

Abstract
Phosphorylation of ion channels, including nicotinic acetylcholine receptors (nAChRs), by protein kinases plays a key role in the modification of synaptic transmission and neuronal excitability. α7 nAChRs are the second most prevalent nAChR subtype in the CNS following α4β2. Serine 365 in the M3–M4 cytoplasmic loop of the α7 nAChR is a phosphorylation site for protein kinase A (PKA). D1/D5 dopamine receptors signal through the adenylate cyclase–PKA pathway and play a key role in working memory and attention in the prefrontal cortex. Thus, we examined whether the dopaminergic system, mediated through PKA, functionally interacts with the α7-dependent cholinergic neurotransmission. In layer 1 interneurons of mouse prefrontal cortex, α7 nicotinic currents were decreased upon stimulation with 8-Br-cAMP, a PKA activator. In HEK 293T cells, dominant negative PKA abolished 8-Br-cAMP's effect of diminishing α7 nicotinic currents, while a constitutively active PKA catalytic subunit decreased α7 currents. In brain slices, the PKA inhibitor KT-5720 nullified 8-Br-cAMP's effect of attenuating α7 nicotinic responses, while applying a PKA catalytic subunit in the pipette solution decreased α7 currents. 8-Br-cAMP stimulation reduced surface expression of α7 nAChRs, but there was no change in single-channel conductance. The D1/D5 dopamine receptor agonist SKF 83822 similarly attenuated α7 nicotinic currents from layer 1 interneurons and this attenuation of nicotinic current was prevented by KT-5720. These results demonstrate that dopamine receptor-mediated activation of PKA negatively modulates nicotinic neurotransmission in prefrontal cortical interneurons, which may be a contributing mechanism of dopamine modulation of cognitive behaviours such as attention or working memory.
Key points
Protein kinases can modify the function of many proteins including ion channels.
However, the role of protein kinase A in modifying nicotinic receptors in the CNS has never been investigated.
We showed through whole-cell recordings of layer 1 prefrontal cortical interneurons that α7 nicotinic responses are negatively modulated by protein kinase A.
Furthermore, we show that stimulation of dopamine receptors can similarly attenuate α7 nicotinic responses through the activation of protein kinase A.
These results suggest how the interaction of the cholinergic and dopaminergic systems may influence neuronal excitability in the brain.
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) are widely distributed throughout the brain and play a key role in mediating neurotransmission in the CNS. The homopentameric α7 nAChR is the second most abundant nAChR subtype in the brain after α4β2 (Clarke et al. 1985; Whiting et al. 1987; Perry et al. 2002; Nashmi & Lester, 2006; Gotti et al. 2006). α7 nAChRs are known to enhance cognitive behaviours such as attention (Levin, 2002; Young et al. 2007; Sydserff et al. 2009) and memory (Thomsen et al. 2010; Castner et al. 2011; Yang et al. 2013). Dysfunction of α7 nAChRs has been implicated in various nervous system disorders. Altered expression of α7 nAChRs has been found in smokers’ brains, as well as those of people stricken with schizophrenia and Alzheimer's disease (Freedman et al. 1995; Wevers et al. 1999; AhnAllen, 2012). Understanding how post-translational modifications to α7 nAChRs alter their function and expression in neurons will enhance our knowledge of the mechanisms that α7 nAChRs contribute to normal behaviours and pathological neural disorders.
Protein kinases play a major role in modifying the activity of ion channels and ultimately neuronal excitability (Porter et al. 1990; Astman et al. 1998; Man et al. 2007; Liu & Murray, 2012; Komal et al. 2014). Protein kinases catalyse the transfer of a phosphate group to the side chain hydroxyl group of a tyrosine, serine or threonine within a protein. Protein kinase A or cAMP-dependent protein kinase (PKA) is a serine kinase (Maller et al. 1978), which assembles as a tetrameric complex consisting of two regulatory and two catalytic subunits (Naira et al. 1985). In neurons, PKA has a wide range of substrates ranging from ion channels to transcription factors (Meyer & Shen, 2000; Zhong et al. 2009). For example, PKA-mediated phosphorylation of glutamate receptors has been shown to affect multiple forms of synaptic plasticity (Esteban et al. 2003). G protein-coupled receptors that couple through Gαs, such as D1/D5 dopamine receptors, are a major receptor system that activates PKA (Beaulieu & Gainetdinov, 2011). Dopamine, which binds and activates D1/D5 dopamine receptors, is released in the prefrontal cortex where it plays a vital role in working memory, motivation and attention (Phillips et al. 2008).
Like other nAChRs, α7 nAChRs are substrates of protein kinases (Vijayaraghavan et al. 1990; Moss et al. 1996; Komal et al. 2014). One major mechanism regulating the function of α7 nAChRs and other nAChR subtypes is the phosphorylation of the major cytoplasmic loop of the receptor, which contains putative phosphorylation sites for protein kinases including PKA and tyrosine kinase (Moss et al. 1996; Charpantier et al. 2005). Electrophysiological studies have shown that the Src family of tyrosine kinases can negatively regulate the function of α7 nAChRs (Cho et al. 2005; Charpantier et al. 2005; Komal et al. 2014). In addition to phosphotyrosine, there is a serine in the M3–M4 cytoplasmic loop of both chick and rat α7 nAChRs that is phosphorylated by PKA but not by CaMKII or protein kinases C or G (Moss et al. 1996). Studies have examined the impact of PKA on nAChR function mainly in the peripheral nervous system (Margiotta et al. 1987) and in the muscle (Green et al. 1991). Interestingly, activation of α7 nAChRs themselves results in a signal transduction, which activates PKA (Dajas-Bailador et al. 2002; Cheng & Yakel, 2014). However, to date there has been no report on the effect of PKA on modulating nAChR activity in the CNS.
In this study, we examined the mechanisms of how PKA activation reduced α7 nAChR currents expressed in cells of the human embryonic kidney cell line HEK 293T and in layer 1 interneurons of the mouse prefrontal and frontal cortices (PFC and FC) using pharmacological and molecular techniques and showed that PKA targets serine 365 of α7 to reduce their surface expression. Furthermore, activation of the D1/D5 dopamine receptors similarly attenuated α7 nAChRs through a PKA-mediated mechanism.
Methods
Ethical approval
All experiments on mice were performed in accordance with the Canadian Council of Animal Care and approved by the Animal Care Committee at the University of Victoria.
cDNA constructs
Mouse α7 cDNA was kindly provided by Jerry Stitzel (University of Colorado, Boulder, CO, USA). Venus fluorescent protein cDNA was provided by Atsushi Miyawaki (Riken Brain Science Institute, Tokyo, Japan) (Nagai et al. 2002). Human RIC-3 cDNA was provided by Neil Millar (University College London, UK) (Lansdell et al. 2005). The cDNA for mouse α7 nAChR subunit tagged with Venus fluorescent protein (α7-Venus) in the M3–M4 cytoplasmic loop was generated previously in the lab (Komal et al. 2014). In mutant α7(S365A) cDNA the serine 365 codon (AGC) was mutated to the alanine codon (GCA) by site-directed mutagenesis (Bio Basic Inc.; Markham, Canada). A dominant negative mouse PKA plasmid M7 pdnPKA-GFP cDNA (Ungar & Moon, 1996) and PKA catalytic subunit α (PKA-Cα) cDNA (Uhler & McKnight, 1987) were obtained from Addgene (cat. nos 16716 and 15310, respectively; Cambridge, USA).
HEK 293T cell culture and transfection
HEK 293T cells were maintained in plating medium consisting of Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mm l-glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. For electrophysiology 5 mm diameter round glass coverslips (Warner Instruments; Hamden, USA) were placed inside 35 mm petri dishes and coated with 1% gelatin type B (EMD Millipore; Etobicoke, Canada) or 1 mg ml−1 poly-dl-lysine (Sigma-Aldrich; St. Louis, USA) for 2 h. For confocal imaging, HEK 293T cells were grown on glass coverslip bottom dishes (MatTek Corporation; Ashland, USA) coated for 2 h with 1% gelatin. Cells were maintained in a 5% CO2 incubator at 37°C. Cells were grown to 40–50% confluency and then transiently transfected with FuGENE Transfection Reagent (Promega; Madison, USA). To each 35 mm dish, 2 μg cDNA of either α7, α7-Venus or their S365A mutant versions, 2 μg cDNA of RIC-3 and 3 μl of FuGENE Transfection Reagent (Promega) were mixed with 250 μl of warmed incomplete medium, which is the plating medium minus FBS. Venus cDNA (0.2 μg) was added to α7 to visually identify transfected HEK 293T cells for whole-cell patch-clamp recordings.
Drugs
8-Bromo-cAMP sodium salt, PHA543613 hydrochloride, KT-5720, SKF 83822 and SKF 81297 were all from Tocris Bioscience (Bristol, UK). Acetylcholine chloride and CNQX disodium salt were from Sigma-Aldrich while tetrodotoxin citrate was purchased from Alomone Labs (Jerusalem, Israel). Protein kinase A catalytic subunit protein, which was added to the pipette solution during slice electrophysiology, was obtained from Sigma-Aldrich.
Whole-cell patch-clamp electrophysiology from cultured cells
HEK 293T cells were visualized with differential interference contrast illumination using an upright microscope (Nikon FN1; Mississauga, Canada) with a CFI APO 40× W NIR objective (0.80 numerical aperture, 3.5 mm working distance). We identified transfected cells with Venus fluorescent protein using fluorescence illumination with a mercury lamp. Whole-cell patch-clamp recordings were performed using a Multiclamp 700B amplifier (Molecular Devices; Sunnyvale, USA), low-pass filtered at 4 kHz, digitized at 10 kHz with a Digidata 1440A (Molecular Devices) and recorded using pCLAMP 10.2 acquisition software (Molecular Devices).
