

Abstract
Calcium signalling is fundamental to the function of the nervous system, in association with changes in ionic gradients across the membrane. Although restoring ionic gradients is energetically costly, a rise in intracellular Ca2+ acts through multiple pathways to increase ATP synthesis, matching energy supply to demand. Increasing cytosolic Ca2+ stimulates metabolite transfer across the inner mitochondrial membrane through activation of Ca2+-regulated mitochondrial carriers, whereas an increase in matrix Ca2+ stimulates the citric acid cycle and ATP synthase. The aspartate–glutamate exchanger Aralar/AGC1 (Slc25a12), a component of the malate–aspartate shuttle (MAS), is stimulated by modest increases in cytosolic Ca2+ and upregulates respiration in cortical neurons by enhancing pyruvate supply into mitochondria. Failure to increase respiration in response to small (carbachol) and moderate (K+-depolarization) workloads and blunted stimulation of respiration in response to high workloads (veratridine) in Aralar/AGC1 knockout neurons reflect impaired MAS activity and limited mitochondrial pyruvate supply. In response to large workloads (veratridine), acute stimulation of respiration occurs in the absence of MAS through Ca2+ influx through the mitochondrial calcium uniporter (MCU) and a rise in matrix [Ca2+]. Although the physiological importance of the MCU complex in work-induced stimulation of respiration of CNS neurons is not yet clarified, abnormal mitochondrial Ca2+ signalling causes pathology. Indeed, loss of function mutations in MICU1, a regulator of MCU complex, are associated with neuromuscular disease. In patient-derived MICU1 deficient fibroblasts, resting matrix Ca2+ is increased and mitochondria fragmented. Thus, the fine tuning of Ca2+ signals plays a key role in shaping mitochondrial bioenergetics.
Irene Llorente-Folch received her PhD from Autonomous University of Madrid in 2013 under the supervision of Prof Jorgina Satrustegui's at the Centre for Molecular Biology “Severo Ochoa” CSIC-UAM and Center for Biomedical Network Research on Rare Diseases and was supported by the Grant MITOLAB-CM. Work in Jorgina Satrustegui's lab has focused on calcium signaling to mitochondria in health and disease with emphasis in the roles played by mitochondrial metabolite transporters which are regulated by extramitochondrial calcium. Michael R. Duchen studied Physiology and Medicine in Oxford and London. He worked in hospital medicine for a few years before embarking on PhD studies in the Physiology department at UCL, with Tim Biscoe as supervisor and mentor. He has stayed at UCL ever since. He developed an interest in mitochondrial cell biology, and his has focussed on understanding the integration of mitochondrial function and cell signaling events in health and disease.

Introduction
In this review, we focus on the role played by Ca2+ ions in the modulation of cellular respiration, as well as the mechanisms involved. This role for Ca2+ is ubiquitous, and most probably can be generalized to all cell types, as will be discussed in the initial part of this review. We then consider in more detail Ca2+-mediated regulation of mitochondrial energy metabolism in neurons as the prototype of the mechanisms involved. Indeed, neurons are responsible for disproportionate oxygen consumption at rest in humans (the brain uses ∼20% of the total oxygen consumed at rest but represents only 2% of body mass; Mink et al. 1981). In addition, neurons are critically and almost exclusively dependent on mitochondrial oxidative phosphorylation (OXPHOS) as a major source of ATP and have a limited capacity to upregulate energy supply through glycolysis when OXPHOS is compromised (Herrero-Mendez et al. 2009). Mitochondria in these cells represent an exclusive target for Ca2+ to guarantee activity-dependent regulation of cellular energy metabolism. Overall, Ca2+-dependent regulation of OXPHOS involves two principal mechanisms: (i) Ca2+ entry into mitochondria through the Ca2+ uniporter (mitochondrial calcium uniporter; MCU) and (ii) Ca2+-dependent activation of mitochondrial metabolite transporters (Ca2+-regulated mitochondrial carriers), where Ca2+ acts on the external surface of the inner mitochondrial membrane. Thus, even though cytosolic and mitochondrial Ca2+ signals are usually tightly coupled, they can also have distinct effects on mitochondrial metabolism, ensuring differential regulation in some cases. Because some of the mechanisms employed by Ca2+ to modulate respiration have only been described in cells other than neurons, we will refer to other cell types (heart, fibroblasts) throughout the review to address these specific mechanisms, which particularly involve those related to the mitochondrial Ca2+ uniporter complex (MCUC) that has only been recently characterized at the molecular level; thus, only a few studies directly address its role in the regulation of OXPHOS.
Cell metabolism and ATP homeostasis
Specialized processes in differentiated cells consume ATP, such as neuronal transmission, muscle contraction, cellular motility and secretion. In addition, energy is required for cellular maintenance and repair to counter the forces of entropy. Events that require the disturbance of ionic gradients across the membrane are also almost invariably associated with Ca2+ signals, either through influx across the plasma membrane or by release from internal stores. Restoring ionic gradients by ion pumps in the plasma membrane [Na+/ Ca2+ exchanger (NCLX); Na+/K+ ATPase pump; Ca2+/H+ ATPase exchanger] and within the organelles (Ca2+/H+ ATPase exchanger) in the endoplasmic reticulum (ER) requires ATP consumption. It has also been known for many years that cells match the rate of ATP production and utilization with little or no measurable change in metabolic intermediates. The maintenance of cellular metabolites during alterations in workload has been termed metabolic homeostasis, and is probably most thoroughly studied in cardiac and skeletal muscle (Balaban 2002; 2006; Glancy et al. 2013).
Neurons are also subject to changes in workload. Most of the energy in the brain is consumed by synaptic transmission (Attwell and Laughlin, 2001; Hall et al. 2012; Harris et al. 2012). In high energy-demanding tissues such as the brain and skeletal and cardiac muscle, the rapid formation of ATP through phosphocreatine and the creatine kinase reaction maintains the distribution of ATP through the cell at almost constant levels and can be important in peak conditions of energy demand (Cerdan et al. 1990; Balaban et al. 2009). However, overall, the major sustainable source of energy is ATP generated by OXPHOS.
Recent studies have revealed that neuronal activity not only contributes significantly to ATP consumption, but also stimulates ATP synthesis through a Ca2+-dependent increase in OXPHOS (Rangaraju et al. 2014). Neuronal activity requires both rapid adaptation of oxidative energy metabolism and a sufficient supply of oxygen and nutrients and, thus, it is very sensitive to altered mitochondrial function (Whittaker et al. 2011; Kann et al. 2012). Furthermore, mitochondrial fission and redistribution to regions of increased metabolic demand have been observed during sustained impulse activity (Sajic et al. 2013), confirming that mitochondrial function is essential for the correct balance of neuronal function in response to an imposed workload.
In neurons using glucose as the main metabolic substrate, an increase in workload is necessarily associated with increased glucose oxidation and augmented oxygen consumption, which is controlled by the mitochondrial proton electrochemical gradient (ΔμH) and mainly used for ATP synthesis (Mitchell & Moyle, 1969).
Regulation of OXPHOS in response to work was initially considered to be carried out by a simple feedback of the ATP hydrolysis products ADP and Pi on mitochondrial ATP synthase (Chance & Williams, 1955; Jacobus et al. 1982). The classical principles of chemiosmotic coupling dictate that increased ATP production by mitochondria is coupled with increased oxygen consumption by the respiratory chain and an increased substrate supply to mitochondria. However, this is not the only mechanism driving changes in mitochondrial function in response to changes in workload. Indeed, it has become clear that Ca2+ regulation of mitochondrial function plays an important role in maintaining ATP homeostasis (McCormack & Denton, 1990; Rizzuto et al. 2012)
Calcium signalling and mitochondrial respiration
Ca2+ is a versatile and ubiquitous intracellular messenger, acting as a mediator of almost all energy demanding processes in mammalian cells. The capacity of mitochondria to take up large quantities of Ca2+ in a membrane potential-dependent manner has been known for decades (Deluca & Engstrom, 1961; Harris, 1977; Nicholls, 1978). Mitochondrial Ca2+ accumulation not only serves both as a Ca2+ buffering system in the cell, but also as a pathway to modulate the energy metabolism of the cell. Ca2+ handling involves a complex dialogue between the mitochondria, the ER, lysosomes, the plasma membrane and the nucleus. Gradients of Ca2+ across the membrane reflect a huge free energy and their maintenance represents a significant energetic burden (Glancy & Balaban, 2012; Rueda et al. 2014).
It is well known that Ca2+-dependent regulation of OXPHOS is mediated through Ca2+ entry into mitochondria through the MCU (Fig. 1). However, the identification of Ca2+-regulated mitochondrial carriers (CaMCs) (del Arco & Satrustegui 1998, del Arco et al. 2000) revealed an additional target of cytosolic Ca2+ signals in neuronal mitochondria. The critical difference between these pathways is that Ca2+-dependent regulation of OXPHOS through the carriers operates by the action of Ca2+ at the outer surface of the inner mitochondrial membrane, rather than in the matrix, and so does not require mitochondrial Ca2+ uptake (Fig. 1). [Ca2+]cyt-activated increase in ATP production by OXPHOS contributes to metabolic homeostasis (i.e. allows the ATP/ADP and NADH/NAD+ levels to remain constant) despite an increase in workload, as reported by Glancy & Balaban (2012).
Figure 1. Schematic representation of Ca2+regulation of mitochondrial respiration.
