

Abstract
An ischaemic stroke occurs during loss of blood flow in the brain from the occlusion of a blood vessel. The ischaemia itself comprises a complex array of insults, including oxygen and glucose deprivation (OGD), glutamate excitotoxicity, acidification/hypercapnia, and loss of sheer forces. A substantial amount of knowledge has accumulated that define the excitotoxic cascade downstream of N-methyl-d-aspartate receptors (NMDARs). While the NMDAR can influence numerous downstream elements, one critical target during ischaemia is the ion channel, pannexin-1 (Panx1). The C-terminal region of Panx1 appears critical for its regulation under a host of physiological and pathological stimuli. We have shown using hippocampal brain slices that Panx1 is activated by NMDARs through Src family kinases. However, it is not yet certain if this involves direct phosphorylation of Panx1 or an allosteric interaction between the channel's C-terminal tail and Src. Interestingly, Panx1 opening during ischaemia and NMDAR over-activation is antagonized by an interfering peptide that comprises amino acids 305–318 of Panx1. Thus, targeting the activation of Panx1 by NMDARs and Src kinases is an attractive mechanism to reduce anoxic depolarizations and neuronal death.
Roger Thompson, PhD is based at the Hotchkiss Brain Institute at the University of Calgary, Calgary Canada. His research background is in electrophysiology, biophysics and cellular imaging. He uses these techniques to investigate the regulation of ion channels by oxygen, including potassium channels and members of the gap junction superfamily. Dr. Thompson is currently a Scholar of the Alberta Heritage Foundation for Medical Research (Alberta Innovates Health Solutions) and is focussed on understanding the synaptic and pathophysiological functions of pannexin ion channels.

Introduction
In 1944 the Brazilian physiologist Aristedes Leao described cortical spreading depression (Leao, 1944). He characterized a slowly propagating wave of depression of electrical activity that spread unidirectionally across the cortex from a pinprick site. Leao later showed a similar spreading depression during global ischaemia following carotid artery ligation (Leao, 1947). This period of electrical negativity persisted for as long as blood flow was interrupted. Van Harreveld reported similar observations in the spinal cord, suggesting that neuronal depolarization was a common event during loss of blood flow (Van Harreveld & Hawes, 1946). It is now appreciated that these anoxic depolarizations are a complex interplay of ion channels and ligand-gated receptors. This review will expand upon the classical view that NMDA receptors are responsible for neuronal death during ischaemia through evidence implicating a novel ion channel, panenxin-1 (Panx1), as key to death and neuronal dysfunction.
Ion fluxes during spreading depression and anoxic depolarization
Dramatic ionic fluxes occur in the brain during anoxic depolarizations that are likely to be both the cause and consequence of Leao's electrical negativity wave. Hansen showed in the late 1970s that ischaemia caused a profound increase in extracellular K+ (Hansen, 1977, 1978). This was based upon the use of K+-selective electrodes to measure anoxic depolarizations and increased the spatial resolution available. The principal findings over the next several decades were that anoxic depolarizations in the partially perfused tissue surrounding the ischaemic core, or penumbra, occurred in waves (Nedergaard & Astrup, 1986). These repetitive wave depolarizations are called peri-infarct depolarizations. The irreversible loss of membrane potential of neurons in the ischaemic core is the anoxic (or terminal) depolarization.
Focal ischaemia induces spreading depolarizations (Nedergaard & Astrup, 1986), which initiate in the infarct rim where it borders the penumbra region. These repeated losses of membrane potential are thought to be important for neurological dysfunctions (Nedergaard, 1987; Takano et al. 2007). Thus, changes in the extracellular milieu during anoxic and peri-infarct depolarizations appear critically linked to neuronal death and dysfunction (Dijkhuizen et al. 1999). On the other hand, there is solid evidence that ATP depletion during ischaemia results in failure of the Na+–K+ pump, which may initiate the anoxic depolarization (Balestrino, 1995; Lipton, 1999). Much of these data are based on the use of oubain to directly block the pump, which can mimic anoxic depolarizations but does not conclusively demonstrate that the Na+–K+ pump is the initial cause. Indeed, other possibilities for initiation of the ischaemia-induced depolarizations have been investigated and include direct activation of Na+ channels (Xie et al. 1995; Risher et al. 2011), exchangers (Stys et al. 1992; Tekkök et al. 2007), activation of non-selective cation channels (Aarts et al. 2003; Gao et al. 2005; Weilinger et al. 2012a), and neurotransmitter-mediated activation of ligand-gated receptors (Rothman, 1984; Rader & Lanthorn, 1989; Madry et al. 2010).