During recordings, cells were perfused continuously at 2 ml min−1 with extracellular solution containing (in mm): 150 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 Hepes, and 10 d-glucose adjusted to pH 7.4. Extracellular recording solution was maintained at 31°C using a temperature controller (TC-344B, Warner Instruments) and in-line heater (SH-27B, Warner Instruments). Standard whole-cell patch-clamp recordings were performed using patch pipettes pulled from borosilicate glass electrodes (1.5 mm o.d. and 1.0 mm i.d., WPI) on a P-97 Flaming/Brown micropipette puller (Sutter Instruments). Patch electrodes had tip resistances between 6 and 11 MΩ and were filled with (in mm): 108 KH2PO4, 4.5 MgCl2, 0.9 EGTA, 9 Hepes, 0.4 CaCl2, 14 creatine phosphate (Tris salt), 4 Mg-ATP, 0.3 GTP (Tris salt), pH 7.4 with KOH. The membrane potential was held at −60 mV and corrected for liquid junction potential. Acetylcholine (ACh, 1 mm) was applied rapidly for 1 s duration using the two-barrel glass θ-tube valve-driven drug applicator (Komal et al. 2011) positioned 300 μm away from the recorded cell. Solution exchange rates were typically less than 500 μs (10–90% peak time) as measured from open tip junction potential changes using 10% extracellular solution.
Whole-cell patch-clamp electrophysiology from brain slices
Postnatal day (P) 10–20 C57BL/6J mice (stock no. 000664, The Jackson Laboratory; Sacramento, USA) of either sex were used for all our electrophysiology experiments. Mice were deeply anaesthetized with isofluorane and decapitated. Brains were removed and placed in ice-cold slicing solution containing (in mm): 250 sucrose, 2.5 KCl, 1.2 NaH2PO4, 1.3 MgCl2, 2.4 CaCl2, 26 NaHCO3 and 11 d-glucose. Coronal slices (320 μm thick) were cut with a vibratome (Leica 1000S; Concord, Canada) from prefrontal and frontal cortices and transferred to an extracellular recording solution-filled incubation chamber in a 31°C water bath for at least 45 min before being transferred to the recording chamber. During recordings, slices were perfused continuously at 2 ml min−1 with extracellular solution containing (in mm): 125 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.3 MgCl2, 2.4 CaCl2, 26 NaHCO3, and 11 d-glucose, aerated with 95% O2 and 5% CO2 during incubation and recording. Layer 1 interneurons of the medial prefrontal and frontal cortices were visualized and targeted for whole-cell recordings using an upright microscope (Nikon FN1) with a CFI APO 40× W NIR objective (0.80 numerical aperture, 3.5 mm working distance) and equipped with infra-red video-assisted differential interference contrast illumination. Patch electrodes had tip resistances between 6 and 11 MΩ and were filled with pipette solution containing (in mm): 130 potassium gluconate, 5 EGTA, 0.5 CaCl2, 2 MgCl2, 10 Hepes, 3 Mg-ATP, 0.2 GTP, and 5 phosphocreatine Tris, pH adjusted to 7.4 with KOH, osmolarity adjusted to 300 mosmol l−1 with sucrose. Whole-cell voltage-clamp recordings were performed at 31°C using a temperature controller (TC-344B, Warner Instruments) and in-line heater (SH-27B, Warner Instruments), with a MultiClamp 700B amplifier (Molecular Devices) and pCLAMP 10.2 software (Molecular Devices). Data were low-pass filtered at 4 kHz and sampled at 10 kHz with a Digidata 1440A data acquisition system (Molecular Devices). The membrane potential was corrected for liquid junction potential, and series resistance was monitored throughout the experiment. Neurons were held at −60 mV. A specific agonist for α7 nAChRs, PHA543613 hydrochloride (100 μm) was applied for 1 s every 60 s using a valve-driven θ-tube drug applicator system (Komal et al. 2011) positioned 600 μm from the recorded cell. To isolate nicotinic responses sodium dependent action potentials were blocked with tetrodotoxin (0.5 μm) and AMPA and kainate glutamatergic responses were inhibited with CNQX (10 μm).
Current fluctuation analysis to estimate single-channel conductance
Single-channel conductance of α7 nAChRs was determined by performing non-stationary current fluctuation analysis on whole-cell mediated α7 nAChR responses from brain slices, elicited via application of 100 μm PHA543613 hydrochloride, at −60 mV. This technique has been previously described by others (Sigworth, 1980; Brown et al. 1998; Komal et al. 2014). We used Clampfit 10.2 software (Molecular Devices) to conduct fluctuation analysis on the whole-cell current traces. In brief, an ensemble of α7 nAChR responses was aligned by the point of maximal rise and averaged. The mean responses was scaled to the peak and substracted from individual responses. The variance of the current at each sample point of each trace was plotted against the mean current of the averaged traces at the same sample point in time. Then a linear fit was performed through the sampled points. The slope of the fit estimated the unitary current, i, of the nicotinic ion channel. The single-channel conductance was calculated using the equation γ = i/(Vh − Erev) where Vh is the holding potential (−60 mV) and Erev is the reversal potential for α7 nAChRs, determined experimentally as (−0.7 mV).
Alexa Fluor 647 α-bungarotoxin labelling of surface α7 nAChRs in HEK 293T cells
We utilized Venus fluorescent protein-tagged α7 nicotinic receptor (α7-Venus) cDNA construct tagged with Venus fluorophore (α7-Venus) and haemagglutinin epitope present in the long intracellular cytoplasmic loop spanning third and fourth transmembrane domains. HEK 293T cells plated on coverslip bottom 35 mm culture dishes were transiently transfected (per dish) with 2 μg each α7-Venus, RIC-3 and 3 μl of FuGENE. On the second day of post-transfection, cells were treated with control 1/1000 volume sterile H2O diluted in cell culture plating medium or 1/1000 volume of 100 mm 8-Br-cAMP stock solution diluted in cell culture plating medium (final concentration 100 μm 8-Br-cAMP) and incubated at 37ºC for 30 min. Cells were washed once with ice-cold PBS (pH 7.4) and then labelled with Alexa Fluor 647 α-bungarotoxin (Fl-BTx, 1 mg ml−1, Life Technologies; Burlington, Canada) at 1:200 dilution for 1 h at 4°C. Subsequently cells were washed and fixed with 2% paraformaldehyde for 10 min at 4°C and washed twice again before imaging. Surface expression of Fl-BTx-labelled α7-Venus receptors vs. mutant α7-Venus (α7(S365A)-Venus) receptors were examined with a Nikon C1si spectral confocal microscope system using a Plan Apo VC 60× 1.4 NA oil immersion objective (0.13 mm working distance). A lambda stack of X–Y images were collected simultaneously with each laser sweep onto an array of 32 photomultiplier tubes and averaged over four laser sweeps. The Venus fluorophore was excited by the 488 nm argon laser line at 5% maximal intensity and imaged between 496.5 and 656.5 nm at 5 nm wavelength intervals. Fl-BTx was excited with a 638 nm laser line at 20% maximal intensity and wavelength emissions were collected between 590 and 750 nm at 5 nm wavelength intervals, averaged over four laser sweeps and Fl-BTx was analysed at the peak emission of Alexa Fluor 647 (670 nm). Images were acquired over a 50 μm × 50 μm field of view at 512 pixels × 512 pixels digital resolution, and 12 bit intensity resolution. The pixel dwell time was set at 5.52 μs and the pinhole was set to medium (60 μm diameter). A single optical slice was imaged through the centre of the cell for the Alexa Fluor 647 α-bungarotoxin image and the α7-Venus image. Using ImageJ v1.43r, software images were analysed for mean signal intensity of Fl-BTx per cell and normalized to the mean Venus intensity per cell to quantify surface expression of α7-Venus nAChRs for both control and 8-Br-cAMP-treated cells.
Alexa Fluor 647 α-bungarotoxin labelling of surface α7 nAChRs on layer 1 interneurons of brain slices
To examine surface α7 nAChR expression in layer 1 interneurons of the medial prefrontal cortex, C57BL/6J mice (P15 and P18) were deeply anaesthetized with isoflurane and decapitated. The brain was removed and embedded in 3% agar. The agar block encompassing the brain was glued on the sectioning platform and submerged in ice-cold NMDG-based slicing solution that contained (in mm): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 Hepes, 25 d-glucose, 5 sodium ascorbate, 3 sodium pyruvate, 0.5 CaCl2, and 10 MgSO4. Coronal slices from the prefrontal cortex were sectioned at 250 μm thickness with a vibratome (Leica 1000S). Brain slices were transferred and incubated for 30 min at room temperature in a Hepes-based holding solution which contained (in mm): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 Hepes, 25 d-glucose, 5 sodium ascorbate, 3 sodium pyruvate, 2 mm CaCl2 and 2 MgSO4. Half the number of slices were incubated for 30 min at 36°C in control extracellular solution which contained (in mm): 119 NaCl, 2.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 12.5 d-glucose, 2 CaCl2 and 2 MgSO4. The remaining slices were incubated for 30 min at 36°C in 8-Br-cAMP solution which consisted of 100 μm 8-Br-cAMP added to extracellular solution.
The slices were then fixed in 4% paraformaldehyde for 24 h, rinsed two times in PBS and then floating brain sections were labelled with Alexa Fluor 647 α-bungarotoxin (1 mg ml−1, Fl-BTx, Life Technologies) at 1:200 in PBS and NeuroTrace 435/455 Blue Fluorescent Nissl Stain (Life Technologies) at 1:300 in PBS. Brain sections were washed twice with PBS, mounted on coated slides (Superfrost Plus Gold, Fisher Scientific; Ottawa, Canada) and coverslipped with Vectashield Mounting Medium (Vector Laboratories; Burlington, Canada).
z-stacks of images were acquired with a Nikon C1si spectral confocal microscope using a 60× oil objective (1.4 NA). Images were acquired at 100 μm × 100 μm field of view and digitized at 512 pixels × 512 pixels with a medium sized pinhole (60 μm diameter) setting. A z-stack of 29 optically sectioned images at 1 μm intervals were gathered over 28 μm closest to the most superficial surface of the brain section. Spectral gain was at 220 and pixel dwell time at 5.52 μs. NeuroTrace 435/455 was excited with the 405 nm laser diode at 1% maximal transmission and the emission was collected between 400 and 560 nm at 5 nm resolution with an average of two sweeps per optical slice, while Fl-BTx was excited with a 560 nm laser diode at 25% maximal transmission and the emission collected between 590 and 750 nm at 5 nm wavelength intervals and an average of four sweeps per optical slice. NeuroTrace and Fl-BTx images were spectrally unmixed and analysed with ImageJ by tracing neuronal cell bodies as regions of interest (ROIs) for every second optical section of each z-stack of the NeuroTrace images. Then the ROIs were transferred to the Fl-BTx stacks of images and the mean fluorescence intensity was calculated for each ROI for the corresponding Fl-BTx z-stack.
