then cloned into the pHMCA5 vector [44]. The resultant plasmids were digested with I-CeuI and PI-SceI, and then cloned into pAdHM34 adenovirus vector [45]. To generate recombinant adenoviruses, 293 cells were transfected with PacI-digested adenovirus vectors using Lipofectamine 2000 [46]. Ten days later, the viruses were released from the cells by freezing and thawing, and then amplified in 293 cells. Recombinant adenoviruses were purified by CsCl gradient ultracentrifugation and titrated using the Adenovirus Titration Kit (Clontech).
Replication-incompetent recombinant adenovirus infection
HeLa cells were infected with empty adenovirus vector or adenovirus expressing U1102 U14 or its deletion mutants at a multiplicity of infection (MOI) of 100. Infections were carried out for 1 h at 37°C in 95% air/5% CO2.
Cell-cycle analysis by flow cytometry
HeLa cells infected with replication-incompetent recombinant adenovirus were trypsinized and washed. Subsequently, cells were fixed overnight in cold 70% ethanol at-20°C. The fixed cells were washed and then incubated for 30 min at room temperature with PBS containing 20 μg/ml propidium iodide (PI), 0.2 mg/ml RNase A, and 0.1% Triton X-100. Flow-cytometric analysis was performed on a Cell Analyzer EC800 (SONY).
Statistical analysis
Data are expressed as mean ± standard deviations (SD). Significance between groups was examined using Student’s t test. P values less than 0.05 were considered to represent statistically significant differences.
Results
Identification of cellular molecules that associate with U14
Previously, we showed that p53 interacts with the HHV-6 U14 protein in HHV-6 infected cells and is incorporated into virions during the late phase of infection. Early in HHV-6 infection, U14 localizes in the nucleus, where it is distributed in a dot-like pattern. Subsequently, U14 translocates to the cytoplasm, possibly to function as a tegument protein during the late phase of infection. To elucidate the function of U14 more precisely, we tried to identify other cellular molecules that associate with it. To this end, we first transfected HHV-6B U14-expressing vector or empty vector into 293T cells and immunoprecipitated the protein from lysates using an anti-U14 monoclonal antibody (Mab). For immunoprecipitations (IP), the Mab was bound to protein G–Sepharose and then cross-linked to protein G. Whole-cell extracts were then incubated with the appropriate protein G–Sepharose-bound antibody. Bound proteins were eluted, and the eluates were prepared for LC/MS analysis. p53 and EDD were mainly detected in the eluates of U14-expressing cell lysates.
To confirm the association between EDD and U14, we transfected HA-tagged U14 protein into 293T cells and immunoprecipitated whole-cell lysates using an anti-HA antibody (to bind tagged U14), anti-U14 Mab, or anti-EDD antibody. The resultant immunoprecipitates were subjected to Western blotting with the anti-U14 Mab or anti-EDD antibody. As shown in Fig 1, EDD co-precipitated with U14, and U14 reciprocally co-precipitated with EDD. Next, the association was examined in lysates of HHV-6A-infected Molt3 cells. As expected, EDD and U14 reciprocally co-precipitated (Fig 1).These results confirmed that U14 associated with EDD, in cells transiently expressing U14 and in cells infected with the HHV-6A virus.
Fig 1. EDD is a cellular molecule that associates with HHV-6 U14.

(A)293T cells were transfected with HA-tagged HHV-6A U14 or empty vector (as a control). Transfected cells were lysed at 72 h post-transfection. The lysates were immunoprecipitated (IP) with anti-HA or anti-EDD antibody, followed by Western blotting (WB) with anti-U14 (Mab BU14) or anti-EDD antibody. (B)Mock- or HHV-6A-infected cell lysates were immunoprecipitated with anti-U14 or anti-EDD antibody followed by Western blotting.
Next, we examined the localization of U14 and EDD in HHV-6A-infected cells. To this end, we infected Molt3 with HHV-6A, cultured the cells with or without phosphonoformic acid (PFA), which inhibits viral DNA synthesis, and fixed the cells at 24 h post-infection (pi). As shown in Fig 2, in the early phase of infection, U14 localized as dots in the nucleus, even in the presence of PFA, as previously reported [9]. EDD also localized in nuclear dots and colocalized with U14 in HHV-6A-infected cells, with or without PFA. By contrast, in mock-infected cells, EDD localized in the nucleus, but as tiny faint dots. These results indicated that, in HHV-6A-infected cells, EDD localizes to prominent nuclear dots.