There is a tendency to look for a singular root cause of a disease because this would support focused drug development. We now appreciate that there are likely to be multiple parallel events involved in anoxic and peri-infarct depolarizations. Our ability to design drugs that are neuroprotective or promote recovery will be rooted in understanding the contributions of different ion channels at different times in the anoxic depolarization. Figure 1 shows a model of the cellular and electrical changes in a neuron (or brain tissue) during ischaemia and identifies where some important ion flux pathways presumably contribute. There are two important points to keep in mind: The first is that to date, inhibition of a single ion channel has failed to prevent the anoxic depolarization, but may drastically delay it. The second is that different brain regions are likely to have distinct balances of key ion channel players. To exemplify this, Andrew's lab showed recently that there is a differential susceptibility of hypothalamic and neocortical neurons to ischaemic injury (Brisson & Andrew, 2012). The exception to this diversity of players may be the NMDA receptor, which has a ubiquitous distribution throughout the brain and has been implicated in anoxic depolarizations and neuronal death (Lipton, 1999; Aarts et al. 2002). In addition, NMDARs have the ability to activate numerous (largely Ca2+-dependent) downstream mechanisms that impact neuronal health.
Figure 1. Schematic representation of the anoxic depolarization in relation to ATP concentration and glutamate release.
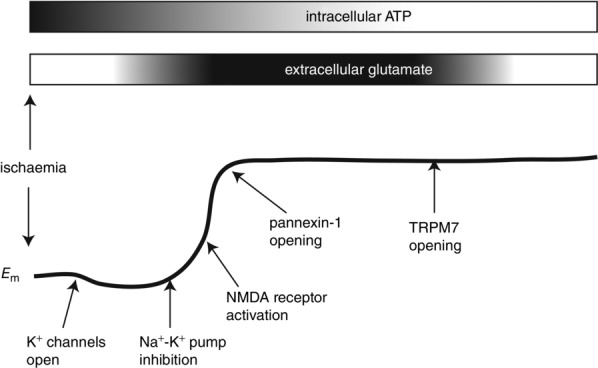
Ischaemia causes depletion of intracellular ATP, which is thought to be important for inhibition of the Na+–K+ pump. Na+–K+ pump inhibition probably initiates membrane depolarization, as depicted in the lower cartoon of Em. The putative timing of activation of NMDARs following a transient increase in glutamate release is indicated. NMDARs probably contribute to depolarization, but neuronal death is proposed to occur following activation of downstream cation channels such as pannexin-1 and TRPM7.
NMDA receptors
The importance of NDMARs in anoxic depolarizations and neuronal death is controversial, probably because different strategies to prevent depolarizations or neuronal death by blocking the receptor have met with variable outcomes – mostly negative (Hoyte et al. 2004). The experimental model, either global or focal ischaemia, may explain part of the controversy (see Lipton, 1999 for a comparison of these insults). On the other hand, the explanation could be related to complexities in the roles of NMDARs originating from the receptor subtype involved, their cellular localization (i.e. synaptic or extrasynaptic), co-agonist and downstream signalling targets.
Functional NMDARs are composed of four subunits, two are obligatory GluN1 subunits and the remaining two arise from GluN2a, GluN2b, GluN2c or GluN2d (Traynelis et al. 2010). It appears that the receptor is usually di-heteromeric, meaning that there are two GluN1 and two subunits from the same GluN2 (i.e. two GluN2a or two GluN2b). However, tri-heteromeric NMDARs are possible and probably exist (Tovar et al. 2013). The activation of NMDARs requires the binding of co-ligands and membrane depolarization. Glutamate binds to the GluN2 subunits, and the co-agonist, glycine or d-serine, bind to the GluN1 subunits (Traynelis et al. 2010). The coincident membrane depolarization functions to ‘kick’ a Mg2+ ion out of the receptor pore, allowing the Na+ and Ca2+ inward currents (Mayer et al. 1984). The influx of Ca2+ through the NMDAR is proposed to be the critical pathway linked to cell death (Tymianski et al. 1993). This is known as the ‘source specific hypothesis’.
NMDAR subunit composition and subcellular localization both appear important for excitotoxicity. There is a strong body of data that suggest GluN2b-containing NMDARs are located primarily extra-synaptically and these receptors are responsible for neuronal death (Liu et al. 2007; Hardingham & Bading, 2010; Gladding & Raymond, 2011; Martel et al. 2012). Subunit specific roles in the anoxic depolarization have not been extensively investigated, probably because of the challenge of separating membrane potential contributions of the different subunits. However, GluN2b receptors appear preferentially linked to NO and ONOO− production during ischaemia (Aarts et al. 2003). This arises from a protein complex whereby post-synpatic density protein-95 (PSD-95) provides a scaffold for assembly of NO synthase and NMDARs. Ca2+ influx through NMDARs activates NO production and subsequent ONOO− initiated cell death (Aarts et al. 2002; Cook et al. 2012). GluN2b activation may also suppress cell survival signalling (see Tymianski, 2011 for a comprehensive review). On the other hand, synaptic GluN2a-containing receptors initiate neuronal survival through the Akt/ERK pathway (Hardingham, 2006). A complete separation of synaptic pro-survival (GluN2a) and extra-synaptic death-inducing (GluN2b) responses is muddied by incomplete subcellular separation of the receptors (Tymianski, 2011).