Statistics
All data are presented as means ± SEM. Significant difference (P < 0.05) between two groups of data were determined using a t test for continuous data meeting parametric assumptions of equal variances and normality. Otherwise, a Wilcoxon rank sum test was performed for non-parametric data. Comparisons between three or more groups were analysed using an analysis of variance (ANOVA) for parametric data followed by post hoc multiple pairwise analysis using either Tukey's HSD tests or Bonferroni corrected t tests. For non-parametric data involving comparison of three or more groups of data a Kruskal–Wallis rank sum test was performed followed by pairwise analyses using Wilcoxon rank sum tests. We also performed two-way ANOVAs on data with more than one treatment group that was repeated over time. All statistical analyses were performed using the R statistical computing language (http://www.R-project.org).
Results
8-Br-cAMP decreases α7 nicotinic receptor currents in HEK 293T cells
Catalytic PKA is activated when cAMP binds to the PKA regulatory subunits. To test whether activation of PKA can modulate α7 nAChR function, we used 8-Br-cAMP, a cell membrane-permeable analogue of cAMP. 8-Br-cAMP, rather than cAMP, was used because 8-Br-cAMP is resistant to hydrolysis by endogenous phosphodiesterases, which normally terminate cAMP signalling. The PKA signalling pathway was triggered by incorporating 8-Br-cAMP in the recording pipette solution. Since previous studies have shown a dose dependent (100 μm to 1 mm) effect of cAMP on other nAChR subtypes in insect ganglionic neurons (Courjaret & Lapied, 2001), in the present study we used a 100 μm concentration of 8-Br-cAMP to examine the effect of activated PKA on α7 nAChRs. We examined the effect of 8-Br-cAMP on α7 nicotinic receptor function by performing whole-cell recordings from HEK 293T cells transiently transfected with mouse α7 nAChRs. Control α7 nAChR responses recorded from HEK 293T cells showed a mean current amplitude of 603 ± 100 pA (n = 21) elicited by 1 mm ACh (1 s) that maintained a steady amplitude with repeated ACh applications (1 min intervals) (Fig.1A, C and D). The α7 nAChR responses showed a significant and progressive attenuation in the peak current over repeated ACh applications when 8-Br-cAMP was present in the micropipette solution, decreasing to a mean current response of 269 ± 82 pA (n = 15) (P = 0.004, Wilcoxon rank sum test) (Fig.1B, C and D) (P = 0.0006, 8-Br-cAMP treatment factor, two-way ANOVA) (Fig.1D). 8-Br-cAMP stimulation resulted in a time dependent decrease in α7 nAChR mediated peak response amplitude over repeated ACh applications with a mean exponential decay time constant of 5.4 ± 1.8 min. This indicates that the 8-Br-cAMP's effect on decreasing α7 nAChR responses was not instantaneous but occurred on a time scale in the order of minutes.
Figure 1. 8-Br-cAMP stimulation attenuates α7 nAChR currents in HEK 293T cells.
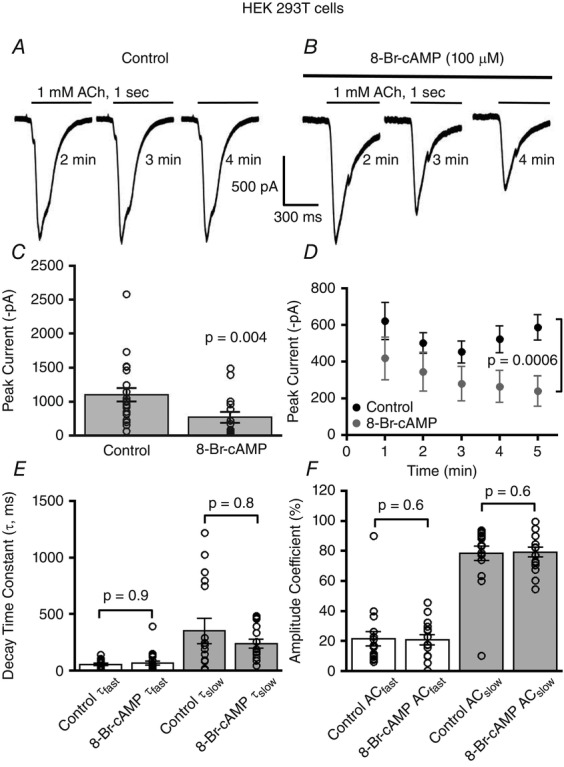
Acetylcholine (1 mm) evoked α7 nAChR whole-cell currents; traces for control (A) and 8-Br-cAMP (100 μm) treated cells showing a decline in α7 currents in the 8-Br-cAMP treated cells (B). C, the mean current amplitude of α7 nAChRs from control (n = 21) differed significantly from 8-Br-cAMP stimulated cells (n = 15) (P = 0.004, Wilcoxon rank sum test). For all the figures, bars represent means ± SEM of responses with each measured cell represented as a circle. D, mean current amplitude of ACh-mediated α7 whole-cell currents at 1 min time intervals for 8-Br-cAMP-stimulated cells was significantly attenuated as compared to control (P = 0.0006, two-way ANOVA). E, decay kinetics of α7 nAChR currents during ACh application showed no change in fast and slow decay time constant for control (n = 18) and 8-Br-cAMP (n = 21) (P = 0.9 for τfast, P = 0.8 for τslow, Wilcoxon rank sum tests) stimulated cells. F, no difference in the percentage composition of amplitude coefficients (AC) is observed for fast and slow decay time components of α7 nAChRs responses for control (n = 18) and 8-Br-cAMP stimulated neurons (n = 15) (P = 0.6, Wilcoxon rank sum tests for both τfast and τslow components). Note that for all figures n represents the total number of cells.
We next examined the effect of 8-Br-cAMP on α7 nAChR gating kinetics by fitting the time course of α7 current decay during ACh application to the sum of two exponential functions. The time course of α7 nAChR decay kinetics recorded from HEK 293T cells showed no significant difference in either the fast (control: 54 ± 10 ms, n = 18 vs. 8-Br-cAMP: 65 ± 19 ms, n = 21) (P = 0.9, Wilcoxon rank sum test) or the slow time constants (control: 351 ± 92 ms, n = 18 vs. 8-Br-cAMP: 238 ± 39 ms, n = 15) (P = 0.8, Wilcoxon rank sum test) between control and 8-Br-cAMP treated cells (Fig.1E). We also found no significant difference in the percentage composition of the fast and the slow decay components of the α7 nicotinic responses between control (amplitude coefficient τfast: 22 ± 5%, n = 18; τslow: 78 ± 5%, n = 18) and 8-Br-cAMP stimulated cells (amplitude coefficient τfast: 21 ± 3%, n = 15; τslow: 79 ± 3%, n = 15) (P = 0.6, Wilcoxon rank sum tests for both τslow and τfast) (Fig.1F). These data indicate that PKA activation via 8-Br-cAMP decreases α7 nAChR function without affecting channel gating kinetics.
8-Br-cAMP stimulation inhibits α7 nicotinic receptor currents in layer 1 cortical interneurons
Layer 1 neurons of the prefrontal cortex are predominantly GABAergic and the majority of these interneurons exhibit robust α7 nicotinic receptor currents (Christophe et al. 2002; Charpantier et al. 2005; Komal et al. 2014). Since we found that in cell lines 8-Br-cAMP activation of PKA attenuates α7 nicotinic receptor function we asked whether the same modulation occurs for α7 nAChRs endogenously expressed in CNS neurons. Accordingly, we targeted layer 1 medial prefrontal cortical neurons for whole-cell recordings. Recordings were performed in the presence of TTX (0.5 μm) and CNQX (10 μm) to block action potential dependent release of neurotransmitters and AMPA and kainate receptors. We examined α7 nAChR responses elicited by the rapid application of α7 nAChR selective agonist PHA543613 (100 μm for 1 s duration, every 1 min) from control and 8-Br-cAMP stimulated neurons, in which the intracellular pipette solution contained 100 μm 8-Br-cAMP. The average peak current of the α7 nAChR responses of the 8-Br-cAMP stimulated neurons (67 ± 4 pA, n = 26) was significantly reduced as compared to the α7 nAChR responses of control treated neurons (104 ± 9 pA, n = 18) (P = 0.008, t test) (Fig.2A–C). Moreover, neurons stimulated with 8-Br-cAMP showed a significantly greater progressive time-dependent decrease in α7 mediated whole-cell currents (n = 13) as compared to α7 nAChR currents recorded from control treated neurons (n = 17) (P = 0.03, 8-Br-cAMP treatment factor, two-way ANOVA), which showed no decrement in amplitude of α7 nAChR currents over time (Fig.2D). The time course of the progressive inhibition of α7 nAChR current responses mediated by 8-Br-cAMP in layer 1 cortical neurons occurred with an exponential decay time constant of 2.2 ± 0.6 min. These results indicate that 8-Br-cAMP mediated negative modulation of α7 nicotinic receptor activity in neurons, just like in HEK cells, occurred on a time scale of minutes, which is more consistent with 8-Br-cAMP targeting PKA and eliciting signal transduction actions of PKA, rather than 8-Br-cAMP non-specifically inhibiting α7 nAChRs directly, which would have occurred almost immediately.