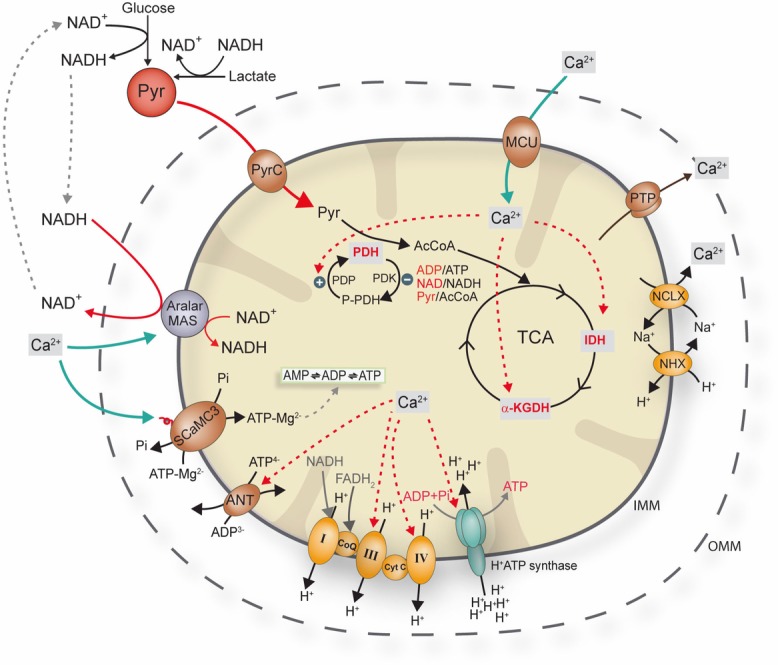
Tricarboxylic acid cycle enzymes are highly sensitive to changes in [Ca2+], which presumably binds directly to isocitrate dehydrogenase (IDH), α-ketoglutarate dehydrogenase (α-KGDH), whereas PDH is activated by the Ca2+-sensitive pyruvate dehydrogenase phosphatase. Complex IV and complex III may also be regulated by intramitochondrial Ca2+. Matrix Ca2+ may also regulate OXPHOS through an effect on the ANT and on the F1Fo-ATP synthase. Extramitochondrial Ca2+ activates Aralar/AGC1-MAS activity and SCaMC-3. P-PDH, phosphorilated pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PDP, pyruvate dehydrogenase phosphatase; Pyr, pyruvate; AcCoA, acetyl coenzyme A; TCA: tricarboxylic acid cycle; NHX, Na+/H+ exchanger;
To maintain substrate supply to mitochondria with an elevated respiratory rate, further control mechanisms acting upstream of mitochondria are required. For example, through its association with calmodulin, Ca2+ activates phosphorylase kinase, which in turn activates glycogen phosphorylase, initiating glycogen breakdown and so increasing glucose supply. This is not only a general pathway in tissues with significant glycogen stores, such as liver or muscle (Picton et al. 1981), but also takes place in astrocytes (Ibrahim et al. 1975; Newman et al. 2011; Müller et al. 2014a,b) and, to some extent, in neurons (Saez I et al. 2014). Thus, Ca2+ stimulates both glycogen breakdown and glucose oxidation, increasing ATP supply (McCormack et al. 1990; Müller b2014).
The physiological importance of respiratory control by Ca2+ in intact neurons and in the CNS in vivo is still largely unknown. Rapid Ca2+-dependent changes in oxygen consumption in response to membrane depolarization have been described in cultured Purkinje neurons (Hayakawa et al. 2005). However, other studies have found no evidence for a role of cytosolic Ca2+ in activity-dependent rises in cerebellar rate of oxygen consumption in the intact brain (Mathiensen et al. 2011). The resolution of these questions is confounded by two opposing actions of Ca2+ because it not only activates ATP production through the stimulation of OXPHOS, but also increases ATP consumption through the increased energy demand required to recover the ionic resting state.
Mitochondrial Ca2+ uptake and its impact on mitochondrial function
The inner mitochondrial membrane maintains a high membrane potential (ΔΨ) (∼150–180 mV negative to the cytosol) and Ca2+ uptake, an electrogenic process, induces a small transient mitochondrial depolarization (Vitorica & Satrustegui, 1985; Duchen, 1992), which correlates with the rising phase of [Ca2+]cyt, reflecting the Ca2+ current across the inner mitochondrial membrane. Because, in most cells, the mitochondrial matrix represents a small proportion of the total cell volume, and there is a strong electrochemical potential favouring the accumulation of Ca2+ into mitochondria, the movement of relatively few Ca2+ ions promotes large concentration changes in the matrix, with obvious signalling potential. Mechanisms of Ca2+ buffering in the matrix are poorly understood. Ca2+ uptake is facilitated by locating mitochondria close to microdomains of high [Ca2+]cyt, allowing for its operation even in the absence of global [Ca2+]cyt signals (Rizzuto et al. 1998; Contreras et al. 2010).
Ca2+ entry into mitochondria requires the recently identified MCU (Baughman et al. 2011; de Stefani et al. 2011), which forms part of a large complex, the MCUC (Rizzuto et al. 2012; Marchi & Pinton, 2014; Pendin et al. 2014), whose components are still not fully resolved. The complex includes MCUb, a dominant negative component of the oligomeric channel (Raffaello et al. 2013), the Ca2+ sensitivity modulators MICU1 and MICU2 (Perocchi et al. 2010; Mallilankaraman et al. 2012b; Bai et al. 2013; Plovanich et al. 2013; Patron et al. 2014), MCU regulator 1 (Mallilankaraman et al. 2012a; but see Paupe et al. 2015) and essential MCU regulator (Sancak et al. 2013). Although the topology of MCU was initially a matter of debate (Drago et al. 2011), its N- and C-terminal domains probably span into the mitochondrial matrix and its nine amino acid linker (i.e. the DIME domain) between the two transmembrane domains faces the intermembrane space (Baughman et al. 2011; Martell et al. 2012). The existence of only two putative transmembrane domains strongly suggests that an active and functional uniporter channel could only be formed by oligomers of MCU.
On the other hand, a K+/H+ antiporter (Froschauer et al. 2005; Nowikovsky et al. 2012; De Marchi et al. 2014; Doonan et al. 2014; Nowikovsky & Bernardi, 2014; Nowikovsky et al. 2004) has also been proposed to mediate Ca2+/H+ exchange; first, as a Ca2+ efflux pathway (Jiang et al. 2009; 2013; Tsai et al. 2014) and, more recently, as a Ca2+ influx pathway alternative to the MCU (Doonan et al. 2014). Furthermore, other routes of Ca2+ entry into mitochondria, such as a rapid mode of uptake (Gunter & Sheu, 2009; Ryu et al. 2010), might be responsible for residual Ca2+ uptake in mitochondria from MCU deficient cells (Pan et al. 2013) and this requires further investigation (Bondarenko et al. 2013; 2014).
Efflux pathways are also essential for equilibrating mitochondrial and cytosolic Ca2+ (Takeuchi et al. 2015). The major pathway is a Na+/Ca2+ exchanger that is distinct from the exchanger at the plasma membrane and was recently characterized as NCLX (Palty et al. 2010), whereas the Ca2+/H+ exchanger remains elusive (Fig. 1).
A number of enzymes in the mitochondrial matrix are regulated by Ca2+. In particular, the citric acid cycle dehydrogenases are extremely sensitive to Ca2+, which presumably binds directly to isocitrate dehydrogenase and α-ketoglutarate dehydrogenase, whereas pyruvate dehydroganase (PDH) is activated by Ca2+-sensitive phosphatase activity (Balaban, 2009; Denton, 2009) (Fig. 1). Ca2+ also modulates F1-FoATPase activity, promoting ATP generation at a given driving force, thus increasing the velocity of ATP production through a post-translational modification whose specific mechanism remains elusive (Balaban et al. 2009; Glancy & Balaban, 2012). Direct Ca2+-dependent activation of F1-ATP synthase by S100A1 has also been implicated in the heart (Borries et al. 2007). The fact that respiration rate increases with workload may also imply Ca2+ activation of some of the respiratory complexes (Balaban et al. 2009; Glancy & Balaban 2012) (Fig. 1). Ca2+ may also regulate OXPHOS through effects on adenine nucleotide translocase (ANT), which mediates the electrogenic exchange of ADP3- with ATP4- between the cytosol and the mitochondrial matrix without modifying the net content of adenine nucleotides (Klingenberg, 2008) (Fig. 1). A rise in intramitochondrial [Ca2+] diminishes ANT activity (Moreno-Sanchez, 1983), lowering the matrix ADP content, which decreases F1-FoATPase activity.
Signalling by extramitochondrial calcium
Aspartate-glutamate carriers (AGCs) and calcium-binding mitochondrial carrier proteins (SCaMCs/APCs)
Aspartate-glutamate carriers and ATP-Mg/Pi transporters (SCaMCs/APCs) (del Arco & Satrústegui, 1998; Palmieri et al. 2001; del Arco & Satrústegui, 2004; Fiermonte et al. 2004; Satrústegui et al. 2007) are the two classes of mitochondrial carriers activated by extramitochondrial Ca2+ (Pardo et al. 2006; Contreras et al. 2007; Traba et al. 2008; Traba et al. 2012).