Oliet and others in Bordeaux recently challenged the notion that extra-synaptic GluN2b receptors are solely responsible for neuronal death (Papouin et al. 2012). They suggest that it is the availability of NMDAR co-agonist for GluN2a- or GluN2b-containing receptors that is important for cell death during O2 and glucose deprivation (OGD). It was posited that astrocytes regulate the availability of co-agonist in the synapse so that synaptic receptors see predominantly d-serine. In contrast, extra-synaptic receptors utilize glycine as their co-agonist. Surprisingly, it is d-serine activation of synaptic NMDARs that is responsible for neuronal death induced by OGD. This interesting mechanism relies on the increased sensitivity of GluN2a receptors for d-serine over glycine, allowing for separation of signalling by the co-agonists.
A third possibility is that the NMDARs are not per se directly responsible for neuronal death during ischaemia and the anoxic depolarization. But rather these receptors initiate downstream signals that recruit other non-selective cation channels that mediate cell death. Evidence for this idea includes recruitment of transient receptor potential (TRP) channels, particularly TRPM7 (Aarts et al. 2003) and TRPM2 (Alim et al. 2013). Our work, and that of others, has expanded these downstream targets to include the unique, large pore ion channel, pannexin-1 (Panx1; Thompson et al. 2006, 2008; Bargiotas et al. 2011; Weilinger et al. 2012b). Therefore, a model is emerging that includes the NMDAR subtypes, co-agonists for NMDARs, coupling to non-selective cation channels, and subcellular localization of these molecular elements.
Pannexin channels and anoxic depolarizations
The pannexin (Panx) family of ion channels was discovered in the early 2000 s. We now know that there are three family members, Panx1–3 (Baranova et al. 2004), with Panx1 being the best characterized experimentally; probably because it has expression in most tissues tested so far (Penuela et al. 2014a). Panx2's expression is largely limited to the brain, where it resides in intracellular locations (Bruzzone et al. 2003; Wicki-Stordeur et al. 2013). Panx3, on the other hand, was found in bone and skin (Bond et al. 2011, p. 3), some cortical arterioles (Lohman et al. 2012a) and skeletal muscle (Pillon et al. 2014). The Panx family are members of the gap junction superfamily. Like their cousins, the intracellular gap junction channel forming connexins, Panxs are permeable to a wide range of molecules. These include cations, anions, second messengers and metabolites (Wang et al. 2007). It is critical to note that in most native tissues (i.e. not in over-expression systems) Panx1 does not form gap junction-like structures, but functions as plasma membrane ion channels (Sosinsky et al. 2011).
In the mid-2000s when I was working as a postdoctoral fellow at the University of British Columbia we made the discovery that Panx1 channels played a role in anoxic depolarizations. We observed that a large hemichannel-like activity was activated in acutely isolated hippocampal neurons exposed to O2–glucose deprivation (OGD; (Thompson et al. 2006). The tools for studying Panx1 at the time were limited so identification was based primarily upon the very large unitary conductance (∼500 pS), pharmacology (block by carbenoxolone, a non-specific gap junction family blocker), and fluorescent dye flux through the channels. Interestingly, OGD potently activated Panx1 channels in isolated neurons when ionotropic glutamate receptors (i.e. NMDARs) were blocked, suggesting that some aspect of OGD may open Panx1 independently of receptor signalling.
A few years after our description of Panx1 activation by OGD, Zhang and colleagues reported that Panx1 was activated (in cultured neurons) by NO (Zhang et al. 2008). This finding was particularly interesting because the highly labile gaseous transmitter NO and its metabolites (i.e. ONOO−) are reportedly important signalling molecules during ischaemia. Several important examples include the activation of transient receptor potential M7 channels and acid-sensitive ion channels (Aarts et al. 2003; Gao et al. 2005). Interestingly, NMDARs are important upstream elements that signal NO production during ischaemia, which may be critical for neurodegenerative disease (Nakamura et al. 2013). NO regulation of Panx1 was suggested because scavengers of NO could prevent dye efflux from cultured neurons exposed to OGD. Thus, Zhang and colleagues suggested that increased NO leads to opening of Panx1. However, the site of action of NO (Nakamura et al. 2013), which presumably is a nitrosylated cysteine somewhere on Panx1, was not identified.