Fig 2. Subcellular localization of U14 and EDD in HHV-6A-infected cells.

HHV-6A- or mock-infected Molt3 cells, with or without PFA, were harvested at 24 h post-infection (pi), fixed, and subjected to IFA using antibodies against U14 and EDD (A) or gQ1(B); nuclear DNA was counterstained with Hoechst 33342. Merged panels show colocalized U14 and EDD in nuclei (A). gQ1 was detected in the cells without PFA, but not with PFA (B). Scale bars: 5 μm (A) and 20μm (B). Single sections are shown.
Localization and association of U14 and EDD
To confirm whether the altered localization of EDD in HHV-6A-infected cells was due only to the association with U14, we transfected the U14 gene into 293T cells and observed the localization of endogenous EDD. At 12 h post-transfection, the cells were fixed and co-stained with anti-EDD and anti-U14 antibodies. As in HHV-6A-infected cells, transfected U14 localized to nuclear dots. In cells transfected with empty vector, EDD appeared only as tiny faint dots (Fig 3, control), but in cells transfected with U14, EDD shifted into larger nuclear dots that colocalized with U14. These results demonstrated that the change of localization of EDD observed in HHV-6A-infected cells was caused by co-expression with U14.
Fig 3. Subcellular localization of U14 and EDD in U14-expressing 293T cells.

293T cells were transfected with HHV-6A U14, its deletion mutants, or empty vector (as a control). Cells were harvested at 12 h post-transfection, fixed, and subjected to IFA using antibodies against HA (for U14) and EDD; nuclear DNA was counterstained with Hoechst 33342. Scale bars: 5 μm. Single sections are shown.
To determine whether co-expression of EDD with U14 is sufficient for the dot-like localization of EDD, we constructed several U14 mutants (Fig 4A) and then analyzed the colocalization of EDD with the mutant proteins. 293T cells were transfected with U14 or its mutants; at 12 h post-transfection, cells were fixed and co-stained with anti-U14 and EDD antibodies. Like wild-type U14, U14Δ429–431 colocalized with EDD in nuclear dots, whereas U14Δ431 or U14Δ429–600 localized mainly in the cytoplasm and did not colocalize with EDD. Localization of EDD was not altered in any of these transfected cells.
Fig 4. Schematic diagram of HHV-6A U14, its deletion mutants, and the localization of each mutant.

(A) The HHV-6A strain U1102 U14 and its deletion mutants (white box) were N-terminally fused to the HA tag (gray box). Numbers indicate positions in the amino acid sequence of U14. (B) 293T cells were transfected with HHV-6A U14, its deletion mutants, or empty vector as a negative control. The cells were harvested at 12 h post-transfection, fixed, and subjected to IFA using antibodies against HA (for U14) and EDD; nuclear DNA was counterstained with Hoechst 33342. Scale bars: 5 μm. Single sections are shown.
Next, we examined co-precipitation of EDD and each U14 mutant by IP followed by Western blotting. As shown in Fig 5, EDD reciprocally co-precipitated with U14 and U14Δ429–431, but not U14Δ431 or U14Δ429–600. These results also suggested that the association between U14 and EDD determines the colocalization of the two proteins, and that the association of EDD with U14 is sufficient for the dot-like localization of EDD. Although the abundant expression of U14Δ431 and U14Δ429–600 (compared with that of wild-type U14) could only be confirmed with IFA (Fig 4), but not with western blotting (indicated as Lysate in Fig 5), the association of EDD with U14Δ431 or U14Δ429–600 could not be detected even in longer exposure of western blotting (lower right panel in Fig 5). These results show that U14Δ431 and U14Δ429–600 didn’t associate with EDD. In addition, U14Δ429–431 did not co-precipitated with p53, although it did co-precipitate with EDD, whereas U14Δ431 co-precipitated with p53 but not with EDD (Fig 5). Thus, the C-terminus of U14 is crucial for the association with EDD, and the interaction of U14 with p53 does not affect the change of EDD localization.
Fig 5. Associations of U14 with EDD and p53.