AGCs are components of the malate–aspartate shuttle (MAS) and, under physiological conditions of polarized mitochondria, comprise the main site of regulation of the shuttle (Satrustegui et al. 2007). AGCs are activated by modest increases of extramitochondrial [Ca2+] at concentrations not far from the resting state. For example, Aralar/AGC1, the isoform prevailing in the brain, has an S0.5 of 324 nm Ca2+ (Palmieri et al. 2001; Pardo et al. 2006; Contreras et al. 2007; Satrustegui et al. 2007). Aralar/AGC1 activation results in the transfer of cytosolic reducing equivalents into the mitochondrial matrix, increasing substrate supply to mitochondria. Moreover, the MAS oxidizes cytosolic NADH, enhancing pyruvate production from lactate and glucose (Safer et al. 1971). Extramitochondrial Ca2+ activation of Aralar/AGC1-MAS activity results in the net transfer of reducing equivalents (NADH) from the cytosol to mitochondria, which increases the state 3 respiration rate when using glutamate plus malate as substrates in the presence of a physiological cytosolic free-Ca2+ concentration (Gellerich et al. 2012, 2013), as well as promotion of Ca2+-dependent pyruvate oxidation in the mitochondria (Gellerich et al. 2012; 2013) (Fig. 1). Interestingly, the mitochondrial pyruvate carrier, recently characterized at the molecular level (Bricker et al. 2012; Herzig et al. 2013), has a low affinity for pyruvate (Km of 0.6 mm in rat liver, Paradies et al. 1983) and so may limit pyruvate oxidation (Schell & Rutter, 2013). This may be important with respect to the role of Aralar/AGC1-MAS activity in increasing pyruvate concentration, favouring its transport into the mitochondrial matrix.
The SCaMCs (del Arco & Satrustegui, 2004) or APCs (Fiermonte et al. 2004) are adenine nucleotide carriers that perform the electroneutral exchange of ATP-Mg2− or HADP− with HPO42− between the cytosol and the mitochondrial matrix (Joyal & Aprille 1992; Nosek & Aprille 1992; Fiermonte et al. 2004). The SCaMCs are activated by extramitochondrial Ca2+ with an S0.5 of activation within the range of the MCU complex of ∼3–4 μm for the brain and liver isoform SCaMC-3/Slc25a23 (Amigo et al. 2013) and of ∼12.7 μm for the tumor cell isoform SCaMC-1/Slc25a24 (Traba et al. 2012). The direction and magnitude of the transport depend on the relative concentrations of ATP-Mg2− or ADPH− and Pi. The Mg2+ and H+ associated with ATP and ADP are essential for transport through the carrier. Although its main substrate is magnesium-bound ATP, the carrier can also exchange free ADP and, to a lesser extent, free AMP (Asimakis & Aprille 1980; Fiermonte et al. 2004). Thus, SCaMC activity regulates the total adenine nucleotide pool in the mitochondrial matrix; the sum of ATP+ADP+AMP (Fig. 1).
By changing the matrix adenine nucleotide content, the ATP-Mg2−/Pi carriers play an important role in the regulation of the mitochondrial metabolic pathways that have adenine nucleotide-dependent enzymes, including pyruvate carboxylase (gluconeogenesis), carbamyl phosphate synthetase (urea cycle), protein synthesis and the import of nuclear encoded proteins into mitochondria (Aprille, 1993; Satrustegui et al. 2007). The mitochondrial adenine nucleotide content increases in adult liver mitochondria upon glucagon treatment as a result of the activity of the liver ATP-Mg2−/Pi carrier SCaMC-3 (Aprille, 1988, 1993, Amigo et al. 2013). The SCaMCs appear to be important for mitochondrial maturation after birth. Indeed, the mitochondrial adenine nucleotide pool increases several-fold in newborn rat liver mitochondria within 3 h after birth, coinciding with the maturation of mitochondrial respiration (Sutton & Pollak, 1978; Valcarce et al. 1988).
Calcium regulation of mitochondrial respiration in intact neurons: basal state
The contribution of Ca2+ signalling to respiration in intact neurons may be inferred from the effects of the disruption of genes involved in the process. In neuronal cultures under basal conditions and in the presence of physiological glucose concentrations, the rate of respiration of cerebrocortical neurons is ∼30% of maximal uncoupled respiration and is driven by a continuous ATP demand (74% is coupled; Llorente-Folch et al. 2013). Spontaneous Ca2+ signals in embryonic neurons in culture decrease in frequency upon maturation (Gu & Spitzer, 1995). Distinct aspects of neuronal differentiation encoded by the frequency of spontaneous intracellular Ca2+ transients (Gu & Spitzer, 1995) remain unknown. A major role for intracellular Ca2+ under these conditions is improbable because basal respiratory activity is not influenced by the presence or absence of extracellular Ca2+ (Llorente-Folch et al. 2013) or by the presence or absence of SCaMC-3 (Llorente-Folch et al. 2013). In addition, although the influence MCU in respiration of cortical neuronal cultures is unknown, total body basal oxygen consumption in MCU knockout (KO) mice on an outbred genetic background was the same as that of control mice (Pan et al. 2013), suggesting that the lack of MCU does not cause major respiratory defects. However, a compensatory effect on basal respiration in MCU KO neurons in this model cannot be ruled out because an MCU KO strain on the C57BL/6 genetic background was embryonic lethal (Murphy et al. 2014).
Basal respiration in neurons diminished by ∼46% in the absence of Aralar/AGC1 (Llorente-Folch et al. 2013), although this may be a consequence of a lack of MAS itself, which decreases pyruvate supply to mitochondria, rather than a lack of Ca2+ signalling through Aralar/AGC1. Indeed, Aralar/AGC1 is not absolutely dependent on Ca2+; in Ca2+-free media, MAS activity is attenuated by ∼70% (Pardo et al. 2006; Contreras et al. 2007) and this may be sufficient to maintain basal respiration in neurons in culture. It must be noted that these considerations apply to the basal state in cultured neurons, and not necessarily to brain neurons in which baseline activity is energetically much higher (Raichle & Mintun, 2006). In addition to a low basal respiratory rate, Aralar/AGC1 KO neurons have also a limited maximal uncoupled respiratory rate, which may be rescued by exogenous pyruvate (Llorente-Folch et al. 2013).
Calcium regulation of mitochondrial respiration in intact neurons: response to workloads
To characterize the role of Ca2+ in the regulation of energy metabolism, the double role of Ca2+ in regulating OXPHOS has to be considered: (i) as an inducer of workload (i.e. as an inducer of ATP utilization to restore Ca2+ levels) and (ii) as a regulator of mitochondrial transporters or dehydrogenases (i.e. as a signal molecule). To dissect these aspects, we applied different stimuli to produce different workloads as a result of an increase in cytosolic Na+ and/or Ca2+. We imposed a high workload using veratridine, a moderate workload with depolarization using isosmotic high K+, and a small workload using carbachol, which mobilizes Ca2+ from ER. With each of these stimuli, the impact of the Ca2+ signals, on the mitochondrial population also differed. Those produced in response to veratridine and K+ stimulation increase matrix Ca2+, whereas that induced by carbachol primarily exerts an action at the external face of the inner mitochondrial membrane. Experiments were conducted in the presence or absence of 2 mm Ca2+ or in cells loaded with BAPTA-AM, a rapid Ca2+ chelator, which allows for a Ca2+-induced workload at the same tine as preventing Ca2+ signalling (Llorente-Folch et al. 2013).
An increase in ATP demand to restore the ionic resting state after a stimulus will in turn stimulate OXPHOS, which might also be regulated by Ca2+ itself. In all cases analysed, the oxygen consumption rate (OCR) was severely reduced in the absence of Ca2+. In particular, the absence of Ca2+ during veratridine stimulation abolishes Ca2+-regulatory mechanism because the veratridine-induced workload is mainly driven by the massive entry of Na+ to the cytosol. Moreover, the fall in cytosolic ATP in Ca2+-free media was even more pronounced than that in the presence of Ca2+, which is attributable to the absence of Ca2+-mediated stimulation of mitochondrial respiration. Consequently, these experiments clearly demonstrated the role of Ca2+ as a regulatory signal to stimulate OXPHOS (Llorente-Folch et al. 2013).
For every Ca2+ ion that enters the mitochondria, 3H+ must enter the matrix to remove it (assuming NCLX stoichiometry 3Na+:1Ca2+ and 1Na+:1H+ for NHE) (Boyman et al. 2013). With the known stoichiometry for mitochondrial ATP production and exchange for ADP (3-4H+/ATP) (Watt et al. 2010), this implies that removing 1 Ca2+ from mitochondria costs approximately 1ATP (i.e. the same as removing it by efflux across the plasma membrane) (Carafoli, 2012). By removing Ca2+ from the media, both Ca2+ signalling and Ca2+-induced workload in response to KCl and carbachol were abolished and so, not unexpectedly, the increase in respiration and the fall in cytosolic ATP was smaller in Ca2+-free media. However, incubation with BAPTA-AM, which maintained the workload but blocked Ca2+ signalling, also decreased the respiratory response. This showed that Ca2+ regulation is required to increase respiration and maintain cytosolic ATP levels in response to any workload (Llorente-Folch et al. 2013).
Mechanisms involved in Ca2+ regulation of mitochondrial respiration upon an increase in workload
To determine the specific role of Ca2+-dependent mitochondrial carriers in the regulation of OXPHOS, we studied the effects of selective removal of SCaMC-3 and Aralar/AGC1 in response to different stimuli aiming to unmask the contribution of matrix versus extra-mitochondrial Ca2+.
Large workloads
Studies in our laboratory have revealed that SCaMC-3 and Aralar/AGC1-MAS are involved in the Ca2+-dependent regulation of mitochondrial respiration at high workloads, such as those imposed by veratridine stimulation, in which the MCU complex and the mitochondrial dehydrogenases pathway also operate.