Figure 2. 8-Br-cAMP diminishes α7 nAChR currents in layer 1 PFC interneurons.
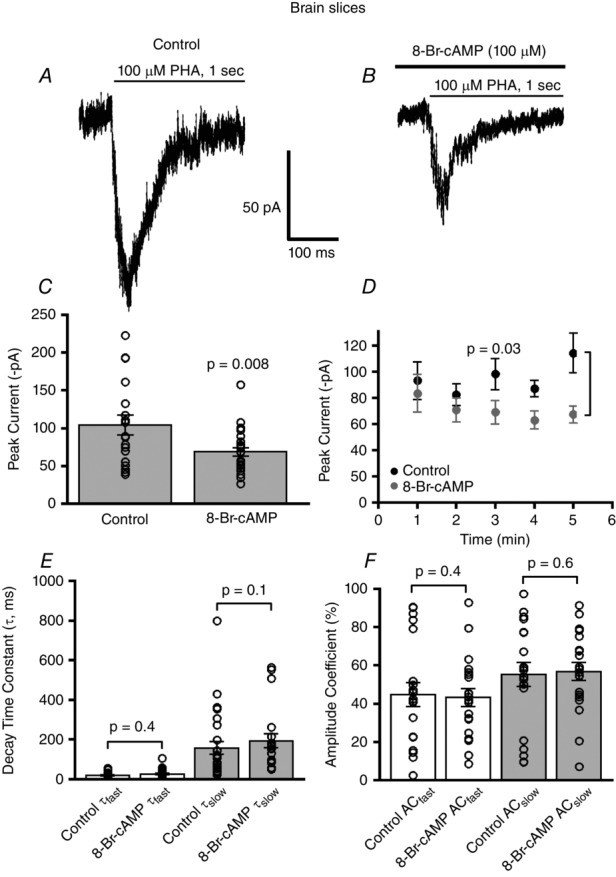
A and B, example traces of PHA543613 (100 μm) activated whole-cell α7 nicotinic responses for control and 8-Br-cAMP dialysed layer 1 interneurons both shown for the 5 min time point. C, there was a significant decrease in the mean current amplitude of α7 nAChRs of 8-Br-cAMP (100 μm) (n = 26)-treated interneurons as compared to control (n = 18) (P = 0.008, Wilcoxon rank sum test). D, time dependent reduction of α7 nAChR responses with 8-Br-cAMP (n = 13) as compared to control (n = 17) (P = 0.03, two-way ANOVA). E, there was no change in desensitization decay kinetics of α7 nAChR currents for fast and slow decay time constants for control (n = 20) and 8-Br-cAMP (n = 20) (P = 0.4 for τfast, P = 0.1 for τslow, Wilcoxon rank sum tests) administered interneurons. F, no difference in the percentage composition of amplitude coefficients (AC) is observed for fast and slow decay time components of α7 nAChR responses for control (n = 20) and 8-Br-cAMP stimulated neurons (n = 20) (P = 0.4 and P = 0.6, for τfast and τslow components, respectively, Wilcoxon rank sum tests).
To examine whether 8-Br-cAMP altered the gating kinetics of α7 nAChRs expressed in neurons, we analysed the current decay kinetics of α7 during the 1 s PHA543613 application by fitting the decay current to a function of two exponentials. There was no significant difference in the fast (control τfast: 20 ± 4 ms, n = 20 vs. 8-Br-cAMP τfast: 26 ± 5 ms, n = 20) (P = 0.4, Wilcoxon rank sum test) or slow decay kinetics (control τslow: 165 ± 42 ms, n = 20 vs. 8-Br-cAMP τslow: 194 ± 36 ms, n = 20) (P = 0.1, Wilcoxon rank sum test) of α7 nicotinic responses recorded from brain slices (Fig.2E). Also, the percentage composition of the fast (control τfast: 45 ± 6%, n = 20 vs. 8-Br-cAMP τfast: 44 ± 5%, n = 20) and slow decay components (control τslow: 55 ± 6%, n = 20 vs. 8-Br-cAMP τslow: 57 ± 5%, n = 20) of the α7 nicotinic responses from layer 1 interneurons, calculated as amplitude coefficients, was not different between control and 8-Br-cAMP treated neurons (P = 0.4 and P = 0.6, t tests for slow and fast τ components, respectively) (Fig.2F). These observations indicate that the 8-Br-cAMP stimulated decrease of α7 nicotinic currents in layer 1 interneurons was not due to alterations in receptor gating kinetics.
In HEK 293T cells 8-Br-cAMP activates PKA to inhibit α7 nAChR function
We further investigated whether 8-Br-cAMP modified α7 nAChR currents through the activation of PKA by performing experiments using a dominant negative form of PKA (PKA-DN) and, alternatively, PKA catalytic subunit α (PKA-Cα), which is a constitutively active form of PKA. The dominant negative form of PKA has a single mutation in one of the cAMP binding sites of the regulatory domain of the enzyme thus resulting in a loss of PKA function. In HEK 293T cells transfected with only α7 nAChRs and recorded using control patch pipette solution, application of ACh (1 mm) elicited an average α7 nAChR response of 564 ± 84 pA (n = 16). With 8-Br-cAMP (100 μm) in the micropipette, there was a significant attenuation of α7 receptor responses to 270 ± 49 pA (n = 26) (P = 0.004, Kruskal–Wallis rank sum test; P = 0.002, Wilcoxon rank sum test post hoc analysis) (Fig.3). In contrast, 8-Br-cAMP was unable to alter α7 nicotinic responses in HEK 293T cells in which α7 nAChRs were co-transfected with the dominant negative form of PKA (390 ± 36 pA, n = 11) as compared to control responses (P = 0.29, Wilcoxon rank sum test). However, the mean α7 nAChR whole-cell currents recorded from PKA-DN expressing cells that were treated with 8-Br-cAMP (390 ± 36 pA, n = 11) was significantly greater than cells only expressing α7 nAChRs and exposed to 8-Br-cAMP (270 ± 49 pA, n = 26) (P = 0.03, Wilcoxon rank sum test) (Fig.3). These results provide additional supporting evidence that 8-Br-cAMP inhibits α7 function through activation of PKA.
Figure 3. PKA inhibits α7 nAChR responses in HEK 293T cells.
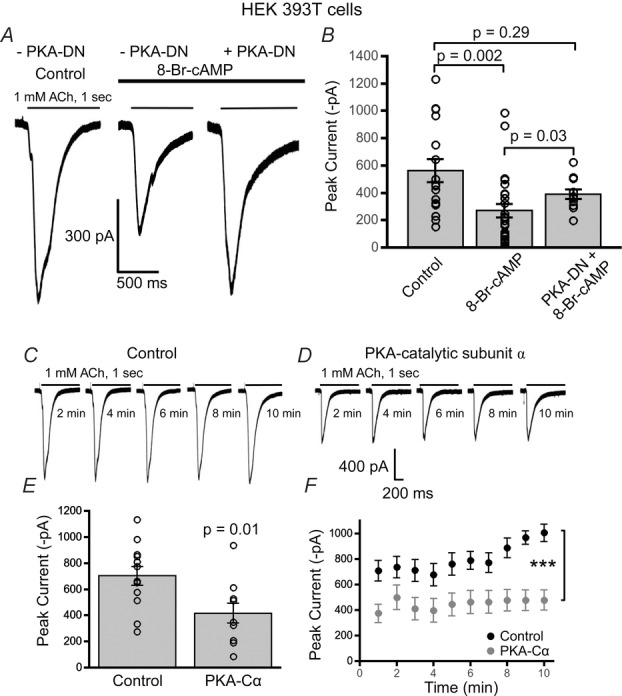
A, example waveforms of ACh (1 mm) evoked α7 nAChR responses from HEK 293T cells under three conditions including: control treatment, 8-Br-cAMP treatment and 8-Br-cAMP treatment with co-transfected dominant negative PKA (PKA-DN). B, 8-Br-cAMP (100 μm) (n = 26) stimulation resulted in a signficant attenuation of the mean α7 nAChR current as compared to control (n = 16) (P = 0.002, Wilcoxon rank sum test). Cells cotransfected with PKA-DN did not display a significant attenuation in nAChR current with 8-Br-cAMP as compared to cells only expressing α7 nAChRs (n = 11) (P = 0.29, Wilcoxon rank sum test). However, 8-Br-cAMP stimulation in α7-only transfected cells significantly attenuated α7 receptor currents as compared to cells co-transfected with PKA-DN (P = 0.03, Wilcoxon rank sum test). Sample traces of ACh (1 mm) evoked α7 nAChR responses from HEK 293T cells transiently expressing only α7 nAChRs (C) and α7 receptors co-expressed with PKA catalytic subunit α (PKA-Cα) (D). E, a significant decrease in the mean current amplitude of α7 nAChRs was observed in cells co-expressing PKA-Cα (n = 9) as compared to control (cells expressing only α7 nAChRs) (n = 12) (P = 0.01, t test). F, PKA-Cα and α7 nAChR co-transfected cells consistently showed reduced α7 nicotinic responses with repeated ACh applications as compared to control (α7 nAChRs only) (***P < 0.001, two-way ANOVA).
To further support that PKA is negatively modulating α7 nAChR function we used an alternative approach to the previous set of experiments. We co-transfected mouse PKA-Cα cDNA with α7 nAChRs in HEK 293T cells. By over-expressing PKA-Cα via transient transfection, we are essentially creating a constitutively active form of PKA since the catalytic subunits would greatly outnumber the PKA regulatory subunits. ACh (1 mm)-mediated whole-cell currents for α7 nicotinic receptor responses co-transfected with PKA-Cα showed a significant decrease in the average peak current amplitude (416 ± 76 pA, n = 9) as compared to control cells having only α7 receptors (702 ± 73 pA, n = 12) (P = 0.01, t test) (Fig.3C–E). Unlike previous experiments in which 8-Br-cAMP in the recording pipette started at an α7 current amplitude equal to control but diminished progressively more than the steady α7 current of the control cells, the PKA-Cα expressing cells (n = 9) already started at a significantly smaller α7 nAChR current and remained steady at the same diminished current amplitude as compared to the control cells (n = 12) (P < 0.001, PKA-Cα treatment factor, two-way ANOVA) (Fig.3F). We also found no difference in the channel gating kinetics by fitting the decay current as a function of two exponentials (data not shown). These results strongly indicate that over-stimulation of PKA activity in cells strongly attenuates α7 nAChR function.