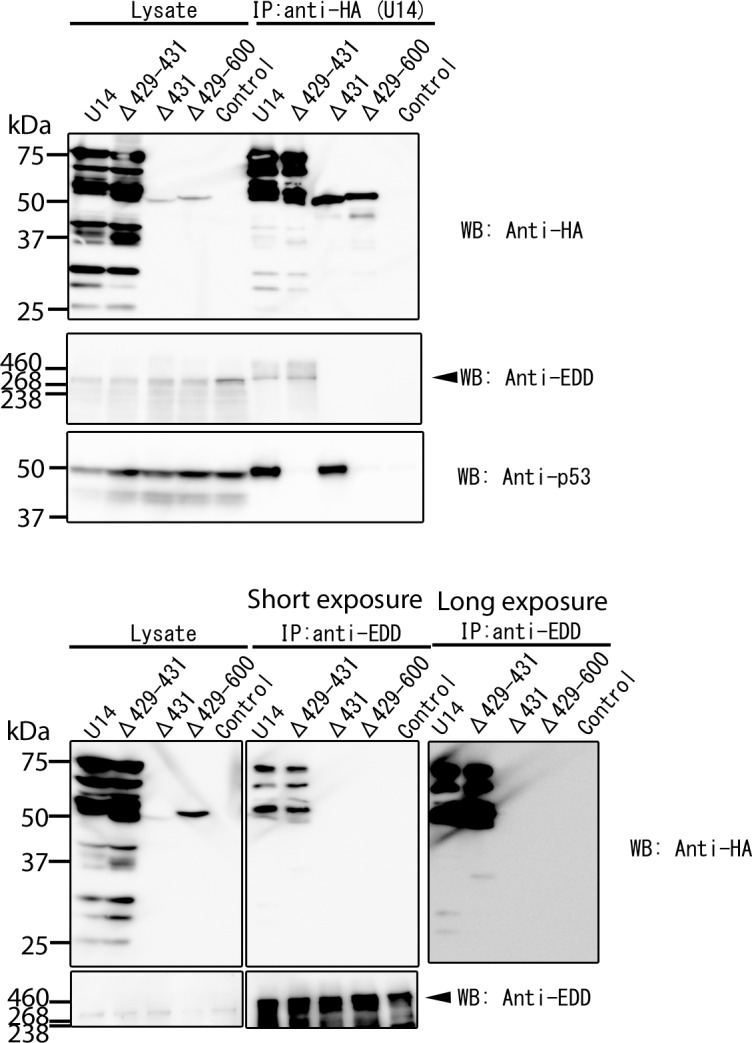
293T cells were transfected with HA-tagged HHV-6A U14, its deletion mutants, or empty vector (as a negative control). Transfected cells were lysed with TNE buffer at 72 h post-transfection. The lysates were immunoprecipitated (IP) with anti-HA antibody (upper) or anti-EDD antibody (lower), and then analyzed by Western blotting with antibody against HA (to detect U14), EDD, or p53. These data show one of three independent experiments and data in the upper and lower panels were from two independent experiments. WB; Western blotting.
U14 induces cell-cycle arrest at G2/M phase
Next, we investigated the functional significance of the U14–EDD association. EDD mediates DNA-damage signal transduction [12], and it is necessary for G1/S and intra-S-phase DNA-damage checkpoint activation and for maintenance of G2/M arrest after double-strand DNA breaks [13]. Therefore, we hypothesized that U14 expressed during the early phase of HHV-6 infection might influence DNA checkpoint activation by associating with EDD in HHV-6-infected cells.