SCaMC-3/APC2
Deficiency of SCaMC-3 decreased the veratridine-induced stimulation of mitochondrial respiration in the presence of Ca2+ (Llorente-Folch et al. 2013). This confirms that SCaMC-3 is recruited at large workloads in which a high cytosolic Ca2+ concentration activates the carrier (Amigo et al. 2013) and the cytosolic ATP/ADP ratio falls. These conditions thermodynamically favour exchange of cytosolic ATP-Mg2− or HADP2− for mitochondrial Pi (Joyal & Aprille, 1992; Aprille, 1993), increasing mitochondrial respiration.
ATP-Mg2−/Pi carrier activity probably favours OXPHOS stimulation by increasing the total adenine nucleotide pool of mitochondria exerting a mass action ratio effect on complex V or the ANT, (Aprille, 1993; Satrustegui et al. 2007; Glancy & Balaban, 2012; Amigo et al. 2013). Moreover, ADP probably comprises the adenine nucleotide transported by SCaMC-3 and may allosterically activate tricarboxylic acid cycle enzymes (Gabriel et al. 1986; Nicholls et al. 1994) and inhibit pyruvate dehydrogenase kinase, preventing PDH inactivation (Hucho, 1974; Pratt & Roche, 1979). Thus, an increase in ADP would promote oxidative metabolism and increase the supply of reduced cofactors to the respiratory chain in response to cytosolic Ca2+ signals. It is also possible that the entry of adenine nucleotides is a protective mechanism against an early opening of the permeability transition pore (PTP) which would cause an immediate failure of OXPHOS. Adenine nucleotides inhibit PTP opening (Chinopoulos & Adam-Vizi, 2012; Traba et al. 2012; Giorgio et al. 2013) and also bind Ca2+ (Haumann et al. 2010), lowering the free matrix Ca2+ concentration, which would decrease the probability of PTP opening. This mechanism has been characterized in transformed cells which over-express SCaMC-1, promoting cell survival by desensitizing mitochondrial permeability transition (Traba et al. 2011).
Aralar/AGC1
Veratridine-induced workload promoted a strong increase in mitochondrial respiration that was severely attenuated, although not completely abolished, in Aralar/AGC1 KO neurons (Llorente-Folch et al. 2013). This showed a major role for the Aralar/AGC1-MAS pathway in response to a high workload-induced stimulation. However, Aralar/AGC1 KO neurons still presented a Ca2+-dependent response, which indicated that the Ca2+-dependent dehydrogenases and SCaMC-3-dependent Ca2+ regulation are also signalling mechanisms engaged under these high workloads. Exogenous pyruvate, which bypasses Aralar/AGC1-MAS activity, rescued the effects of the lack of Aralar/AGC1 on respiration, clearly showing that the major role of Aralar/AGC1-MAS is to provide pyruvate to mitochondria (Llorente-Folch et al. 2013). These results also reveal that the accumulation of Ca2+ into mitochondria through the MCUC is not sufficient to fully activate mitochondrial respiration and that Aralar/AGC1-MAS activity, by providing pyruvate into mitochondria, is unambiguously required.
Small workloads
Aralar/AGC1-MAS pathway is the only Ca2+-regulated mechanism responsible for upregulation of respiration in response to small Ca2+ signals produced by carbachol (Llorente-Folch et al. 2013). Small Ca2+ signals generated by activation of G-protein-coupled receptors and Ca2+ release from intracellular stores did not reach mitochondria in neurons (Pardo et al. 2006; Llorente-Folch et al. 2013) but increased neuronal mitochondrial NAD(P)H mediated by activation of Aralar/AGC1-MAS (Pardo et al. 2006). Carbachol stimulation resulted in a small increase in ATP demand and coupled respiration in intact neurons which, not unexpectedly, was absolutely dependent on Aralar/AGC1-MAS (Llorente-Folch et al. 2013).
Moderate workloads
Depolarization in response to isosmotic high K+ promoted an increase in cytosolic Ca2+ that reached the mitochondrial matrix and produced a moderate fall in cytosolic ATP levels (Llorente-Folch et al. 2013) (Fig. 2). This in turn stimulated mitochondrial respiration (Llorente-Folch et al. 2013) (Fig. 3A and E).
Figure 2. Changes of cytosolic and mitochondrial.

Ca2+ and cytosolic ATP in cortical neurons in response to potassiumChanges in [Ca2+]cyt, in Fura-2 loaded neurons (A–C) or [Ca2+]mit, in neurons transfected with Mit GEM-GECO1 probe (D–F) obtained by stimulation with 30 mm KCl in 2 mm Ca2+ medium (A and D), Ca2+ free medium in the presence of the Ca2+ chelator EGTA (B and E), or in 50 μm BAPTA-AM preloaded neurons, an intracellular Ca2+ chelator that preserves the workload preventing Ca2+ signalling, in 2 mm Ca2+ medium (C and F). Recordings from at least 60 cells per condition and two independent experiments were used for cytosolic Ca2+ imaging and a minimum of 15 cells and eight independent experiments were used for mitochondrial Ca2+ imaging. Individual cell recordings (in grey) and average (thick black trace) were shown. G–I, cytosolic ATP levels after a switch from HCSS medium to isosmotic high K+ medium in which 30 mm NaCl was replaced by 30 mm KCl either in 2 mm Ca2+ medium (G) or 100 μm EGTA medium (H). Comparison between the two previously mentioned conditions is shown in (I). The drop in ATP values with respect to basal levels 200 s after high K+ stimulation was 18.6 ± 0.3% in the presence and 9 ± 0.1% in the absence of Ca2+ (**P = 0.009, two-tailed unpaired Student's t test). Data are expressed as the mean ± SEM (modified from Llorente-Folch et al. 2013).
Figure 3. OCR responses to potassium in Aralar/AGC1-deficient neurons.

Cellular OCR was measured using a Seahorse XF24 Extracellular Flux Analyser (Seahorse Bioscience, North Billerica, MA, USA) (Qian and Van Houten, 2010). Cortical neurons were plated in XF24 V7 cell culture at 1.0 × 105 cells/well and incubated for 9–10 days in a 37ºC, 5% CO2 incubator in serum-free B27-supplemented neurobasal medium with high levels of glucose. To study OCR, cells were equilibrated for 1 h in 2.5 mm glucose HCSS in the presence of 2 mm CaCl2. Next, neurons were either maintained in the same medium or stimulated with 30 mm KCl in 2.5 mm glucose in Ca2+-containing isosmotic HCSS medium in which 30 mm NaCl was replaced by 30 mm KCl at the start of the respirometry experiments. Calibration of respiration took place after the vehicle (veh) injection in port A. Substrates were prepared in the same medium in which the experiment was conducted and were injected from the reagent ports automatically to the wells at the times indicated. Mitochondrial function in neurons was determined through sequential addition of 6 μm oligomycin (Olig), 0.5 mm 2,4-dinitrophenol (DNP) and 1 μm antimycin/1 μm rotenone (A/R). This allowed the determination of basal oxygen consumption (BS), oxygen consumption linked to ATP synthesis (ATP), non-ATP linked oxygen consumption (leak), mitochondrial uncoupled respiration (MUR) and non-mitochondrial oxygen consumption (NM) (Qian and Van Houten, 2010; Brand and Nicholls, 2011). Respiratory profiles are shown for control (A) and Aralar/AGC1-deficient neurons (B) upon K+ stimulation, in neurons pre-treated or not with 2 mm DCA for 1 h, in the presence of 2 mm Ca2+. Respiratory profiles are shown for control (C) and Aralar/AGC1-deficient neurons (D) upon K+ stimulation, with or without the addition of 2 mm pyruvate just before starting the experiment, in the presence of 2 mm Ca2+. Stimulation of mitochondrial respiration (E) and maximal uncoupled respiration (MUR) (F) is shown upon K+ stimulation after 2 mm DCA pre-teatment or 2 mm pyruvate addition. Data correspond to four or five and two to four independent experiments in wild-type and Aralar/AGC1 KO cultured neurons, respectively (one-way ANOVA, *P ≤ 0.05; **P ≤ 0.01). KCl, isosmotic high K+, 30 mm; Pyr, pyruvate, 2 mm.
Interestingly, Ca2+-dependent regulation of mitochondrial respiration by high K+-induced depolarization was completely abolished in the absence of Aralar/AGC1 and the addition of external pyruvate rescued the lack of Aralar/AGC1 (Fig. 3B and D). Paradoxically, the lack of response of Aralar/AGC1 KO neurons to K+-depolarization occurred despite increased cytosolic and mitochondrial Ca2+ concentrations and increased mitochondrial NAD(P)H levels (Pardo et al. 2006), which would be expected to increase respiration. Moreover, wild-type and Aralar/AGC1 KO neurons showed a similar activation of PDH after 5 min in isosmotic 30 mm KCl (1.20 ± 0.09-fold versus 1.21 ± 0.06-fold increase in the PDH/P-PDH ratio in wild-type and Aralar/AGC1 KO neuronal cultures, respectively) (Fig. 4A–C). Thus, the absence of stimulation of mitochondrial respiration in Aralar/AGC1 KO neurons is not a result of differences in PDH dephosphorylation compared to wild-type. Treatment with the PDH kinase inhibitor dichloroacetic acid (DCA), 1 h before K+ stimulation, also causes a similar maximal PDH dephosphorylation in wild-type and Aralar/AGC1 KO neurons (1.35 ± 0.01-fold versus 1.39 ± 0.05-fold increase in PDH/P-PDH ratio in wild-type and Aralar/AGC1 KO neurons, respectively) (Fig. 4A–C). However, DCA treatment increases basal respiration only in wild-type and not in Aralar/AGC1 KO neurons, and this increase in respiration caused by DCA was not increased any further by 30 mm KCl (Fig. 3A). However, DCA treatment did not change basal or K+-stimulated respiration in Aralar/AGC1 KO neurons (Fig. 3B and E).