PKA activation inhibits α7 nicotinic currents in layer 1 PFC neurons
To confirm that activation of PKA in neurons would also inhibit α7 nicotinic currents we performed whole-cell recordings from layer 1 PFC interneurons in brain slices. The PKA catalytic subunit (200 U ml−1) was included in the patch electrode solution during the recording of PHA543613 (100 μm)-elicited α7 responses. We found that neurons dialysed with the active form of the PKA catalytic subunit caused a significant decrease in the α7 nicotinic responses (56 ± 6.7 pA, n = 15) from layer 1 interneurons as compared to responses recorded with control patch electrode solutions (101 ± 9 pA, n = 30) (P = 0.003, Kruskal–Wallis rank sum test; P = 0.002, Wilcoxon rank sum test, post hoc analysis) (Fig.4). The decrease in α7 nAChR current responses with catalytic PKA (56 ± 7 pA, n = 15) (P = 0.002, Wilcoxon rank sum test) was similar to that of 8-Br-cAMP (100 μm) stimulation (62 ± 3 pA, n = 23). However, having the PKA blocker KT-5720 (200 nm) in the patch recording solution (111 ± 15 pA, n = 14) (P = 0.7, Wilcoxon rank sum test) abrogated any decrease with 8-Br-cAMP application. These observations suggest that endogenous PKA in CNS neurons plays an important role in regulating cholinergic synaptic transmission by inhibiting α7 nicotinic responses in layer 1 cortical neurons.
Figure 4. PKA activation and inhibition have opposing effects on modulating α7 nAChR currents in PFC interneurons.
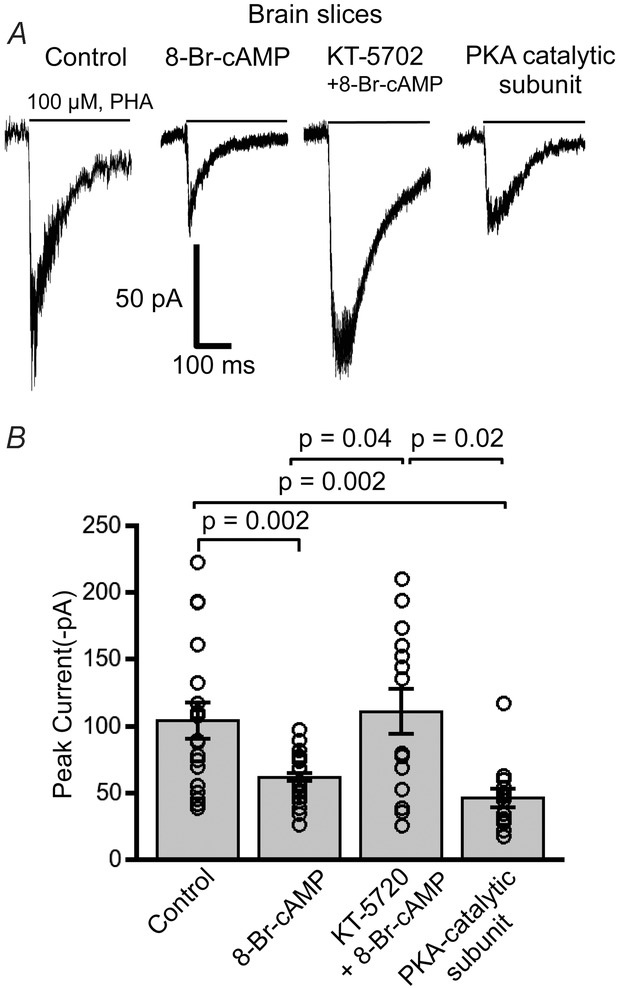
A, example traces of PHA543613 (100 μm) evoked α7 nAChR whole-cell current responses from layer 1 interneurons for control, 8-Br-cAMP (100 μm), combined KT-5702 (200 nm, PKA inhibitor) with 8-Br-cAMP, and PKA catalytic subunit (200 U ml−1). B, plot illustrating differences in the mean current amplitude for the four test conditions. α7 receptor responses were significantly attenuated with 8-Br-cAMP (n = 23) as compared to control responses (n = 30) (P = 0.002, Wilcoxon rank sum test). Dialysing neurons with PKA catalytic subunit in the patch pipette (n = 15) similarly diminished α7 current responses (P = 0.002, Wilcoxon rank sum test). The effect of 8-Br-cAMP in inhibiting α7 nAChR responses was abolished with the PKA inhibitor KT-5702 (n = 14), which showed α7 responses that were significantly greater than 8-Br-cAMP (P = 0.04, Wilcoxon rank sum test) and the PKA catalytic subunit-activated neurons (P = 0.02, Wilcoxon rank sum test).
PKA activation does not alter α7 nAChR single-channel conductance
A possible mechanism for the PKA mediated inhibition of α7 nAChR responses could be decreased intrinsic channel activity. To evaluate if 8-Br-cAMP affected α7 nAChR single-channel conductance, we performed ionic current fluctuation analysis on whole-cell recorded α7 nicotinic currents from HEK 293T cells and brain slices. Non-stationary current fluctuation analysis provides a good estimate of single-channel conductance of ion channels (Sigworth, 1980, 1981; Gill et al. 1995; Komal et al. 2014). We tested whether in HEK 293T cells and cortical interneurons 8-Br-cAMP mediated stimulation of PKA decreased α7 nAChR single-channel conductance. The variance of the current fluctuations of ACh (1 mm) mediated α7 whole-current responses was plotted against the mean current of the whole-cell current at each time point. The slope of the relationship gives the unitary current and the single-channel conductance (γ) was calculated as the unitary current divided by the electrochemical driving force. Example current fluctuation analyses are shown for control (γ = 57 pS) and 8-Br-cAMP (γ = 58 pS) treated PFC interneurons from brain slices (Fig.5A and B). On average there was no significant difference in α7 nAChR single-channel conductance of control treated (43 ± 9 pS, n = 18) vs. 8-Br-cAMP treated layer 1 interneurons (34 ± 10 pS, n = 11) (P = 0.6, Wilcoxon rank sum test) (Fig.5C). Also, we found no significant difference in the mean single-channel conductance of α7 receptor responses recorded from HEK 293T cells between control (63 ± 16 pS, n = 15) and 8-Br-cAMP treatment (74 ± 16 pS, n = 15) (P = 0.6, Wilcoxon rank sum test) (Fig.5D). Therefore, the 8-Br-cAMP mediated decrease in α7 nAChR mediated whole-cell currents is not due to alterations in single-channel conductance.
Figure 5. 8-Br-cAMP stimulation does not alter α7 nAChR single-channel conductance.
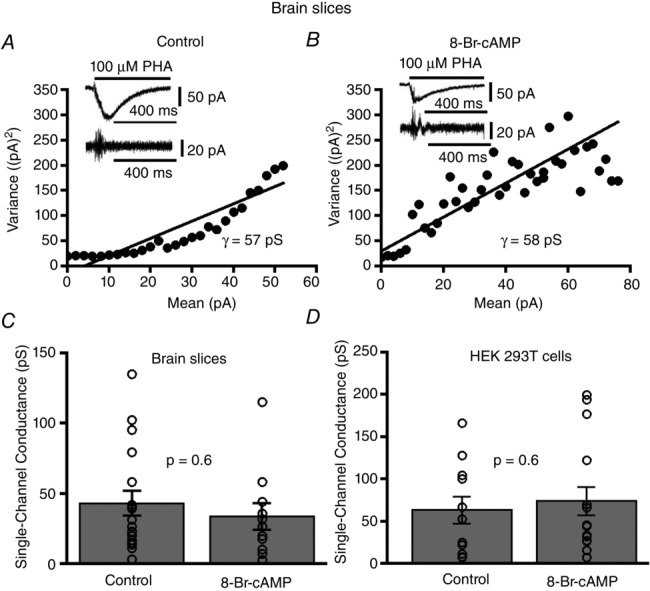
A, non-stationary current fluctuation analyses depicting mean current–variance relationship performed with PHA543613 (100 μm) elicited α7 nAChR responses from layer 1 interneurons for control and (B) 8-Br-cAMP (100 μm, in the recording pipette) stimulated interneurons. Inset shows whole-cell recorded α7 nicotinic current waveforms and their respective AC filtered waveforms for both control and 8-Br-cAMP stimulation. C, no significant change in the mean single-channel conductance is observed for α7 responses between control (n = 18) and 8-Br-cAMP treated cortical interneurons (n = 11) (P = 0.6, Wilcoxon rank sum test). D, no significant difference in the mean single-channel conductance for α7 responses is observed between control (n = 11) and 8-Br-cAMP treated HEK 293T cells (n = 15) (P = 0.6, Wilcoxon rank sum test).