To study the function of U14 in the cell cycle, we overexpressed U14 in HeLa cells using a replication-incompetent recombinant adenovirus vector. We then performed cell-cycle analyses of empty adenovirus-infected and U14-expressing adenovirus-infected cells at 12, 31 and 34 h post-infection (pi), using propidium iodide (PI) staining to measure DNA content. The empty adenovirus—infected cells were used as control. These analyses revealed that empty adenovirus-infected cells maintained normal cell-cycle profiles during infection, even at 34 hpi (Fig 6). However, U14-expressing cells exhibited a dramatic reduction in the proportion of cells in G1 phase, and a significant increase in the proportion of cells in G2/M phase, relative to empty adenovirus-infected cells. These results show that U14 expression induces cell cycle arrest at G2/M phase, and then induces cell-cycle arrest in G2/M phase. Next, we performed cell-cycle analyses using control-, U14-, U14Δ429–431-, U14Δ431-, and U14Δ429–600-expressing cells (Fig 7). As expected, as shown in Fig 7A and 7B, U14Δ429–431-expressing cells significantly decreased the proportion of cells in G1 phase and increased the proportion of cells in S and G2/M, relative to control cells; however, in U14Δ431- and U14Δ429–600-expressing cells, the reduction in the G1 population was less pronounced, although the G2/M population did increase slightly, indicating that G2/M progression from G1 and S was less strongly induced in U14Δ431- or U14Δ429–600-expressing cells. Expression of U14, U14Δ431, and U14Δ429–600 proteins was confirmed by Western blots performed on the corresponding cell lysates (Fig 7C). These results indicate that the U14 protein can induce cell-cycle arrest in G2/M phase, and that the U14–EDD association plays a key role in this arrest.
Fig 6. U14 induces cell-cycle arrest in G2/M phase.

HeLa cells were empty adenovirus vector-infected (described as control) or infected with adenovirus expressing U14. Subsequently, cells were fixed at the indicated time points and stained with propidium iodide (PI). Flow-cytometric analyses of DNA content were performed by measuring the levels of PI. Numbers in each graph represent percentages of cells in the G0/G1, S, and G2/M phases of the cell cycle. Data represent one of two independent experiments.
Fig 7. C-terminus of U14 is crucial for cell-cycle arrest in G2/M phase.

HeLa cells were infected with empty adenovirus vector (described as control), adenovirus expressing U14, or its deletion mutants (U14Δ429–431, U14Δ431, or U14Δ429–600) at a multiplicity of infection (MOI) of 100. Subsequently (34 hpi), cells were fixed and stained with PI. (A) FACS analyses of DNA content were performed by measuring the levels of PI in U14-expressing cells. Numbers in each graph represent percentages of cells in the G0/G1, S, and G2/M phases of the cell cycle. Data represent one of three independent experiments. (B) The percentages of control or U14-expressing adenovirus-infected cells in G1, S, and G2/M phases at 34 hpi. Data are shown as means ± SD from three independent experiments. Asterisks indicate significant differences (** p < 0.01). (C) Cell lysates shown above (collected 34 hpi) were used for Western blotting, and the expression level of each protein was examined.
Discussion/Conclusions
Previously, we showed that HHV-6 U14 can interact with p53 [9]. In this study, we identified EDD as a novel U14- associating protein. EDD is an E3 ubiquitin-protein ligase that regulates S phase and G2/M DNA-damage checkpoints, [13]. Furthermore, EDD itself physically interacts with p53, and this interaction blocks phosphorylation of p53 by ATM [14]. EDD inhibits ATM-mediated phosphorylation of p53-Ser15 and suppresses the induction of p53 target genes during DNA damage, but this effect does not require its E3 ligase activity. Thus, through binding to p53, EDD inhibits p53 phosphorylation by ATM and plays a role in ensuring smooth G1/S progression [14].
When HHV-6 U14 is expressed in the early phase of infection, it localizes in nuclear dots; later in infection, however, it is localized mainly in the cytoplasm and is incorporated into virus particles as a tegument protein [9]. When EDD is co-expressed with U14, it also localizes in nuclear dots and colocalizes with U14; by contrast, in the absence of U14, EDD appears in the nucleus as fainter and much smaller dots. In HHV-6-infected cells, EDD also appears in prominent nuclear dots containing U14, especially during the early stages of infection. Although U14 translocates to the cytoplasm during the late phase of infection, EDD likely maintains its localization in the nucleus and no longer colocalizes with U14. Based on its localization and association patterns during the early phase of infection, we hypothesized that U14 modulates the cell cycle by associating with EDD. Therefore, we investigated whether U14 could induce cell-cycle arrest using replication-incompetent recombinant adenovirus vector in HeLa cells. As shown in Fig 6, we observed cell-cycle arrest at G2/M phase in U14-expressing cells.