Figure 4. Pyruvate dehydrogenase complex dynamics after K+- depolarization in wild-type and Aralar/AGC1 KO primary cortical neurons.
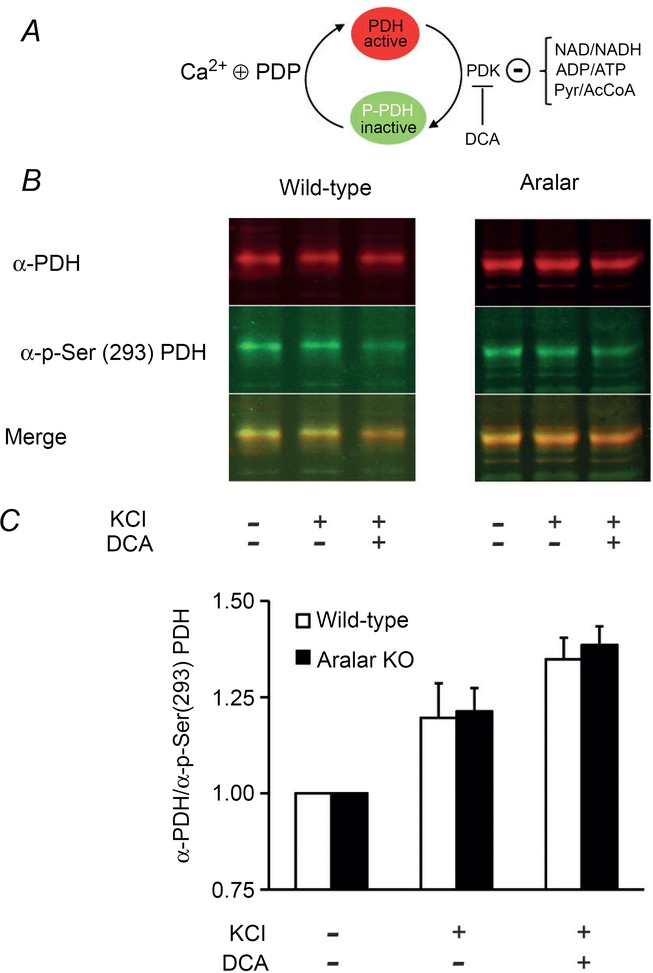
A, scheme depicting the complex dynamics of pyruvate dehydrogenase. PDH-E1 subunit is active in its dephosphorylated state. Pyruvate dehydrogenase kinase (PDK), whose activity is negatively controlled by NAD/NADH, ADP/ATP and Pyr/AcCoA ratios, phosphorylates the enzyme in the Ser(293) residue, inactivating the complex. On the other hand, pyruvate dehydrogenase phosphatase (PDP), which is positively regulated by intramitochondrial Ca2+, dephosphorylates PDH-E1, recovering the active form. DCA inhibits PDK, favouring the active form of PDH. B, representative western blot against PDH (anti-PDH subunit E1, mouse monoclonal antibody, dilution 1:5000; Invitrogen, Carlsbad, CA, USA) and p-Ser(293) PDH (anti-PDH, rabbit polyclonal antibody, dilution 1:2000; Novus Biologicals, Littleton, CO, USA) and IRDye secondary antibodies optimized for use with Oddysey (800 CW goat anti-rabbit IgG and 680 RD goat anti-mouse IgG, dilution 1:50000; Li-Cor, Lincoln, NE, USA). Neurons were obtained under control conditions, or after 5 min of exposure to isosmotic 30 mm KCl, or under these same conditions prior to pre-treatment for 1 h with 2 mm DCA. Merged image combines both green and red fluorescence to denote the PDH/phosphorylated pyruvate dehydrogenase (P-PDH) ratio. C, western-blotting quantification expressed as the fold increase in anti-PDH/anti-p-Ser(293) PDH ratio compared to control. Data correspond to three to five independent experiments in wild-type and Aralar/AGC1 KO cultured neurons, respectively (Student's t test, *P < 0.05). Pyr, pyruvate; AcCoA, acetyl coenzyme A; KCl: isosmotic high K+, 30 mm.
The findings in wild-type neurons are consistent with a role of matrix Ca2+ in ‘pulling’ pyruvate into mitochondria during K+-induced stimulation of respiration (Rueda et al. 2014). By activating PDH phosphatase and enzyme dephosphorylation, matrix Ca2+ will activate PDH and the decarboxylation of pyruvate, which will pull pyruvate from the cytosol to mitochondria across the pyruvate carrier by mass action ratio effects. The finding that activation of PDH by DCA results in an increase in OCR similar to that obtained by K+-depolarization is consistent with a role of PDH activity in driving pyruvate into mitochondria.
However, K+-induced or DCA-induced activation of respiration does not take place in Aralar/AGC1 KO neurons even though PDH is activated under both conditions (Fig. 4A–C). The lack of Aralar/AGC1-MAS activity and the diminished pyruvate levels in Aralar/AGC1 KO neurons (Pardo et al. 2011) reveal that activation of PDH is insufficient to increase pyruvate entry in mitochondria under this workload.
MCUC in the regulation of mitochondrial respiration
The role of the MCUC in basal respiration or workload-induced respiration in neurons is still unknown. MCU is clearly responsible for glutamate/NMDA-induced Ca2+ entry in neuronal mitochondria, and knockdown of endogenous MCU decreased NMDA-induced increases in mitochondrial Ca2+ in neurons and slightly attenuated the sensitivity to excitotoxicity (Qiu et al. 2013). However, this approach did not completely eliminate NMDA-induced increases in mitochondrial Ca2+, either as a result of the insufficient silencing efficiency or the presence of other pathways for Ca2+ uptake (Qiu et al. 2013). Thus, the consequences of MCU silencing on neuronal respiration remain to be determined.
On the other hand, the description of an MCU KO mouse with an unexpectedly mild phenotype (Pan et al. 2013) and of human diseases as a result of mutations in MICU1 (the Ca2+ sensitive regulatory subunits of MCUC) may shed light of the role of MCUC in the regulation of mitochondrial respiration. Mitochondria from MCU KO mice had no Ru360-inhibitable Ca2+ uptake in mitochondria, with no functional compensation for the rapid entry of Ca2+ into the matrix because MCU KO mitochondria do not take up any measurable Ca2+ over a 10–20 min period (Pan et al. 2013; Murphy et al. 2014). However mitochondrial Ca2+ levels were depleted but not completely absent, suggesting a possible slow mechanism for Ca2+ uptake independent of MCU (Murphy et al. 2014).
As discussed earlier, MCU KO mice did not present alterations in basal metabolism at the whole animal level, or any defect in respiration in mouse embryonic fibroblast cultures, even though the Ca2+ content in skeletal muscle mitochondria was decreased and the PDH phosphorylation state was increased (Pan et al. 2013). Consistent with a role of MCU at high workloads, skeletal muscle peak performance was slightly decreased (∼15%) in MCU KO mice. Whether compensatory mechanisms explain the mild effect of MCU deficiency on mitochondrial metabolism remains to be established. Indeed, MCU KO generated a viable phenotype only in a mixed background as a hypomorph, although this has proven to be embryonic lethal in pure inbred strains, including the C57BL/6 background (Murphy et al. 2014).
By contrast to the mild phenotype of the MCU KO mouse, loss of function mutations of MICU1, the first genetic human disease to be identified involving mutations of the MCUC, were associated with proximal myopathy, learning difficulties and a progressive extrapyramidal movement disorder in children (Logan et al. 2014). Genetic analysis identified two mutations, a splice acceptor site mutation, c.1078-1G>C, and a splice donor site mutation, c.741 + 1G>A, in MICU1 in a total of 15 affected individuals from seven families, resulting in nonsense-mediated decay and loss of protein. Cellular and mitochondrial Ca2+ homeostasis were analysed in primary skin fibroblasts from patients showing that MICU1 deficiency caused loss of the physiological cooperative sigmoid regulation of mitochondrial Ca2+ concentration (Szabadkai & Duchen, 2008), increasing the basal Ca2+ load in the organelle with a significant increase in the velocity of mitochondrial Ca2+ uptake in response to a rise in cytosolic Ca2+ concentration. The increased resting mitochondrial Ca2+ concentration was associated with a highly fragmented mitochondrial network. The data suggested that loss of MICU1 leads to chronic activation of the MCU channel even at resting cytosolic Ca2+ concentrations, highlighting the functional importance of the sigmoid dependence of mitochondrial Ca2+ uptake on extramitochondrial [Ca2+]. The expression of MICU1 effectively results in a threshold effect at low, submicromolar extramitochondrial [Ca2+] and cooperative Ca2+-mediated activation of Ca2+ uptake kinetics at higher (> μm) extramitochondrial Ca2+ levels. Such a dual role of MICU1 has been previously shown in cellular models (Mallilankaraman et al. 2012a; Csordas et al. 2013) and the molecular details of the regulatory mechanism were recently refined further by the description of MICU1/MICU2 heterodimers (Patron et al. 2014). Importantly, although all of the studies agree that MICU1 (and consequently MICU2) loss leads to a significant increase in basal resting mitochondrial [Ca2+], no clear metabolic consequences have been demonstrated. This might be a result of the highly glycolytic phenotype of human fibroblasts (Logan et al. 2014) and cancer cell lines (Mallilankaraman et al. 2012a; Csordas et al. 2013), although, similarly, primary hepatocytes, where cellular energy metabolism relies more on OXPHOS, have also shown unaltered basal respiration after knockdown of MICU1 (Csordas et al. 2013). By contrast, in the hepatocyte model, Ca2+-dependent activation of oxygen consumption was blunted in the absence of MICU1. These findings might indicate the relatively minor importance of basal Ca2+ levels on mitochondrial metabolism (in contrast to previous findings; Cardenas et al. 2010) or can be attributed to a more complex role of Ca2+ in mitochondrial metabolism.