There are several interesting candidate cysteine residues on Panx1 that could be sites of nitrosylation. Most of them have been systematically mutated and different groups report different overall effects on channel opening. Whilst this has been reviewed recently (Penuela et al. 2014b), C346 is a notable target because mutating C346 to serine opened Panx1 channels and caused cell death (Bunse et al. 2010). In agreement with this, mutation of C346 and C40 to alanine made Panx1 insensitive to NO donor-induced closing (Lohman et al. 2012b). Taken together, the work from Zoidl's lab (Bunse et al. 2010) and Isakson's lab (Lohman et al. 2012) suggested that NO acts at C346 to close the channel, which contrasts with the report of Zhang and colleagues (2008) who suggest NO opens Panx1. Thus, the potential regulation of Panx1 by NO during ischaemia is an open question. Perhaps these conflicting data may be explained by a ‘priming’ step where Panx1 is regulated by another factor such as phosphorylation prior to becoming susceptible to the effects of NO or the method of NO delivery (donor versus physiological or pathological stimulation) is critical.
NMDA receptors activate Panx1
After our report that Panx1 was activated during OGD, accounting for much of the large depolarizing inward current, we turned our attention to the possibiltity that Panx1 could also be directly regulated by the NMDA receptor. This was because of the important roles of NMDARs have in anoxic depolarizations. In 2008 I used a 0 mm extracellular Mg2+ model of interictal spiking to show that NMDARs induced Panx1 opening that increased both the amplitude and frequency of the spikes (Thompson et al. 2008). Directly activating NMDARs with short applications (i.e. 10 min) of exogenous NMDA activated a pathway for dye flux into hippocampal neurons in brain slices. Thus, we were able to show that NMDAR activation opened Panx1, which contributed to plasticity responses in neurons and increased membrane permeability to small molecules (Thompson & Macvicar, 2008; Thompson et al. 2008).
We initially hypothesized that the link between NMDARs and Panx1 was increased [Ca2+]i through the NMDAR itself (Thompson et al. 2008). Prior to our study, it had been suggested that increases in [Ca2+]i via P2Y purinergic receptors opened Panx1 (Locovei et al. 2006). However, in our hands a role for [Ca2+]i was not obvious because high concentrations (10 mm) of the Ca2+ chelator BAPTA failed to block NMDAR-induced Panx1 opening (Thompson et al. 2008).
So how is NMDARs activating Panx1? Several groups have reported that the Panx1 C-terminal region is critical for channel gating and a diversity of mechanisms have been proposed. These include modulation of cysteine residues (Bunse et al. 2010; Lohman et al. 2012b) and caspase cleavage of the Panx1 C-terminal (Chekeni et al. 2010). Bayliss’ group at the University of Virginia have reported that caspase-3/7 cleaves Panx1 at (or near) residues 376–379 (DWD) during apoptosis (Chekeni et al. 2010; Sandilos et al. 2012). The fascinating consequence is a constitutively open Panx1 channel that releases ATP to signal the immune system that the cell is apoptotic. We tested the possibility that NMDAR over-activation during excitotoxicty, which induces anoxic depolarization-like responses, was mediated by caspase cleavage of Panx1. The pan-caspase blocker zVAD failed to affect NMDAR-induced Panx1 activation and we did not detect a change in Panx1 abundance or mobility shift via Western blots (Weilinger et al. 2012b). Therefore, in our hands caspase 3/7 activation of Panx1 does not appear to play a role during initiation or maintenance of the anoxic depolarization, but we cannot rule out a role for this mechanisms in the hours or days after an ischaemic event.
In keeping with the theme that the Panx1 C-terminal region is important for regulating the channel, we investigated the possible modulation of activity by kinases. It is well known that NMDARs directly interact with Src family kinases (SFKs) (Salter & Kalia, 2004) so we postulated that SFKs may be important activators of Panx1. We reported that NMDAR-activation of Panx1 involves SFKs probably acting at Y308 in the Panx1 C-terminal (Weilinger et al. 2012b). The region around Y308 is a putative consensus site for SFKs. We demonstrated that during exposure of hippocampal slices to anoxia, SFKs are activated by NMDARs. Panx1 activation was prevented by the SFK antagonist PP2 and by a rationally designed interfering peptide comprising amino acids 305–318 of Panx (Weilinger et al. 2012b). The mechanism of action of SFKs on Panx1 is currently unknown, but could involve phosphorylation of Y308 or allosteric interactions of SFKs with the Panx1 C-terminus. One intriguing possibility is that NMDARs, SFKs and Panx1 exist as a molecular complex, which could restrict SFK signalling to a microdomain.
Panx1 and neuronal death
The pieces are falling into place to suggest that Panx1 plays a role in the anoxic depolarization and subsequent neuronal death. The Schwaninger and Monyer groups tested this idea using global Panx1 knockout mice and a permanent middle cerebral artery occlusion stroke model (Bargiotas et al. 2011, 2012). They found that knockout of Panx1 alone was not sufficient to significantly reduce lesion size and improve neurological outcomes, and the co-knockout of Panx2 was required. There are several explanations for why both Panx channels may contribute. Permanent middle cerebral artery occlusion is a different insult from transient vessel occlusions and in vitro models of stroke (i.e. OGD). In early studies on Panx1 and Panx2 it was reported that they could form heteromeric channels in Xenopus oocytes, suggesting that these heteromers could be critical (Bruzzone et al. 2003). Finally, Panx1 and Panx2 could play redundant roles during stroke by either providing critical sources of intracellular Ca2+ (influx and release from stores respectively) or being able to compensate for the deletion of the other channel.