Data obtained using deletion mutants of U14 showed that the association of U14 with EDD enabled cell-cycle into G2/M phase. One deletion mutant of U14, U14Δ429–431, which associated with EDD but not p53, could still induce cell arrest at G2/M phase. Thus, the cell-cycle arrest induced by U14 may not require p53. In addition, neither U14Δ431 nor U14Δ429–600 associated with EDD, although both of them could associate with p53, and both proteins induced cell arrest at G2/M phase much more weakly than wild-type U14. These results indicate that the C-terminal region of U14 is important for the binding with EDD and consequently for U14-induced cell arrest at G2/M phase, and also demonstrate that p53 could not be required for cell-cycle arrest induced by U14. Previously, we showed that U14 could interact with p53 in both HHV-6-infected and U14-expressing cells [9]. Although the role of the U14–p53 interaction remains unclear, it is possible that U14 induces the stabilization and functional inactivation of p53. Consistent with this, HHV-6 can induce cell-cycle arrest at G1/S or G2/M phase [20,21,22,23,24,25], and the HHV-6 DR6 protein has the ability to inhibit G2/M transition independently of p53 [31]; furthermore, HHV-6B U19 inhibits p53-dependent cell death [32]. Therefore, it is possible that U14 promotes cell-cycle arrest at G2/M independently of p53. Taken together, the results of this study demonstrate that U14 acts as an important cell-cycle modulator in HHV-6 infection.
Acknowledgments
We thank H. Oyaizu and M. Higuchi (Osaka University) for technical assistance, J. Miyazaki (Osaka University) for providing reagents, and H. Yamada (Kobe University) for the CBMCs. This work was supported by a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS).
Data Availability
All relevant data are within the paper.
Funding Statement
This work was supported by a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS).
References
- 1. Ablashi D, Agut H, Alvarez-Lafuente R, Clark DA, Dewhurst S, DiLuca D, et al. Classification of HHV-6A and HHV-6B as distinct viruses. Arch Virol. 2014;159(5):863–70. Epub 2013/11/07. 10.1007/s00705-013-1902-5 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Okuno T, Takahashi K, Balachandra K, Shiraki K, Yamanishi K, Takahashi M, et al. Seroepidemiology of human herpesvirus 6 infection in normal children and adults. J Clin Microbiol. 1989;27(4):651–3. Epub 1989/04/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yamanishi K, Okuno T, Shiraki K, Takahashi M, Kondo T, Asano Y, et al. Identification of human herpesvirus-6 as a causal agent for exanthem subitum. Lancet. 1988;1(8594):1065–7. Epub 1988/05/14. doi: S0140–6736(88)91893–4 [pii]. . [DOI] [PubMed] [Google Scholar]
- 4. Clark DA. Human herpesvirus 6 and human herpesvirus 7: emerging pathogens in transplant patients. Int J Hematol. 2002;76 Suppl 2:246–52. Epub 2002/11/15. . [DOI] [PubMed] [Google Scholar]
- 5. Zerr DM, Corey L, Kim HW, Huang ML, Nguy L, Boeckh M. Clinical outcomes of human herpesvirus 6 reactivation after hematopoietic stem cell transplantation. Clin Infect Dis. 2005;40(7):932–40. Epub 2005/04/13. doi: CID34999 [pii] 10.1086/428060 . [DOI] [PubMed] [Google Scholar]
- 6. Dominguez G, Dambaugh TR, Stamey FR, Dewhurst S, Inoue N, Pellett PE. Human herpesvirus 6B genome sequence: coding content and comparison with human herpesvirus 6A. J Virol. 1999;73(10):8040–52. Epub 1999/09/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gompels UA, Nicholas J, Lawrence G, Jones M, Thomson BJ, Martin ME, et al. The DNA sequence of human herpesvirus-6: structure, coding content, and genome evolution. Virology. 1995;209(1):29–51. Epub 1995/05/10. doi: S0042–6822(85)71228–7 [pii] 10.1006/viro.1995.1228 . [DOI] [PubMed] [Google Scholar]