Ultimately, these data show that the MICU1-mediated sigmoid Ca2+ activation of mitochondrial Ca2+ uptake serves as a signal-to-noise discriminator, protecting cells from chronic futile Ca2+ cycling, which represents an important energy drain. In the absence of MICU1/MICU2, increased uptake of Ca2+ even at rest would represent chronic activation of a futile Ca2+ cycle, dissipating the proton motive force and preventing OXPHOS (Fig. 5). The net result on cellular metabolism in this case might not be evident from measurements of membrane potential and O2 fluxes, particularly in cells where the reverse mode of the ATP synthase (Campanella et al. 2009) is also involved in maintaining the resting membrane potential. Thus, although the results from MCU KO mice and MICU1 deficient models suggest that intramitochondrial Ca2+ signalling makes a relatively small contribution in metabolism, its tight regulation and its conservation under evolutionary pressure are evident from the pathology of the human MICU1 deficiency, affecting primarily muscle and neurons. The precise influence of the MCU-mediated pathway on respiration of brain neurons awaits clarification using the models now available.
Figure 5. Consequences of MICU1/2 loss on mitochondrial Ca2+ homeostasis, shape and metabolic function.
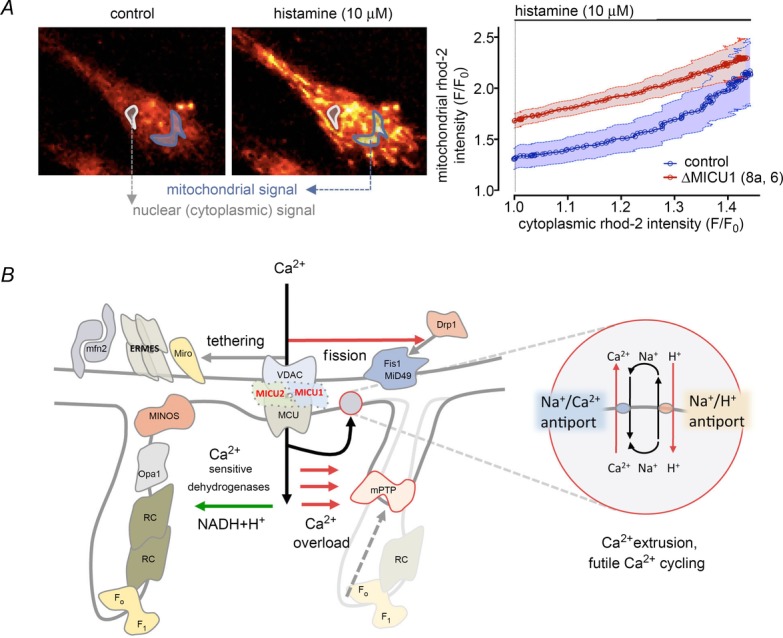
A, relationship between cytosolic and mitochondrial [Ca2+] during evoked Ca2+ signals. MICU1/2 defective cells have an increased basal mitochondrial Ca2+ load, which follows linearly the increase in cytoplasmic [Ca2+] (reproduced with permission from Logan et al. 2014). B, increased mitochondrial Ca2+ load can activate mitochondrial metabolism by stimulating Ca2+-dependent dehydrogenases of the mitochondrial matrix. On the other hand, chronic Ca2+ load in the mitochondrion might also have also a metabolic cost by resulting in futile Ca2+ cycling (inset) and opening of the mitochondrial permeability transition pore (mPTP), both leading to depolarization. In addition, we observed increased mitochondrial fission, which might impact on the metabolic capacity of the organelle. In the human fibroblast model of the disease, no changes in ER mitochondria tethering were observed. mfn2, mitofusin 2; ERMES, ER-mitochondria encounter structures; Miro, mitochondrial Rho GTPase; MINOS: MINOS/MitOS/MICOS complexes; RC, respiratory supercomplexes; F1/FO, F1FO ATPase.
On the other hand, Aralar/AGC1-MAS activity by itself or through stimulation by extramitochondrial Ca2+ has a strong relevance both under basal conditions and upon stimulation. Aralar/AGC1-MAS deficiency significantly compromises glucose oxidation, basal respiration and maximal uncoupled respiration (Llorente-Folch et al. 2013). Moreover, the Aralar/AGC1-MAS pathway, by regulation of pyruvate supply to mitochondria, plays a paramount role in the response to the small, moderate and strong signals that we have investigated (Llorente-Folch et al. 2013, Rueda et al. 2014). The roles played by MAS activity itself with respect to Ca2+ regulation through Ca2+ binding to Aralar/AGC1 remain to be established.
Glossary
- AGC
aspartate-glutamate carrier
- ANT
adenine nucleotide translocase
- CaMC
Ca2+-regulated mitochondrial carrier
- DCA
′dichloroacetic acid
- ER
endoplasmic reticulum
- KO
knockout
- MAS
malate–aspartate shuttle
- MCU
mitochondrial calcium uniporter
- MCUC
mitochondrial calcium uniporter complex
- NCLX
Na+/Ca2+ exchanger
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- PDH
pyruvate dehydroganase
- PTP
permeability transition pore
- SCaMC/APC
calcium-binding mitochondrial carrier protein
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
All authors approved the final version of the manuscript submitted for publication.
References
- Amigo I, Traba J, Gonzalez-Barroso MM, Rueda CB, Fernandez M, Rial E, Sanchez A, Satrustegui J, del Arco A. Glucagon regulation of oxidative phosphorylation requires an increase in matrix adenine nucleotide content through Ca2+-activation of the mitochondrial ATP-Mg/Pi carrier SCaMC-3. J Biol Chem. 2013;288:7791–7802. doi: 10.1074/jbc.M112.409144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprille JR. Regulation of the mitochondrial adenine nucleotide pool size in liver: mechanism and metabolic role. FASEB J. 1988;2:2547–2556. doi: 10.1096/fasebj.2.10.3290024. [DOI] [PubMed] [Google Scholar]
- Aprille JR. Mechanism and regulation of the mitochondrial ATP-Mg/P(i) carrier. J Bioenerg Biomembr. 1993;25:473–481. doi: 10.1007/BF01108404. [DOI] [PubMed] [Google Scholar]
- Asimakis GK, Aprille JR. In vitro alteration of the size of the liver mitochondrial adenine nucleotide pool: correlation with respiratory functions. Arch Biochem Biophys. 1980;203:307–316. doi: 10.1016/0003-9861(80)90181-2. [DOI] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Bai Y, Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK. MICU2, a paralog of MICU1, Resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE. 2013;8:e55785. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol. 2002;34:1259–1271. doi: 10.1006/jmcc.2002.2082. [DOI] [PubMed] [Google Scholar]
- Balaban RS. Maintenance of the metabolic homeostasis of the heart: developing a systems analysis approach. Ann NY Acad Sci. 2006;1080:140–153. doi: 10.1196/annals.1380.013. [DOI] [PubMed] [Google Scholar]
- Balaban RS. The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta. 2009;1787:1334–1341. doi: 10.1016/j.bbabio.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko AI, Jean-Quartier C, Malli R, Graier WF. Characterization of distinct single-channel properties of Ca2+ inward currents in mitochondria. Pflugers Arch. 2013;465:997–1010. doi: 10.1007/s00424-013-1224-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko AI, Jean-Quartier C, Parichatikanond W, Alam MR, Waldeck-Weiermair M, Malli R, Graier WF. Mitochondrial Ca(2+) uniporter (MCU)-dependent and MCU-independent Ca(2+) channels coexist in the inner mitochondrial membrane. Pflugers Arch. 2014;466:1411–1420. doi: 10.1007/s00424-013-1383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman L, Williams GS, Khananshvili D, Sekler I, Lederer WJ. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol. 2013;59:205–213. doi: 10.1016/j.yjmcc.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cell. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS, Rutter J. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100. doi: 10.1126/science.1218099. (6090): doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerries M, Most P, Gledhill JR, Walker JE, Katus HA, Koch WJ, Aebi U, Schoenenberger CA. Ca2+-dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol. 2007;27:4365–4373. doi: 10.1128/MCB.02045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella M, Parker N, Tan CH, Hall AM, Duchen MR. IF(1): setting the pace of the F(1)F(o)-ATP synthase. Trends Biochem Sci. 2009;34:343–350. doi: 10.1016/j.tibs.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Carafoli E. The interplay of mitochondria with calcium: an historical appraisal. Cell Calcium. 2012;52:1–8. doi: 10.1016/j.ceca.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142((2)):270–83. doi: 10.1016/j.cell.2010.06.007. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerdan S, Künnecke B, Seelig J. Cerebral metabolism of [1,2-13C2]acetate as detected by in vivo and in vitro 13C NMR. J Biol Chem. 1990;265((22)):12916–26. [PubMed] [Google Scholar]
- Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem. 1955;217:383. [PubMed] [Google Scholar]
- Chinopoulos C, Adam-Vizi V. Modulation of the mitochondrial permeability transition by cyclophilin D, moving closer to F(0)-F(1) ATP synthase. Mitochondrion. 2012;12:41–45. doi: 10.1016/j.mito.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Contreras L, Drago I, Zampese E, Pozzan T. Mitochondria: the calcium connection. Biochem Biophys Acta. 2010;1797:607–618. doi: 10.1016/j.bbabio.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, Satrústegui J. Ca2+ activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J Biol Chem. 2007;282:7098–7106. doi: 10.1074/jbc.M610491200. [DOI] [PubMed] [Google Scholar]
- Csordás G, Golenár T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnóczky G. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013;17((6)):976–87. doi: 10.1016/j.cmet.2013.04.020. doi: 10.1016/j.cmet.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Arco A, Satrustegui J. Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J Biol Chem. 1998;273:23327–23334. doi: 10.1074/jbc.273.36.23327. [DOI] [PubMed] [Google Scholar]
- del Arco A, Agudo M, Satrustegui J. Characterization of a second member of the subfamily of calcium-binding mitochondrial carriers expressed in human non-excitable tissues. Biochem J. 2000;345:725–732. doi: 10.1042/0264-6021:3450725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Arco A, Satrústegui J. Identification of a novel human subfamily of mitochondrial carriers with calcium-binding domains. J Biol Chem. 2004;279:24701–24713. doi: 10.1074/jbc.M401417200. [DOI] [PubMed] [Google Scholar]
- Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A. 1961;47:1744–1750. doi: 10.1073/pnas.47.11.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, Demaurex N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J Biol Chem. 2014;289:20377–85. doi: 10.1074/jbc.M113.540898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787:1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doonan PJ, Chandramoorthy HC, Hoffman NE, Zhang X, Cárdenas C, Shanmughapriya S, Rajan S, Vallem S, Chen X, Foskett JK, Cheung JY, Houser SR, Madesh M. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. 2014;28:4936–4949. doi: 10.1096/fj.14-256453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago I, Pizzo P, Pozzan T. After a century mitochondrial in- and efflux machineries reveal themselves. EMBO J. 2011;30:4119–4125. doi: 10.1038/emboj.2011.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J. 1992;283:41–50. doi: 10.1042/bj2830041. (Pt 1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000;28:339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- Fiermonte G, De Leonardis F, Todisco S, Palmieri L, Lasorsa FM, Palmieri F. Identification of the mitochondrial ATP-Mg/Pi transporter bacterial expression, reconstitution, functional characterization, and tissue distribution. J Biol Chem. 2004;279:30722–30730. doi: 10.1074/jbc.M400445200. [DOI] [PubMed] [Google Scholar]
- Froschauer E, Nowikovsky K, Schweyen RJ. Electroneutral K+/H+ exchange in mitochondrial membrane vesicles involves Yol027/Letm1 proteins. Biochim Biophys Acta. 2005;1711:41–48. doi: 10.1016/j.bbamem.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Gabriel JL, Zervos PR, Plaut GW. Activity of purified NAD-specific isocitrate dehydrogenase at modulator and substrate concentrations approximating conditions in mitochondria. Metabolism. 1986;35:661–667. doi: 10.1016/0026-0495(86)90175-7. [DOI] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina Z, Trumbekaite S, Korzeniewski B, Gaynutdinov T, Seppet E, Vielhaber S, Heinze HJ, Striggow F. Cytosolic Ca2+ regulates the energization of isolated brain mitochondria by formation of pyruvate through the malate-aspartate shuttle. Biochem J. 2012;443:747–755. doi: 10.1042/BJ20110765. [DOI] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina Z, Gainutdinov T, Muth K, Seppet E, Orynbayeva Z, Vielhaber S. The control of brain mitochondrial energization by cytosolic calcium: the mitochondrial gas pedal. IUBMB Life. 2013;65:180–190. doi: 10.1002/iub.1131. [DOI] [PubMed] [Google Scholar]
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. 2013;52:2793–2809. doi: 10.1021/bi3015983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–2973. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Spitzer NC. Distinct aspects of neuronal differentiation encoded by frequency of spontaneous Ca2+ transients. Nature. 1995;375:784–787. doi: 10.1038/375784a0. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim Biophys Acta. 2009;1787:1291–1308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Klein-Flügge MC, Howarth C, Attwell D. Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J Neurosci. 2012;32:8940–8951. doi: 10.1523/JNEUROSCI.0026-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–777. doi: 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Harris EJ. The uptake and release of calcium by heart mitochondria. Biochem J. 1977;168:447–456. doi: 10.1042/bj1680447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haumann J, Dash RK, Stowe DF, Boelens AD, Beard DA, Camara AK. Mitochondrial free [Ca2+] increases during ATP/ADP antiport and ADP phosphorylation, exploration of mechanisms. Biophys J. 2010;99:997–1006. doi: 10.1016/j.bpj.2010.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Nemoto T, Iino M, Kasai H. Rapid Ca2+-dependent increase in oxygen consumption by mitochondria in single mammalian central neurons. Cell Calcium. 2005;37:359–370. doi: 10.1016/j.ceca.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. 2009;11:747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, Martinou JC. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;6(337(6090)):93–96. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- Hucho F. Regulation of the mammalian pyruvate dehydrogenase multienzyme complex by Mg2+ and the adenine nucleotide pool. Eur J Biochem. 1974;46:499–505. doi: 10.1111/j.1432-1033.1974.tb03643.x. [DOI] [PubMed] [Google Scholar]
- Ibrahim MZ. Glycogen and its related enzymes of metabolism in the central nervous system. Adv Anat Embryol Cell Biol. 1975;52:3–89. doi: 10.1007/978-3-642-86875-7. [DOI] [PubMed] [Google Scholar]
- Jacobus WE, Moreadity RW, Vandegaer KM. Mitochondrial respiratory control: evidence against the regulation of respiration by extramitochondrial phosphorylation potentials or by ATP/ADP ratios. J Biol Chem. 1982;257:2397–2402. [PubMed] [Google Scholar]
- Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144–147. doi: 10.1126/science.1175145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Zhao L, Clish CB, Clapham DE. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc Natl Acad Sci U S A. 2013;110:E2249–E2254. doi: 10.1073/pnas.1308558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyal JL, Aprille JR. The ATP-Mg/Pi carrier of rat liver mitochondria catalyzes a divalent electroneutral exchange. J Biol Chem. 1992;267:19198–19203. [PubMed] [Google Scholar]
- Kann O. The energy demand of fast neuronal network oscillations: insights from brain slice preparations. Front Pharmacol. 2012;10(2):90. doi: 10.3389/fphar.2011.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 2008;1778:1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Llorente-Folch I, Rueda CB, Amigo I, del Arco A, Saheki T, Pardo B, Satrústegui J. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J Neurosci. 2013;33:13957–13971. doi: 10.1523/JNEUROSCI.0929-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, Bonthron DT, Pysden KA, Whyte T, Munteanu I, Foley AR, Wheway G, Szymanska K, Natarajan S, Abdelhamed ZA, Morgan JE, Roper H, Santen GW, Niks EH, van der Pol WL, Lindhout D, Raffaello A, De Stefani D, den Dunnen JT, Sun Y, Ginjaar I, Sewry CA, Hurles M, Rizzuto R UK10K Consortium. Duchen MR, Muntoni F, Sheridan E. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet. 2014;46:188–193. doi: 10.1038/ng.2851. [DOI] [PubMed] [Google Scholar]
- Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, Csordás G, Madireddi P, Yang J, Müller M, Miller R, Kolesar JE, Molgó J, Kaufman B, Hajnóczky G, Foskett JK, Madesh M. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012a;14:1336–1343. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, Hoffman NE, Gandhirajan RK, Molgó J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012b;151:630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular componets, structure and physiopathological implications. J Physiol. 2014;592:829–839. doi: 10.1113/jphysiol.2013.268235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol. 2012;30:1143–1148. doi: 10.1038/nbt.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiesen C, Caesar K, Thomsen K, Hoogland TM, Witgen BM, Brazhe A, Lauritzen M. Activity-dependent increases in local oxygen consumption correlate with postsynaptic currents in the mouse cerebellum in vivo. J Neurosci. 2011;31:18327–18337. doi: 10.1523/JNEUROSCI.4526-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Mink JW, Blumenschine RJ, Adams DB. Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Physiol. 1981;241:R203–R212. doi: 10.1152/ajpregu.1981.241.3.R203. Regul Integr Comp Physiol. [DOI] [PubMed] [Google Scholar]
- Mitchell P, Moyle J. Estimation of membrane potential and pH difference across the cristae membrane of rat liver mitochondria. Eur J Biochem. 1969;7:471–484. doi: 10.1111/j.1432-1033.1969.tb19633.x. [DOI] [PubMed] [Google Scholar]
- Moreno-Sánchez R. Regulation of oxidative phosphorylation in mitochondria by external free Ca2+ concentrations. J Biol Chem. 1985;260:4028–4034. [PubMed] [Google Scholar]
- Moreno-Sánchez R. Inhibition of oxidative phosphorylation by a Ca2+-induced diminution of the adenine nucleotide translocator. Biochim Biophys Acta. 1983;724:278–285. doi: 10.1016/0005-2728(83)90146-9. [DOI] [PubMed] [Google Scholar]
- Müller MS, Fox R, Schousboe A, Waagepetersen HS, Bak LK. Astrocyte glycogenolysis is triggered by store-operated calcium entry and provides metabolic energy for cellular calcium homeostasis. Glia. 2014a;62:526–534. doi: 10.1002/glia.22623. [DOI] [PubMed] [Google Scholar]