PKA targets serine 365 in the M3–M4 cytoplasmic loop of α7 nAChRs to modulate α7 nAChR function
A previous study showed that a serine residue located in the chick and rat neuronal α7 nAChR is phosphorylated only by protein kinase A and not by protein kinase C, cGMP-dependent protein kinase, or calcium/calmodulin-dependent protein kinase (Moss et al. 1996). Through PROSITE analysis of the mouse α7 nAChR cDNA we found a single putative PKA phosphorylation site at serine 365. Therefore, in order to examine whether PKA targets serine 365 directly to modulate α7 nAChR function we produced a mutant form of the α7 nAChR cDNA, in which we mutated serine 365 to alanine (α7(S365A)). Therefore, in HEK 293T cells expressing mutant α7(S365A) receptors we compared the effects of control solution vs. 8-Br-cAMP on α7 nAChR mediated whole-cell currents elicited by 1 mm ACh application. Unlike previous results with wild-type α7 nAChRs, for the mutant α7(S365A) receptors there was no significant attenuation of the nicotinic currents from 8-Br-cAMP stimulated cells (809 ± 134 pA, n = 12) as compared to control treatment (826 ± 123 pA, n = 15) (P = 0.5, t test) (Fig.6A–C). Repeated application of ACh (every 1 min) showed a consistent amplitude of mutant α7(S365A) receptor currents over 10 min that were not significantly different between recordings performed with 8-Br-cAMP in the patch electrode solution and control patch solutions (P = 0.7, 8-Br-cAMP treatment factor, two-way ANOVA) (Fig.6D). These results imply that serine 365 in the M3–M4 cytoplasmic loop of α7 nAChRs forms the major regulatory site for PKA in modulating α7 receptor function.
Figure 6. PKA targets serine 365 of α7 nAChRs to modulate channel function.
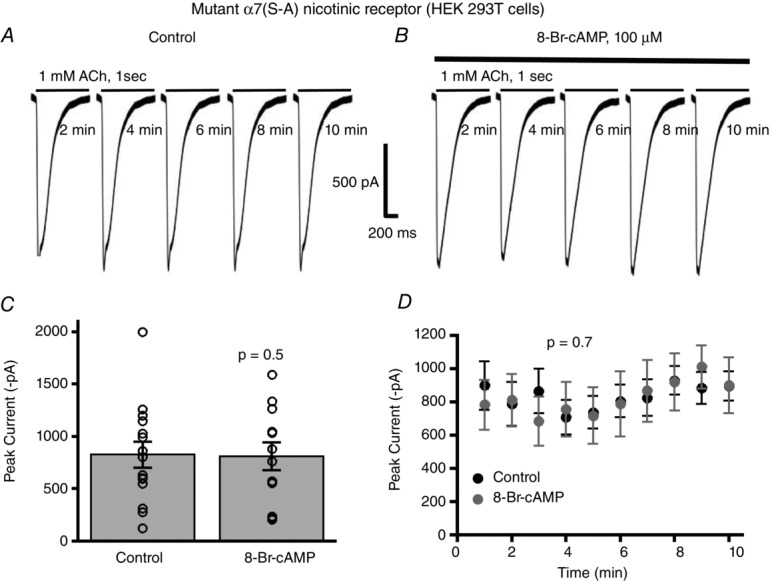
A, whole-cell current traces of α7 nAChRs with the serine 365 to alanine mutation (α7(S–A)) show no alterations in the amplitude of the ACh (1 mm) elicited nicotinic currents with 8-Br-cAMP stimulation of HEK 293T cells (B). C, there was no significant difference in ACh mediated mutant α7(S–A) responses between control (n = 15) and 8-Br-cAMP stimulated cells (n = 12) (P = 0.5, t test). D, repeated applications of ACh with 1 min intervals showed no time-dependent effect of 8-Br-cAMP stimulation on mutant α7(S–A) nAChR responses (n = 12) as compared to control (n = 15) (P = 0.7, two-way ANOVA).
PKA targets serine 365 to decrease α7 nicotinic receptor surface expression
Since our results ruled out the possibility that PKA modulation of α7 was due to changes in single-channel conductance or gating kinetics, we explored whether a potential mechanism of the negative regulation of α7 nAChR function may be due to decreased surface receptor expression following PKA stimulation. We performed surface labelling of fluorescent protein tagged α7 nAChRs (α7-Venus) transfected in HEK 293T cells using Alexa Fluor 647 α-bungarotoxin (Fl-BTx) labelling under non-permeabilized conditions. Spectral confocal images were obtained and the mean intensity of Fl-BTx labelling was normalized to the mean intensity of Venus fluorescence on a per cell basis to calculate the amount of surface expression of α7 nAChRs. We found that 8-Br-cAMP (100 μm for 30 min)-stimulated HEK 293T cells transiently transfected with α7-Venus nAChRs showed significantly less Fl-BTx labelling of surface α7 nAChRs (0.21 ± 0.03, n = 31) as compared to control (0.37 ± 0.05, n = 28) (P = 0.01, Wilcoxon rank sum test) (Fig.7A–F and M). The total cellular Venus fluorophore intensity remained the same for control and 8-Br-cAMP treated dishes (data not shown).
Figure 7. PKA stimulation decreases surface expression of α7 nAChRs.

A–F, confocal images of HEK 293T cells transfected with α7-Venus. Surface labelling of α7-Venus was performed with Alexa Fluor 647 α-bungarotoxin (Fl-BTx) under non-permeabilizing conditions. M, 8-Br-cAMP stimulation (100 μm for 30 min) (n = 31) resulted in a significant decrease in the amount of Fl-BTx labelled surface α7-Venus receptors as compared to control treatment (n = 28) (P = 0.01, Wilcoxon rank sum test). G–L, confocal images of HEK 293T cells transfected with mutant α7-Venus in which the putative PKA phosphorylated site (S365) was mutated to alanine (α7(S–A)-Venus). N, 8-Br-cAMP stimulation (100 μm for 30 min) (n = 13) showed no significant change in Fl-BTx bound surface mutant α7-Venus nAChRs as compared to control treatment (n = 25) (P = 0.5, Wilcoxon rank sum test). O–T, confocal images of surface Fl-BTx labelled (red) α7 nAChRs on layer 1 interneurons (blue NeuroTrace) from medial prefrontal cortex. U, interneurons from brain slices treated with 100 μm 8-Br-cAMP (n = 897 ROIs, 190 neurons, 2 mice) showed significantly diminished surface Fl-BTx labelling than control (n = 912 ROIs, 214 neurons, 2 mice) (P < 0.0001, Wilcoxon rank sum test).
Next we tested whether PKA mediates its effect of diminishing surface receptor expression by targeting serine 365 of the α7 nAChR. Using the mutant receptor, α7(S365A)-Venus, transfected in HEK 293T cells we found no significant difference in the surface expression of mutant α7(S365A)-Venus nAChRs with 30 min control treatment (0.06 ± 0.01, n = 25) and mutant α7(S365A)-Venus nAChRs with 30 min of 100 μm 8-Br-cAMP incubation (0.07 ± 0.02, n = 13) (P = 0.5, Wilcoxon rank sum test) (Fig.7G–L and N). These results indicate that PKA targets serine 365 of the M3–M4 cytoplasmic loop of α7 nAChRs to down-regulate the surface expression of receptors.
To examine whether stimulation of PKA also diminishes α7 nAChR surface expression in neurons we performed Alexa Fluor 647 α-bungarotoxin labelling of surface α7 nAChRs in layer 1 medial prefrontal cortical neurons of brain slices prepared similarly to those for electrophysiological recordings. Brain slices were incubated for 30 min at 36°C either in control bubbled extracellular solution or 100 μm 8-Br-cAMP in bubbled extracellular solution. Alexa Fluor 647 α-bungarotoxin labelled surface α7 nAChRs, while NeuroTrace 435/455 labelled neurons, under non-permeabilized conditions of fixed brain slices. z-stacks of spectral confocal images were obtained from layer 1 of the medial prefrontal cortex (Fig.7O–T). There was a significant decrease in Fl-BTx labelling of surface α7 nAChRs in layer 1 NeuroTrace labelled interneurons of slices incubated in 8-Br-cAMP (27.8 ± 0.3%, n = 897 ROIs, 190 neurons, 2 mice) treated solutions as compared to control (100.0 ± 1.2%, n = 912 ROIs, 214 neurons, 2 mice) (P < 0.0001, Wilcoxon rank sum test) (Fig.7O–U).
Endogenous activation of PKA pathway via D1/D5 dopamine receptor stimulation decreases α7 nAChR currents in layer 1 interneurons
Although we have used pharmacological and molecular techniques to directly activate PKA, we wanted to determine whether an endogenous signalling pathway in the CNS can activate PKA in layer 1 interneurons of the prefrontal cortex. Furthermore, we sought to determine whether stimulation of that endogenous signal transduction system would display the same type of modulation of α7 nAChR currents as witnessed via direct stimulation of PKA using exogenous pharmacology. G protein coupled receptors that couple to Gαs stimulate the adenylate cyclase–PKA signal transduction pathway. Specifically D1 and D5 subtypes of G protein coupled dopamine receptors signal through Gαs. Thus, we investigated whether these dopamine receptors are a likely modulator of neuronal function through the actions of PKA.
To determine if native α7 nAChR activity can be modulated by D1/D5 dopamine receptors, we utilized the D1/D5 dopamine receptor specific agonist SKF 83822 (10 μm) in the bath and monitored PHA543613 (100 μm)-mediated α7 peak current responses from layer 1 interneurons of the prefrontal or frontal cortical slices, in the presence of TTX (0.5 μm) and CNQX (10 μm). Following stimulation of dopamine D1/D5 receptors, we found that α7 nicotinic currents were significantly reduced as compared to control responses (without SKF in the bath) (control: 107 ± 17 pA, n = 8 vs. SKF 83822: 55 ± 12 pA, n = 7) (P = 0.04, ANOVA; P = 0.03, t test with Bonferroni correction post hoc analysis) (Fig.8B). To validate that the effect of the D1/D5 agonist was due to activation of the cAMP–PKA pathway, the PKA inhibitor KT-5720 (200 nm) was included in the recording pipette before the application of SKF 83822 in the bath. Under these conditions of combined KT-5720 and SKF 83822 the α7 responses (95 ± 12 pA, n = 6) were significantly elevated from SKF 83822 stimulated responses (55 ± 12 pA, n = 7) (P = 0.04, t test with Bonferroni correction) but were comparable to that of control (107 ± 17 pA, n = 8) (Fig.8B). To further support that the effect witnessed with SKF 83822 was due to activation of dopaminergic receptors, we used another dopamine D1/D5 receptor specific agonist SKF 81297 (8 μm) in the bath and found a similar attenuation in the α7 nicotinic responses (control: 129 ± 17 pA, n = 16 vs. SKF 81297: 78 ± 10 pA, n = 19) (P = 0.02, Wilcoxon rank sum test) (Fig.8C). To rule out the possibility that the effect of the D1/D5 agonists was due to non-specific off-target effects on α7 nicotinic receptor function, we performed whole-cell recording from HEK 293T cells transfected only with α7 nAChRs and found no difference in the peak current amplitude of α7 nAChR responses in the presence of either D1/D5 agonists SKF 83822 or SKF 81297, as compared to control solution (data not shown). These results indicate that dopamine receptor stimulation down-regulates α7 nAChR function in CNS neurons through activation of the cAMP–PKA signalling pathway.