- 8. Isegawa Y, Mukai T, Nakano K, Kagawa M, Chen J, Mori Y, et al. Comparison of the complete DNA sequences of human herpesvirus 6 variants A and B. J Virol. 1999;73(10):8053–63. Epub 1999/09/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takemoto M, Koike M, Mori Y, Yonemoto S, Sasamoto Y, Kondo K, et al. Human herpesvirus 6 open reading frame U14 protein and cellular p53 interact with each other and are contained in the virion. J Virol. 2005;79(20):13037–46. Epub 2005/09/29. doi: 79/20/13037 [pii] 10.1128/JVI.79.20.13037-13046.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Callaghan MJ, Russell AJ, Woollatt E, Sutherland GR, Sutherland RL, Watts CK. Identification of a human HECT family protein with homology to the Drosophila tumor suppressor gene hyperplastic discs. Oncogene. 1998;17(26):3479–91. Epub 1999/02/25. 10.1038/sj.onc.1202249 . [DOI] [PubMed] [Google Scholar]
- 11. Henderson MJ, Russell AJ, Hird S, Munoz M, Clancy JL, Lehrbach GM, et al. EDD, the human hyperplastic discs protein, has a role in progesterone receptor coactivation and potential involvement in DNA damage response. J Biol Chem. 2002;277(29):26468–78. Epub 2002/05/16. 10.1074/jbc.M203527200 M203527200 [pii]. . [DOI] [PubMed] [Google Scholar]
- 12. Henderson MJ, Munoz MA, Saunders DN, Clancy JL, Russell AJ, Williams B, et al. EDD mediates DNA damage-induced activation of CHK2. J Biol Chem. 2006;281(52):39990–40000. Epub 2006/11/01. doi: M602818200 [pii] 10.1074/jbc.M602818200 . [DOI] [PubMed] [Google Scholar]
- 13. Munoz MA, Saunders DN, Henderson MJ, Clancy JL, Russell AJ, Lehrbach G, et al. The E3 ubiquitin ligase EDD regulates S-phase and G(2)/M DNA damage checkpoints. Cell Cycle. 2007;6(24):3070–7. Epub 2007/12/13. doi: 5021 [pii]. . [DOI] [PubMed] [Google Scholar]
- 14. Ling S, Lin WC. EDD inhibits ATM-mediated phosphorylation of p53. J Biol Chem. 2011;286(17):14972–82. Epub 2011/03/09. 10.1074/jbc.M110.182527 M110.182527 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davy C, Doorbar J. G2/M cell cycle arrest in the life cycle of viruses. Virology. 2007;368(2):219–26. Epub 2007/08/07. doi: S0042–6822(07)00387-X [pii] 10.1016/j.virol.2007.05.043 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fehr AR, Yu D. Control the host cell cycle: viral regulation of the anaphase-promoting complex. J Virol. 2013;87(16):8818–25. Epub 2013/06/14. 10.1128/JVI.00088-13 JVI.00088–13 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Izumi T, Io K, Matsui M, Shirakawa K, Shinohara M, Nagai Y, et al. HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc Natl Acad Sci U S A. 2010;107(48):20798–803. Epub 2010/11/13. 10.1073/pnas.1008076107 1008076107 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reitsma JM, Savaryn JP, Faust K, Sato H, Halligan BD, Terhune SS. Antiviral inhibition targeting the HCMV kinase pUL97 requires pUL27-dependent degradation of Tip60 acetyltransferase and cell-cycle arrest. Cell Host Microbe. 2011;9(2):103–14. Epub 2011/02/16. 10.1016/j.chom.2011.01.006 S1931–3128(11)00027–8 [pii]. . [DOI] [PubMed] [Google Scholar]
- 19. Zhao RY, Elder RT. Viral infections and cell cycle G2/M regulation. Cell Res. 2005;15(3):143–9. Epub 2005/03/23. 10.1038/sj.cr.7290279 . [DOI] [PubMed] [Google Scholar]
- 20. De Bolle L, Hatse S, Verbeken E, De Clercq E, Naesens L. Human herpesvirus 6 infection arrests cord blood mononuclear cells in G(2) phase of the cell cycle. FEBS Lett. 2004;560(1–3):25–9. Epub 2004/02/28. 10.1016/S0014-5793(04)00035-3 S0014579304000353 [pii]. . [DOI] [PubMed] [Google Scholar]