Regardless of a requirement for Panx2 in addition to Panx1, the mechanisms of cell death are still unknown. There is strong evidence that neuronal death during ischaemia requires mitochondrial dysfunction, probably due to Ca2+ overload (Vergun et al. 1999; Lemasters et al. 2009). Interestingly, despite the reports that NMDARs are the critical source of Ca2+ for neuronal death, it has been reported in cultured cortical neurons that NMDAR antagonists did not prevent mitochondrial dysfunction during experimental ischaemia if the magnitude of the mitochondrial depolarization (and presumably the Ca2+ influx) was large (Vergun et al. 1999). Furthermore, the use-dependent NMDAR antagonist (i.e. pore blocker) MK801 does not substantially prevent neuronal death until cells are exposed to OGD for >3 h (Aarts et al. 2003). This suggests that if Ca2+ influx through NMDARs is important for neuronal death this probably only occurs after several hours of activation. One possibility is that NMDAR–SFK activation of Panx1 is responsible for a rapid (i.e. 10 s or minutes) influx of the critical Ca2+ that induces mitochondrial dysfunction and death (Fig. 2). Another possibility is that the NMDAR–SFK–Panx1 complex is closely associated with specific pools of mitochondria, such as synaptic mitochondrial (Naga et al. 2007).
Figure 2. Schematic depiction of Panx1 and a model for the role of NMDA receptors and pannexin-1 (Panx1) in ischaemia-induced neuronal death.
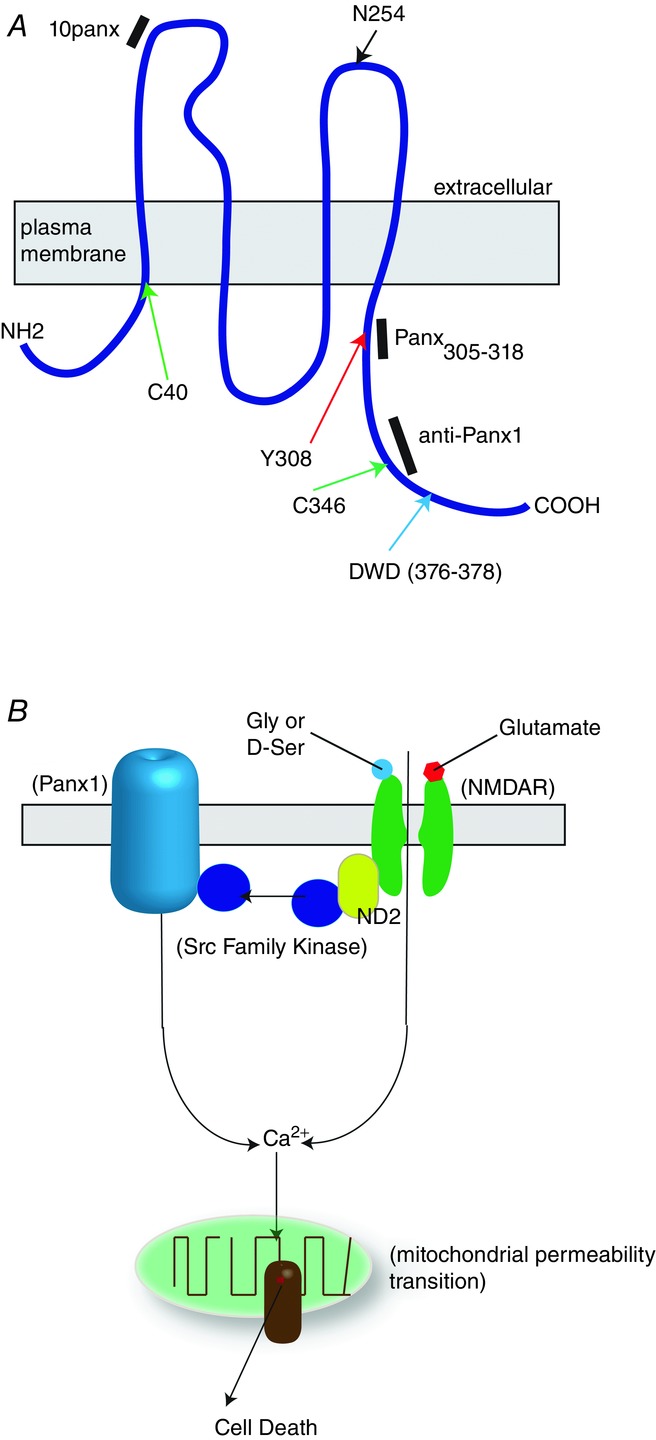
A, schematic representation of a single Panx1 peptide showing the location of known posttranslational modifications that alter function. The black bars show the known sites of action of three antagonists that block the anoxic depolarization. Panx305–318 and 10panx are small peptides and anti-Panx1 is a polyclonal antibody. Green arrows represent putative S-nitrosylation sites, red arrows putative phosphorylation (by Src kinases), blue arrow the caspase 3/7 cleavage site and the black arrow is the known site for N-linked glycosylation. B, functional Panx1 channels are hexameric. Interruption of blood flow (ischaemia) induces glutamate and glycine (gly)/d-serine (D-ser) release. This causes over-activation of the NMDAR, which is a putative pathway for excitotoxic calcium influx. Additionally, NMDARs can activate Panx1 via Src family kinases, leading to exasperated calcium influx. Calcium buffering by mitochondria can lead to permeability transition and cell death. ND2 is NADH dehydrogenase 2. Theoretically, any strategy to disrupt this putative complex could be effective at reducing anoxic depolarizations.