Figure 8. Activation of D1/D5 dopamine receptors inhibits α7 nAChR activity in PFC interneurons.
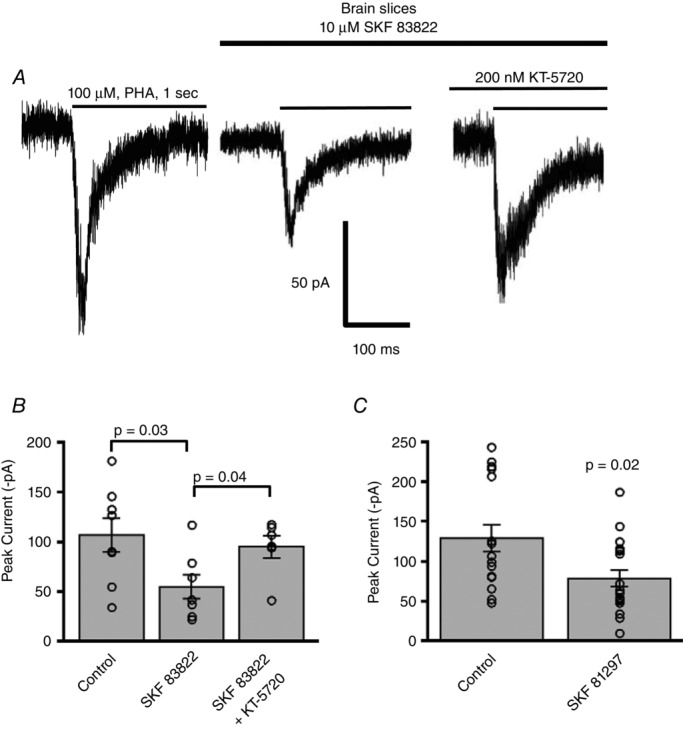
A, example traces of α7 nAChR responses elicited by PHA543613 (100 μm) in PFC layer 1 cortical neurons showing responses under control, SKF 83822 and combined SKF 83822 and KT-5720 conditions. B, there was a significant decrease in α7 nAChR responses in neurons treated with the D1/D5 dopamine receptor agonist SKF 83822 (n = 7) as compared to control (n = 8) (P = 0.03, t test with Bonferroni correction). Meanwhile the PKA inhibitor KT-5720, when co-applied with SKF 83822, blocked any effect of the SKF compound so that the α7 nAChR responses with KT-5720 (n = 6) were significantly elevated as compared to SKF 83822 alone (n = 7) (P = 0.04, t test with Bonferroni correction). C, we verified our results using another D1/D5 dopamine receptor agonist. The D1/D5 dopamine receptor agonist SKF 81297 (n = 19) also resulted in a significant inhibition of α7 nAChR currents as compared to control (n = 16) (P = 0.02, Wilcoxon rank sum test).
Discussion
We have shown in mouse layer 1 cortical neurons that α7 nAChR function is negatively modulated by cAMP dependent protein kinase through activation of D1/D5 dopamine receptors. 8-Br-cAMP attenuated α7 nAChR currents but did not change the desensitization kinetics nor the single-channel conductance of α7 nAChRs. Serine 365 in the cytoplasmic M3–M4 domain of the α7 nAChR is a key molecular target of PKA, since 8-Br-cAMP treatment of HEK 293T cells expressing mutant α7(S365A) no longer resulted in a decrease of nicotinic current as was the case with wild-type α7 nAChRs. We also provide evidence that PKA activation inhibits α7 nAChR function. Co-transfecting dominant negative PKA with α7 or preincubating the recorded neurons with PKA antagonist KT-5720 abrogated the effects of 8-Br-cAMP, while transfecting PKA catalytic subunit in cells or inclusion in the recording pipette attenuated α7 nAChR currents in cell lines and in cortical interneurons, respectively. Fl-αBTx labelling showed that the PKA mediated down-regulation of α7 nicotinic currents is due to a decrease in surface expression of α7 nAChRs following PKA targeting serine 365 of α7 nAChRs. Finally, we demonstrated that activation of the dopaminergic neurotransmitter system, specifically D1/D5 dopamine receptors, can similarly inhibit α7 nicotinic responses via activation of PKA downstream to the activation of the D1/D5 receptors.
Mechanism of attenuation of α7 nAChR function by PKA
To our knowledge this is the first study that has demonstrated a direct effect of the cAMP–PKA pathway on modulating nAChR function in CNS neurons. Although dopamine receptors have previously been shown to modulate ion channel function, neuronal excitability and synaptic plasticity (Zhou & Hablitz, 1999; Trantham-Davidson et al. 2004; Yang & Dani, 2014), this is the first study to show that activation of dopamine receptors can modify nAChR activity.
Previous studies have investigated the effect of 8-Br-cAMP on nAChR function in bovine adrenal chromaffin cells and chick ganglionic neuronal culture (Margiotta et al. 1987; Dubin et al. 1992). Dubin and colleagues (1992) found no effect of 8-Br-cAMP on nAChR responses, though Margiotta and colleagues (1987) found potentiation of nicotinic responses following 6 h treatment of 8-Br-cAMP on cultured chick ganglionic neurons. The discrepancy among the results could be due to the differences in the nAChR subunit composition, concentration of 8-Br-cAMP and time scale of 8-Br-cAMP incubation. α3β4 nAChRs constitute the main nAChR subtype found in adrenal chromaffin cells and chick ganglionic neurons (Sala et al. 2008), and α7 nAChRs do not contribute to catecholamine secretion from chromaffin cells (Sala et al. 2008). Moreover, both of these studies used a 20-fold higher concentration of 8-Br-cAMP (2 mm) and monitored nAChR responses on a longer time scale ranging from 6 to 48 h. We have examined the effects of 8-Br-cAMP on an acute time scale of minutes. Under physiological conditions the level of PKA activation determines the net effect on receptor function as demonstrated in the case of GABAA receptors (Cai et al. 2002). It could be that different concentrations of 8-Br-cAMP could have opposing effects on receptor function. In our study, we monitored the modulation of α7 nAChR function directly in CNS neurons using 100 μm 8-Br-cAMP. Even though we do not know the endogenous concentration of cAMP in layer 1 cortical neurons during stimulation of the adenylate cyclase pathway, our experiments with the D1/D5 dopamine receptor agonists addresses this issue since the D1/D5 dopamine receptor signals through the Gαs, cAMP and PKA signal transduction pathway. By using the D1/D5 dopamine receptor agonists SKF 83822 and SKF 81297 we would stimulate the neurons to produce physiological levels of cytoplasmic cAMP. The activation of the dopaminergic system with SKF compounds resulted in the same effect of attenuation of α7 nAChR currents as did pharmacological stimulation of PKA with 8-Br-cAMP or our molecular approaches in targeting PKA. This reinforces our conclusion that the endogenous activation of PKA negatively modulates α7 nAChR activity.
We found that the mechanism of attenuation of α7 responses with PKA activation was solely due to a loss of surface expression of α7 receptors, with no change in single-channel conductance. This is in contrast to Margiotta and colleagues (Margiotta et al. 1987), who showed an increase in nicotinic current in chick ganglionic neurons that was not due to alterations in expression of surface receptors, single-channel conductance or gating kinetics. The difference, again, is probably due to the difference in nAChR subunit composition in ganglionic neurons as compared to layer 1 cortical neurons. Furthermore, they studied more prolonged effects of 8-Br-cAMP (3–48 h), while we observed the brief effects of 8-Br-cAMP over several minutes.
Unlike in our previous study in which we found that tyrosine phosphorylation of α7 nAChRs resulted in decreased single-channel conductance and reduced surface receptor expression (Komal et al. 2014), we found that PKA stimulation did not alter single-channel conductance or gating kinetics of α7 nAChRs but only decreased surface receptor expression. One reason for the lack of effect of 8-Br-cAMP-mediated PKA activation on single-channel conductance could be due to the fact that the putative PKA phosphorylation site, serine 365, lies close to the M3 transmembrane domain in the M3–M4 cytoplasmic loop of the channel, which is remote from the pore-lining amphipathic helix situated close to the M4 transmembrane domain. The amphipathic helix lines the cytoplasmic side portals of Cys-loop receptors and forms part of the ion permeation pathway (Unwin, 2005; Hales et al. 2006). This is unlike tyrosine 442, which is situated within the cytoplasmic amphipathic helix pre-M4, and therefore could potentially sterically hinder ion flow. Thus, it appears that PKA, as well as tyrosine kinases, can decrease α7 nAChR function in CNS neurons, although through slightly different mechanisms. Our electrophysiology data (Fig.6) and Fl-αBTx labelling of surface receptors (Fig.7) of mutant α7(S365A) nAChRs clearly demonstrate that the serine 365 residue forms the major target of PKA to modulate nAChR function by altering surface expression of the receptor.