- 21. Li L, Gu B, Zhou F, Chi J, Feng D, Xie F, et al. Cell cycle perturbations induced by human herpesvirus 6 infection and their effect on virus replication. Arch Virol. 2014;159(2):365–70. Epub 2013/09/10. 10.1007/s00705-013-1826-0 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li L, Gu B, Zhou F, Chi J, Wang F, Liu G, et al. Human herpesvirus 6A infects human embryonic fibroblasts and induces G2/M arrest and cell death. J Med Virol. 2012;84(4):657–63. Epub 2012/02/18. 10.1002/jmv.23226 . [DOI] [PubMed] [Google Scholar]
- 23. Li L, Gu B, Zhou F, Chi J, Wang F, Peng G, et al. Human herpesvirus 6 suppresses T cell proliferation through induction of cell cycle arrest in infected cells in the G2/M phase. J Virol. 2011;85(13):6774–83. Epub 2011/04/29. 10.1128/JVI.02577-10 JVI.02577–10 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mlechkovich G, Frenkel N. Human herpesvirus 6A (HHV-6A) and HHV-6B alter E2F1/Rb pathways and E2F1 localization and cause cell cycle arrest in infected T cells. J Virol. 2007;81(24):13499–508. Epub 2007/10/05. doi: JVI.01496–07 [pii] 10.1128/JVI.01496-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oster B, Bundgaard B, Hollsberg P. Human herpesvirus 6B induces cell cycle arrest concomitant with p53 phosphorylation and accumulation in T cells. J Virol. 2005;79(3):1961–5. Epub 2005/01/15. doi: 79/3/1961 [pii] 10.1128/JVI.79.3.1961-1965.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oster B, Kaspersen MD, Kofod-Olsen E, Bundgaard B, Hollsberg P. Human herpesvirus 6B inhibits cell proliferation by a p53-independent pathway. J Clin Virol. 2006;37 Suppl 1:S63–8. Epub 2007/02/06. doi: S1386–6532(06)70014–2 [pii] 10.1016/S1386-6532(06)70014-2 . [DOI] [PubMed] [Google Scholar]
- 27. Oster B, Kofod-Olsen E, Bundgaard B, Hollsberg P. Restriction of human herpesvirus 6B replication by p53. J Gen Virol. 2008;89(Pt 5):1106–13. Epub 2008/04/19. 10.1099/vir.0.83262-0 89/5/1106 [pii]. . [DOI] [PubMed] [Google Scholar]
- 28. Oster B, Bundgaard B, Hupp TR, Hollsberg P. Human herpesvirus 6B induces phosphorylation of p53 in its regulatory domain by a CK2- and p38-independent pathway. J Gen Virol. 2008;89(Pt 1):87–96. Epub 2007/12/20. doi: 89/1/87 [pii] 10.1099/vir.0.83136-0 . [DOI] [PubMed] [Google Scholar]
- 29. Sharon E, Volchek L, Frenkel N. Human herpesvirus 6 (HHV-6) alters E2F1/Rb pathways and utilizes the E2F1 transcription factor to express viral genes. Proc Natl Acad Sci U S A. 2014;111(1):451–6. Epub 2013/12/18. 10.1073/pnas.1308854110 1308854110 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takemoto M, Mori Y, Ueda K, Kondo K, Yamanishi K. Productive human herpesvirus 6 infection causes aberrant accumulation of p53 and prevents apoptosis. J Gen Virol. 2004;85(Pt 4):869–79. Epub 2004/03/25. . [DOI] [PubMed] [Google Scholar]
- 31. Schleimann MH, Hoberg S, Solhoj Hansen A, Bundgaard B, Witt CT, Kofod-Olsen E, et al. The DR6 protein from human herpesvirus-6B induces p53-independent cell cycle arrest in G2/M. Virology. 2014;452–453:254–63. Epub 2014/03/13. 10.1016/j.virol.2014.01.028 S0042–6822(14)00041–5 [pii]. . [DOI] [PubMed] [Google Scholar]
- 32. Kofod-Olsen E, Moller JM, Schleimann MH, Bundgaard B, Bak RO, Oster B, et al. Inhibition of p53-dependent, but not p53-independent, cell death by U19 protein from human herpesvirus 6B. PLoS One. 2013;8(3):e59223 Epub 2013/04/05. 10.1371/journal.pone.0059223 PONE-D-12–34687 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mori Y, Yang X, Akkapaiboon P, Okuno T, Yamanishi K. Human herpesvirus 6 variant A glycoprotein H-glycoprotein L-glycoprotein Q complex associates with human CD46. J Virol. 2003;77(8):4992–9. Epub 2003/03/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mori Y. Recent topics related to human herpesvirus 6 cell tropism. Cell Microbiol. 2009;11(7):1001–6. Epub 2009/03/18. 10.1111/j.1462-5822.2009.01312.x CMI1312 [pii]. . [DOI] [PubMed] [Google Scholar]