- Müller MS. Functional impact of glycogen degradation on astrocytic signalling. Biochem Soc Trans. 2014b;142:1311–1315. doi: 10.1042/BST20140157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Pan X, Nguyen T, Liu J, Holmström KM, Finkel T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem Biophys Res Commun. 2014;449:384–385. doi: 10.1016/j.bbrc.2014.04.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman LA, Korol DL, Gold PE. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS ONE. 2011;6:e28427. doi: 10.1371/journal.pone.0028427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls BJ, Rigoulet M, Denton RM. Comparison of the effects of Ca2+, adenine nucleotides and pH on the kinetic properties of mitochondrial NAD(+)-isocitrate dehydrogenase and oxoglutarate dehydrogenase from the yeast Saccharomyces cerevisiae and rat heart. Biochem J. 1994;303:461–465. doi: 10.1042/bj3030461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Calcium transport and porton electrochemical potential gradient in mitochondria from guinea-pig cerebral cortex and rat heart. Biochem J. 1978;170:511–522. doi: 10.1042/bj1700511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosek MT, Aprille JR. ATP-Mg/Pi carrier activity in rat liver mitochondria. Arch Biochem Biophys. 1992;296:691–697. doi: 10.1016/0003-9861(92)90628-a. [DOI] [PubMed] [Google Scholar]
- Nowikovsky K, Pozzan T, Rizzuto R, Scorrano L, Bernardi P. Perspectives on: SGP symposium on mitochondrial physiology and medicine: the pathophysiology of LETM1. J Gen Physiol. 2012;139:445–454. doi: 10.1085/jgp.201110757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowikovsky K, Bernardi P. LETM1 in mitochondrial cation transport. Front Physiol Feb. 2014;26(5):83. doi: 10.3389/fphys.2014.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowikovsky K, Froschauer EM, Zsurka G, Samaj J, Reipert S, Kolisek M, Wiesenberger G, Schweyen RJ. The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf-Hirschhorn syndrome. J Biol Chem. 2004;16:30307–30315. doi: 10.1074/jbc.M403607200. [DOI] [PubMed] [Google Scholar]
- Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrústegui J, Palmieri F. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001;20:5060–5069. doi: 10.1093/emboj/20.18.5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradies G, Capuano F, Palombini G, Galeotti T, Papa S. Transport of pyruvate in mitochondria from different tumor cells. Cancer Res. 1983;43:5068–5071. [PubMed] [Google Scholar]
- Pardo B, Contreras L, Serrano A, Ramos M, Kobayashi K, Iijima M, Saheki T, Satrustegui J. Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J Biol Chem. 2006;281:1039–1047. doi: 10.1074/jbc.M507270200. [DOI] [PubMed] [Google Scholar]
- Pardo B, Rodrigues TB, Contreras L, Garzón M, Llorente-Folch I, Kobayashi K, Saheki T, Cerdan S, Satrústegui J. Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J Cereb Blood Flow Metab. 2011;31((1)):90–101. doi: 10.1038/jcbfm.2010.146. doi: 10.1038/jcbfm.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Estafani D, Rizzuto R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter exerting positive effects on MCU activity. Mol Cell. 2014;53:726–737. doi: 10.1016/j.molcel.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. 2015;21:109–116. doi: 10.1016/j.cmet.2014.12.004. [DOI] [PubMed] [Google Scholar]
- Pendin D, Greotti E, Pozzan T. The elusive importance of being a mitochondrial calcium uniporter. Cell Calcium. 2014;55:139–145. doi: 10.1016/j.ceca.2014.02.008. [DOI] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XS, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picton C, Klee CB, Cohen P. The regulation of muscle phosphorylase kinase by calcium ions, calmodulin and troponin-C. Cell Calcium. 1981;2:281–294. doi: 10.1016/0143-4160(81)90021-x. [DOI] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE. 2013;8:e55785. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Van Houten B. Alterations in bioenergetics due to changes in mitochondrial DNA copy number. Methods. 2010;51:452–427. doi: 10.1016/j.ymeth.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, Hardingham GE. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun. 2013;4:2034. doi: 10.1038/ncomms3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt ML, Roche TE. Mechanism of pyruvate inhibition of kidney pyruvate dehydrogenasea kinase and synergistic inhibition by pyruvate and ADP. J Biol Chem. 1979;254:7191–7196. [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32:2362–2376. doi: 10.1038/emboj.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, Mintun MA. Brain work and brain imaging. Annu Rev Neurosci. 2006;29:449–476. doi: 10.1146/annurev.neuro.29.051605.112819. [DOI] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan TA. Activity-driven ATP synthesis is required for synaptic function. Cell. 2014;156:825–835. doi: 10.1016/j.cell.2013.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contact with the endoplasmic reticulum as determinants of mitochondrial calcium responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rueda CB, Llorente-Folch I, Amigo I, Contreras L, González-Sánchez P, Martínez-Valero P, Juaristi I, Pardo B, del Arco A, Satrústegui J. Ca(2+) regulation of mitochondrial function in neurons. Biochim Biophys Acta. 2014;1837:1617–1624. doi: 10.1016/j.bbabio.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Ryu SY, Beutner G, Dirksen RT, Kinnally KW, Sheu SS. Mitochondrial ryanodine receptors and other mitochondrial Ca2+ permeable channels. FEBS Lett. 2010;584:1948–1955. doi: 10.1016/j.febslet.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF, Martínez-Pons C, Milán M, Guinovart JJ. Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. Cereb Blood Flow Metab. 2014;34:945–955. doi: 10.1038/jcbfm.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safer B, Smith CM, Williamson JR. Control of the transport of reducing equivalents across the mitochondrial membrane in perfused rat heart. J Mol Cell Cardiol. 1971;2:111–124. doi: 10.1016/0022-2828(71)90065-4. [DOI] [PubMed] [Google Scholar]
- Sajic M, Mastrolia V, Lee CY, Trigo D, Sadeghian M, Mosley AJ, Gregson NA, Duchen MR, Smith KJ. Impulse conduction increases mitochondrial transport in adult mammalian peripheral nerves in vivo. PLoS Biol. 2013;11:e1001754. doi: 10.1371/journal.pbio.1001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342:1379–1382. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satrustegui J, Pardo B, del Arco A. Mitochondrial transporters as novel targets for intracellular calcium signalling. Physiol Rev. 2007;87:29–67. doi: 10.1152/physrev.00005.2006. [DOI] [PubMed] [Google Scholar]
- Schell JC, Rutter J. The long and winding road to the mitochondrial pyruvate carrier. Cancer Metab. 2013;1:6. doi: 10.1186/2049-3002-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton R, Pollak JK. The increasing adenine nucleotide concentration and the maturation of rat liver mitochondria during neonatal development. Differentiation. 1978;12((1)):15–21. doi: 10.1111/j.1432-0436.1979.tb00985.x. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology. 2008;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- Takeuchi A1, Kim B, Matsuoka S. The destiny of Ca2+ released by mitochondria. J Physiol Sci. 2015;65((1)):11–24. doi: 10.1007/s12576-014-0326-7. doi: 10.1007/s12576-014-0326-7. Epub 2014 Jul 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traba J, Froschauer EM, Wiesenberger G, Satrustegui J, del Arco A. Yeast mitochondria import ATP through the calcium-dependent ATP-Mg/Pi carrier Sal1p, and are ATP consumers during aerobic growth in glucose. Mol Microbiol. 2008;69:570–585. doi: 10.1111/j.1365-2958.2008.06300.x. [DOI] [PubMed] [Google Scholar]
- Traba J, Satrustegui J, del Arco A. Adenine nucleotide transporters in organelles: novel genes and functions. Cell Mol Life Sci. 2011;68:1183–1206. doi: 10.1007/s00018-010-0612-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traba J, del Arco A, Duchen MR, Szabadkai G, Satrustegui J. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca(2+) buffering. Cell Death Differ. 2012;19:650–660. doi: 10.1038/cdd.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Jiang D, Zhao L, Clapham D, Miller C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J Gen Physiol. 2014;143:67–73. doi: 10.1085/jgp.201311096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcarce C, Navarrete RM, Encabo P, Loeches E, Satrústegui J, Cuezva JM. Postnatal development of rat liver mitochondrial functions. The roles of protein synthesis and of adenine nucleotides. J Biol Chem. 1988;263:7767–7775. [PubMed] [Google Scholar]
- Vitorica J, Satrústegui J. The role of ADP in the modulation of the calcium-efflux pathway in rat brain mitochondria. Biochem J. 1985;225:41–49. doi: 10.1042/bj2250041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci U S A. 2010;107:16823–16827. doi: 10.1073/pnas.1011099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker RG, Turnbull DM, Whittington MA, Cunningham MO. Impaired mitochondrial function abolishes gamma oscillations in the hippocampus through an effect on fast-spiking interneurons. Brain. 2011;134:e180. doi: 10.1093/brain/awr018. [DOI] [PMC free article] [PubMed] [Google Scholar]