Conclusions and future directions
The anoxic depolarization is a complex neuronal response to ischaemia. It is likely to be a multiphasic recruitment of pumps, voltage-gated ion channels, non-selective cation channels and ligand-gated iontropic receptors. NMDA receptors are playing a critical role, but it is unclear if this is always a direct role or through initiation of downstream signalling. Key downstream targets of the NMDAR are TRPM7 and pannexin-1 channels. Our current understanding suggests that TRPM7 and Panx1 have distinct mechanisms of activation by the NMDAR, involving NO and SFKs, respectively. However, crosstalk of these players has not been rigorously investigated.
While there is still much to learn about the process of the anoxic depolarization and neuronal death, we are moving closer to making tailored interfering strategies with the promise of neuroprotection. One of these strategies is the Panx308 peptide (Weilinger et al. 2012b) that prevents Panx1 activation by SFKs, which we are currently testing in in vivo models of stroke. A similar strategy for block of PSD-95-binding proteins such as nitric oxide synthase is showing promise in clinical trials for stroke treatment (Hill et al. 2012). Future strategies aimed at neuroprotection by disruption or recovery of the anoxic depolarization are likely to be effective if they inhibit multiple targets, including Panx1.
Glossary
- NMDAR
N-methyl-d-aspartate receptor
- OGD
oxygen and glucose deprivation
- Panx1
pannexin-1
- SFK
Src family kinase
- TRP
transient receptor potential (channel)
Additional information
Competing interests
None declared.
Funding
Funding was received from the Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, Heart and Stroke Foundation of Canada. Additionally, R.J.T. is the recipient of a Scholar award from Alberta Innovates - Health Solutions.
References
- Aarts M, Iihara K, Wei W-L, Xiong Z-G, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang Y-T, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor–PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Alim I, Teves L, Li R, Mori Y, Tymianski M. Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J Neurosci. 2013;33:17264–17277. doi: 10.1523/JNEUROSCI.1729-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrino M. Pathophysiology of anoxic depolarization: new findings and a working hypothesis. J Neurosci Methods. 1995;59:99–103. doi: 10.1016/0165-0270(94)00199-q. [DOI] [PubMed] [Google Scholar]
- Baranova A, Ivanov D, Petrash N, Pestova A, Skoblov M, Kelmanson I, Shagin D, Nazarenko S, Geraymovych E, Litvin O, et al. The mammalian pannexin family is homologous to the invertebrate innexin gap junction proteins. Genomics. 2004;83:706–716. doi: 10.1016/j.ygeno.2003.09.025. [DOI] [PubMed] [Google Scholar]
- Bargiotas P, Krenz A, Hormuzdi SG, Ridder DA, Herb A, Barakat W, Penuela S, von Engelhardt J, Monyer H, Schwaninger M. Pannexins in ischemia-induced neurodegeneration. Proc Natl Acad Sci U S A. 2011;108:20772–20777. doi: 10.1073/pnas.1018262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargiotas P, Krenz A, Monyer H, Schwaninger M. Functional outcome of pannexin-deficient mice after cerebral ischemia. Channels (Austin) 2012;6:453–456. doi: 10.4161/chan.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond SR, Lau A, Penuela S, Sampaio AV, Underhill TM, Laird DW, Naus CC. Pannexin 3 is a novel target for Runx2, expressed by osteoblasts and mature growth plate chondrocytes. J Bone Miner Res. 2011;26:2911–2922. doi: 10.1002/jbmr.509. [DOI] [PubMed] [Google Scholar]
- Brisson CD, Andrew RD. A neuronal population in hypothalamus that dramatically resists acute ischemic injury compared to neocortex. J Neurophysiol. 2012;108:419–430. doi: 10.1152/jn.00090.2012. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, Hormuzdi SG, Barbe MT, Herb A, Monyer H. Pannexins, a family of gap junction proteins expressed in brain. Proc Natl Acad Sci U S A. 2003;100:13644–13649. doi: 10.1073/pnas.2233464100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunse S, Schmidt M, Prochnow N, Zoidl G, Dermietzel R. Intracellular cysteine 346 is essentially involved in regulating Panx1 channel activity. J Biol Chem. 2010;285:38444–38452. doi: 10.1074/jbc.M110.101014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, et al. Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DJ, Teves L, Tymianski M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature. 2012;483:213–217. doi: 10.1038/nature10841. [DOI] [PubMed] [Google Scholar]