- 35. Mori Y, Yagi H, Shimamoto T, Isegawa Y, Sunagawa T, Inagi R, et al. Analysis of human herpesvirus 6 U3 gene, which is a positional homolog of human cytomegalovirus UL 24 gene. Virology. 1998;249(1):129–39. Epub 1998/09/19. doi: S0042–6822(98)99305–9 [pii] 10.1006/viro.1998.9305 . [DOI] [PubMed] [Google Scholar]
- 36. Mori Y, Akkapaiboon P, Yang X, Yamanishi K. The Human Herpesvirus 6 U100 Gene Product Is the Third Component of the gH-gL Glycoprotein Complex on the Viral Envelope. Journal of Virology. 2003;77(4):2452–8. 10.1128/jvi.77.4.2452-2458.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Akkapaiboon P, Mori Y, Sadaoka T, Yonemoto S, Yamanishi K. Intracellular processing of human herpesvirus 6 glycoproteins Q1 and Q2 into tetrameric complexes expressed on the viral envelope. J Virol. 2004;78(15):7969–83. Epub 2004/07/16. 10.1128/JVI.78.15.7969-7983.2004 78/15/7969 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ota M, Serada S, Naka T, Mori Y. MHC class I molecules are incorporated into human herpesvirus-6 viral particles and released into the extracellular environment. Microbiol Immunol. 2014;58(2):119–25. Epub 2013/12/18. 10.1111/1348-0421.12121 . [DOI] [PubMed] [Google Scholar]
- 39. Kawabata A, Jasirwan C, Yamanishi K, Mori Y. Human herpesvirus 6 glycoprotein M is essential for virus growth and requires glycoprotein N for its maturation. Virology. 2012;429(1):21–8. Epub 2012/04/28. 10.1016/j.virol.2012.03.027 S0042–6822(12)00188–2 [pii]. . [DOI] [PubMed] [Google Scholar]
- 40. Nakamura T, Kuromitsu J, Oda Y. Evaluation of comprehensive multidimensional separations using reversed-phase, reversed-phase liquid chromatography/mass spectrometry for shotgun proteomics. J Proteome Res. 2008;7(3):1007–11. Epub 2008/02/06. 10.1021/pr7005878 . [DOI] [PubMed] [Google Scholar]
- 41. Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18(6):911–7. Epub 2012/05/23. 10.1038/nm.2749 nm.2749 [pii]. . [DOI] [PubMed] [Google Scholar]
- 42. Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193–9. Epub 1991/12/15. . [DOI] [PubMed] [Google Scholar]
- 43. Koshizuka T, Ota M, Yamanishi K, Mori Y. Characterization of varicella-zoster virus-encoded ORF0 gene—comparison of parental and vaccine strains. Virology. 2010;405(2):280–8. Epub 2010/07/06. 10.1016/j.virol.2010.06.016 S0042–6822(10)00398–3 [pii]. . [DOI] [PubMed] [Google Scholar]
- 44. Sakurai F, Kawabata K, Yamaguchi T, Hayakawa T, Mizuguchi H. Optimization of adenovirus serotype 35 vectors for efficient transduction in human hematopoietic progenitors: comparison of promoter activities. Gene Ther. 2005;12(19):1424–33. Epub 2005/06/10. doi: 3302562 [pii] 10.1038/sj.gt.3302562 . [DOI] [PubMed] [Google Scholar]
- 45. Mizuguchi H, Hayakawa T. Adenovirus vectors containing chimeric type 5 and type 35 fiber proteins exhibit altered and expanded tropism and increase the size limit of foreign genes. Gene. 2002;285(1–2):69–77. Epub 2002/06/01. doi: S0378111902004109 [pii]. . [DOI] [PubMed] [Google Scholar]
- 46. Kawabata K, Sakurai F, Yamaguchi T, Hayakawa T, Mizuguchi H. Efficient gene transfer into mouse embryonic stem cells with adenovirus vectors. Mol Ther. 2005;12(3):547–54. Epub 2005/06/14. doi: S1525–0016(05)00185–1 [pii] 10.1016/j.ymthe.2005.04.015 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.