- Dijkhuizen RM, Beekwilder JP, van der Worp HB, Berkelbach van der Sprenkel JW, Tulleken KA, Nicolay K. Correlation between tissue depolarizations and damage in focal ischemic rat brain. Brain Res. 1999;840:194–205. doi: 10.1016/s0006-8993(99)01769-2. [DOI] [PubMed] [Google Scholar]
- Gao J, Duan B, Wang D-G, Deng X-H, Zhang G-Y, Xu L, Xu T-L. Coupling between NMDA receptor and acid-sensing ion channel contributes to ischemic neuronal death. Neuron. 2005;48:635–646. doi: 10.1016/j.neuron.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci. 2011;48:308–320. doi: 10.1016/j.mcn.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Hansen AJ. Extracellular potassium concentration in juvenile and adult rat brain cortex during anoxia. Acta Physiol Scand. 1977;99:412–420. doi: 10.1111/j.1748-1716.1977.tb10394.x. [DOI] [PubMed] [Google Scholar]
- Hansen AJ. The extracellular potassium concentration in brain cortex following ischemia in hypo- and hyperglycemic rats. Acta Physiol Scand. 1978;102:324–329. doi: 10.1111/j.1748-1716.1978.tb06079.x. [DOI] [PubMed] [Google Scholar]
- Hardingham GE. Pro-survival signalling from the NMDA receptor. Biochem Soc Trans. 2006;34:936–938. doi: 10.1042/BST0340936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MD, Martin RH, Mikulis D, Wong JH, Silver FL, Terbrugge KG, Milot G, Clark WM, Macdonald RL, Kelly ME, et al. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012;11:942–950. doi: 10.1016/S1474-4422(12)70225-9. [DOI] [PubMed] [Google Scholar]
- Hoyte L, Barber PA, Buchan AM, Hill MD. The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol Med. 2004;4:131–136. doi: 10.2174/1566524043479248. [DOI] [PubMed] [Google Scholar]
- Leao A. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Leao A. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol. 1947;10:409–414. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen A-L. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locovei S, Wang J, Dahl G. Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett. 2006;580:239–244. doi: 10.1016/j.febslet.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Lohman AW, Billaud M, Straub AC, Johnstone SR, Best AK, Lee M, Barr K, Penuela S, Laird DW, Isakson BE. Expression of pannexin isoforms in the systemic murine arterial network. J Vasc Res. 2012a;49:405–416. doi: 10.1159/000338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman AW, Weaver JL, Billaud M, Sandilos JK, Griffiths R, Straub AC, Penuela S, Leitinger N, Laird DW, Bayliss DA, Isakson BE. S-nitrosylation inhibits pannexin 1 channel function. J Biol Chem. 2012b;287:39602–39612. doi: 10.1074/jbc.M112.397976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madry C, Haglerød C, Attwell D. The role of pannexin hemichannels in the anoxic depolarization of hippocampal pyramidal cells. Brain. 2010;133:3755–3763. doi: 10.1093/brain/awq284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel M-A, Ryan TJ, Bell KFS, Fowler JH, McMahon A, Al-Mubarak B, Komiyama NH, Horsburgh K, Kind PC, Grant SGN, et al. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron. 2012;74:543–556. doi: 10.1016/j.neuron.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Naga KK, Sullivan PG, Geddes JW. High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J Neurosci. 2007;27:7469–7475. doi: 10.1523/JNEUROSCI.0646-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S-I, Lipton SA. Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M. Neuronal injury in the infarct border: a neuropathological study in the rat. Acta Neuropathol. 1987;73:267–274. doi: 10.1007/BF00686621. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Astrup J. Infarct rim: effect of hyperglycemia on direct current potential and [14C]2-deoxyglucose phosphorylation. J Cereb Blood Flow Metab. 1986;6:607–615. doi: 10.1038/jcbfm.1986.108. [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet J-P, Oliet SHR. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Penuela S, Harland L, Simek J, Laird DW. Pannexin channels and their links to human disease. Biochem J. 2014a;461:371–381. doi: 10.1042/BJ20140447. [DOI] [PubMed] [Google Scholar]
- Penuela S, Simek J, Thompson RJ. Regulation of pannexin channels by post-translational modifications. FEBS Lett. 2014b;588:1411–1415. doi: 10.1016/j.febslet.2014.01.028. [DOI] [PubMed] [Google Scholar]
- Pillon NJ, Li YE, Fink LN, Brozinick JT, Nikolayev A, Kuo M-S, Bilan PJ, Klip A. Nucleotides released from palmitate challenged muscle cells through pannexin-3 attract monocytes. Diabetes. 2014;63:3815–3826. doi: 10.2337/db14-0150. [DOI] [PubMed] [Google Scholar]
- Rader RK, Lanthorn TH. Experimental ischemia induces a persistent depolarization blocked by decreased calcium and NMDA antagonists. Neurosci Lett. 1989;99:125–130. doi: 10.1016/0304-3940(89)90276-0. [DOI] [PubMed] [Google Scholar]
- Risher WC, Lee MR, Fomitcheva IV, Hess DC, Kirov SA. Dibucaine mitigates spreading depolarization in human neocortical slices and prevents acute dendritic injury in the ischemic rodent neocortex. PLoS One. 2011;6:e22351. doi: 10.1371/journal.pone.0022351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman S. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J Neurosci. 1984;4:1884–1891. doi: 10.1523/JNEUROSCI.04-07-01884.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Sandilos JK, Chiu Y-H, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, Bayliss DA. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J Biol Chem. 2012;287:11303–11311. doi: 10.1074/jbc.M111.323378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosinsky GE, Boassa D, Dermietzel R, Duffy HS, Laird DW, MacVicar B, Naus CC, Penuela S, Scemes E, Spray DC, et al. Pannexin channels are not gap junction hemichannels. Channels (Austin) 2011;5:193–197. doi: 10.4161/chan.5.3.15765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+–Ca2+ exchanger. J Neurosci. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian G-F, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10:754–762. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- Tekkök SB, Ye Z, Ransom BR. Excitotoxic mechanisms of ischemic injury in myelinated white matter. J Cereb Blood Flow Metab. 2007;27:1540–1552. doi: 10.1038/sj.jcbfm.9600455. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, MacDonald JF, MacVicar BA. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science. 2008;322:1555–1559. doi: 10.1126/science.1165209. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Macvicar BA. Connexin and pannexin hemichannels of neurons and astrocytes. Channels (Austin) 2008;2:81–86. doi: 10.4161/chan.2.2.6003. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–927. doi: 10.1126/science.1126241. [DOI] [PubMed] [Google Scholar]
- Tovar KR, McGinley MJ, Westbrook GL. Triheteromeric NMDA receptors at hippocampal synapses. J Neurosci. 2013;33:9150–9160. doi: 10.1523/JNEUROSCI.0829-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M. Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat Neurosci. 2011;14:1369–1373. doi: 10.1038/nn.2951. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Harreveld A, Hawes RC. Asphyxial depolarisation in the spinal cord. Am J Physiol. 1946;147:669–684. doi: 10.1152/ajplegacy.1946.147.4.669. [DOI] [PubMed] [Google Scholar]
- Vergun O, Keelan J, Khodorov BI, Duchen MR. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol. 1999;519:451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ma M, Locovei S, Keane RW, Dahl G. Modulation of membrane channel currents by gap junction protein mimetic peptides: size matters. Am J Physiol Cell Physiol. 2007;293:C1112–C1119. doi: 10.1152/ajpcell.00097.2007. [DOI] [PubMed] [Google Scholar]
- Weilinger NL, Maslieieva V, Bialecki J, Sridharan SS, Tang PL, Thompson RJ. Ionotropic receptors and ion channels in ischemic neuronal death and dysfunction. Acta Pharmacol Sin. 2012a;34:39–34. doi: 10.1038/aps.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilinger NL, Tang PL, Thompson RJ. Anoxia-induced NMDA receptor activation opens pannexin channels via SRC family kinases. J Neurosci. 2012b;32:12579–12588. doi: 10.1523/JNEUROSCI.1267-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicki-Stordeur LE, Boyce AKJ, Swayne LA. Analysis of a pannexin 2-pannexin 1 chimeric protein supports divergent roles for pannexin C-termini in cellular localization. Cell Commun Adhes. 2013;20:73–79. doi: 10.3109/15419061.2013.791681. [DOI] [PubMed] [Google Scholar]
- Xie Y, Zacharias E, Hoff P, Tegtmeier F. Ion channel involvement in anoxic depolarization induced by cardiac arrest in rat brain. J Cereb Blood Flow Metab. 1995;15:587–594. doi: 10.1038/jcbfm.1995.72. [DOI] [PubMed] [Google Scholar]
- Zhang L, Deng T, Sun Y, Liu K, Yang Y, Zheng X. Role for nitric oxide in permeability of hippocampal neuronal hemichannels during oxygen glucose deprivation. J Neurosci Res. 2008;86:2281–2291. doi: 10.1002/jnr.21675. [DOI] [PubMed] [Google Scholar]