Neurotransmitters such as dopamine, noradrenaline (norepinephrine) and serotonin target G protein coupled receptors and signal through cAMP mediated PKA pathways (Seamans et al. 2001; Cai et al. 2002; Yan, 2002; Seamans & Yang, 2004). We have shown that activation of the D1/D5 subclass of dopamine receptors can negatively modulate α7 nAChR function in layer 1 prefrontal and frontal cortical interneurons. The agonists used are not selective enough to distinguish between D1 and D5 receptors though both receptor subtypes are found in the prefrontal cortex (Gensat Brain Atlas; http://www.gensat.org). These different neurotransmitter modulators have been shown to regulate other ionotropic receptors such as GABAA and glutamate receptors, thereby modulating neurotransmission (Flores-Hernandez et al. 2000; Wang & O'Donnell, 2001). In pyramidal neurons of the prefrontal cortex serotonin was shown to modulate post-synaptic GABAA receptors depending on the strength of the PKA activation level (Cai et al. 2002). In addition, neuronal activity itself is considered one potent mechanism of PKA activation (Dunn et al. 2006). It is reasonable to speculate that the other neuromodulators in addition to dopamine, such as noradrenaline or serotonin, would also have a net impact on α7 nAChR function under physiological conditions.
Physiological relevance of PKA in neurotransmission and synaptic plasticity
Dopamine receptor regulation of α7 nAChR function in CNS neurons through activation of PKA may have further physiological ramifications. Postsynaptic α7 nAChRs are involved in fast synaptic transmission where they modulate the firing properties of neurons (Frazier et al. 1998; Jones & Wonnacott, 2004; Komal et al. 2014). Therefore, it is crucial to understand the cellular mechanisms of ion channel regulation in order to have a better understanding of neural function and excitability in the brain (Alkondon et al. 1998). It is well documented that PKA activation in neurons contributes to long-term potentiation (LTP) in the dentate gyrus of the hippocampus (Nguyen & Kandel, 1996; Wu et al. 2006). Since α7 nAChRs have been shown to contribute towards LTP (Mansvelder & McGehee, 2000; Matsuyama et al. 2000; Ondrejcak et al. 2012), a cellular mechanism for learning and memory, PKA modulation of α7 nAChR function may influence synaptic plasticity and memory formation in different brain regions.
Layer 1 cortical interneurons can influence layer 2/3 pyramidal neuronal excitability by either inhibiting pyramidal neurons directly via GABAergic neurotransmission or, conversely, by increasing pyramidal neuronal excitability through disynaptic disinhibition (Christophe et al. 2002; Arroyo et al. 2012). Similarly, layer 1 cortical interneurons can also directly inhibit or disinhibit layer 5 pyramidal neuronal firing depending on the layer 1 interneuronal subtype (Jiang et al. 2013). The elongated neurogliaform interneurons of layer 1 form inhibitory synapses to greater than 60% of layer 5 pyramidal neurons in the cortex (Jiang et al. 2013). If we assume a monosynaptic inhibition of pyramidal neurons, then the action of PKA on inhibiting α7 nAChR activity would putatively lead to an overall increase in pyramidal neuronal excitability. One mechanism of PKA stimulation is through activation of G protein coupled receptors that signal through the cAMP–adenylate cyclase pathway such as the D1/D5 dopamine receptors. Dopamine signalling in the prefrontal cortex is known to play an important role in executive cognitive function such as motivation and working memory (Phillips et al. 2008). We have shown that stimulation of the D1/D5 dopamine receptors and the downstream activation of PKA attenuate α7 nAChR currents on layer 1 prefrontal cortical interneurons. As we have previously shown (Komal et al. 2014), a decrease in α7 activity would decrease excitability of layer 1 interneurons. We predict that this would result in less inhibitory input onto pyramidal neurons and enhance pyramidal neuronal excitability. It was also shown that dopamine release through ventral tegmental area afferents in the PFC results in ‘up states’ of pyramidal neuronal excitability (Lewis & O'Donnell, 2000). These ‘up states’ are plateau depolarizations of the membrane potential that result in burst firing. Perhaps PKA modulation of α7 nAChRs through dopamine receptor activation could be a contributing mechanism of these ‘up states’ of neuronal excitability.
Through the action of dopamine, α7 nAChRs may be an important regulator of working memory and motivation. Previous studies have portrayed a critical role for α7 nAChRs in behavioural tasks involving attention (Young et al. 2004; Hoyle et al. 2006). Although in our study we examined postsynaptic α7 nAChRs, α7 receptors are also located presynaptically, where they facilitate neurotransmitter release (Mansvelder & McGehee, 2000; Zhou et al. 2001; Jones & Wonnacott, 2004; Cheng & Yakel, 2014). The attenuation of α7 nAChR function by PKA activation could also potentially affect the release of neurotransmitters such as dopamine and glutamate as shown in the rat prefrontal cortex (Livingstone et al. 2010) and modulate postsynaptic excitability. However, due to multiple isoforms of PKA expressed in neurons and the difference in their subcellular localization, it remains unclear which isoform combination of regulatory and catalytic subunits of PKA has the major effect of modulating α7 nAChRs as well as other ion channels (Meinkoth et al. 1990).
These data indicate how PKA activation via D1/D5 dopamine receptors can attenuate α7 nicotinic responses in PFC layer 1 neurons. This has important implications with regard to how the interaction of two neurotransmitter systems, namely ACh and dopamine, may converge to alter not only neuronal excitability but ultimately cognitive behaviours such as motivation, attention and working memory.
Acknowledgments
The excellent technical assistance of Clifford Donovan, Yuebo Yang, Marcus Saunders, Nora Penty and Qi Huang is gratefully acknowledged. We thank Ariel Sullivan, Carl Jensen, Dean Rysstad and all other members of the mouse facility at the University of Victoria for providing excellent mouse husbandry. We thank Dr Kerry Delaney for helpful discussions.
Glossary
- Fl-BTx
Alexa Fluor 647 α-bungarotoxin
- FC
frontal cortex
- nAChRs
nicotinic acetylcholine receptors
- PFC
prefrontal cortex
- PKA
protein kinase A or cAMP-dependent protein kinase
- PKA-Cα
PKA catalytic subunit α
- PKA-DN
dominant negative form of PKA
- ROI
region of interest
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
P.K. and R.N. designed the experiments and wrote the paper. P.K. performed and analysed most of the experiments. M.K. J.E. and R.N. performed and analysed some experiments. A.R. performed some experiments and edited the manuscript. All authors approved the final version of the manuscript.
Funding
This research was supported by a Natural Sciences and Engineering Research Council of Canada Discovery Grant, a Heart and Stroke Foundation of Canada Grant, a Canadian Foundation for Innovation grant, a British Columbia Knowledge Development Fund and a Natural Sciences and Engineering Research Council of Canada Research Tools and Instrumentation Grant.
References
- AhnAllen CG. The role of the α7 nicotinic receptor in cognitive processing of persons with schizophrenia. Curr Opin Psychiatry. 2012;25:103–108. doi: 10.1097/YCO.0b013e3283503637. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Albuquerque EX. α-Bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Res. 1998;810:257–263. doi: 10.1016/s0006-8993(98)00880-4. [DOI] [PubMed] [Google Scholar]
- Arroyo S, Bennett C, Aziz D, Brown SP, Hestrin S. Prolonged disynaptic inhibition in the cortex mediated by slow, non-α7 nicotinic excitation of a specific subset of cortical interneurons. J Neurosci. 2012;32:3859–3864. doi: 10.1523/JNEUROSCI.0115-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astman N, Gutnick MJ, Fleidervish IA. Activation of protein kinase C increases neuronal excitability by regulating persistent Na+ current in mouse neocortical slices. J Neurophysiol. 1998;80:1547–1551. doi: 10.1152/jn.1998.80.3.1547. [DOI] [PubMed] [Google Scholar]
- Beaulieu J-M, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Brown AM, Hope AG, Lambert JJ, Peters JA. Ion permeation and conduction in a human recombinant 5-HT3 receptor subunit (h5-HT3A. J Physiol. 1998;507:653–665. doi: 10.1111/j.1469-7793.1998.653bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Flores-Hernandez J, Feng J, Yan Z. Activity-dependent bidirectional regulation of GABAA receptor channels by the 5-HT4 receptor-mediated signalling in rat prefrontal cortical pyramidal neurons. J Physiol. 2002;540:743–759. doi: 10.1113/jphysiol.2001.013391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castner SA, Smagin GN, Piser TM, Wang Y, Smith JS, Christian EP, Mrzljak L, Williams GV. Immediate and sustained improvements in working memory after selective stimulation of α7 nicotinic acetylcholine receptors. Biol Psychiatry. 2011;69:12–18. doi: 10.1016/j.biopsych.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Charpantier E, Wiesner A, Huh K-H, Ogier R, Hoda J-C, Allaman G, Raggenbass M, Feuerbach D, Bertrand D, Fuhrer C. α7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J Neurosci. 2005;25:9836–9849. doi: 10.1523/JNEUROSCI.3497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Yakel JL. Presynaptic α7 nicotinic acetylcholine receptors enhance hippocampal mossy fiber glutamatergic transmission via PKA activation. J Neurosci. 2014;34:124–133. doi: 10.1523/JNEUROSCI.2973-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C-H, Song W, Leitzell K, Teo E, Meleth AD, Quick MW, Lester RAJ. Rapid upregulation of α7 nicotinic acetylcholine receptors by tyrosine dephosphorylation. J Neurosci. 2005;25:3712–3723. doi: 10.1523/JNEUROSCI.5389-03.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophe E, Roebuck A, Staiger JF, Lavery DJ, Charpak S, Audinat E. Two types of nicotinic receptors mediate an excitation of neocortical layer I interneurons. J Neurophysiol. 2002;88:1318–1327. doi: 10.1152/jn.2002.88.3.1318. [DOI] [PubMed] [Google Scholar]
- Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A. Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-α-bungarotoxin. J Neurosci. 1985;5:1307–1315. doi: 10.1523/JNEUROSCI.05-05-01307.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courjaret R, Lapied B. Complex intracellular messenger pathways regulate one type of neuronal α-bungarotoxin-resistant nicotinic acetylcholine receptors expressed in insect neurosecretory cells (dorsal unpaired median neurons) Mol Pharmacol. 2001;60:80–91. [PubMed] [Google Scholar]
- Dajas-Bailador FA, Soliakov L, Wonnacott S. Nicotine activates the extracellular signal-regulated kinase 1/2 via the α7 nicotinic acetylcholine receptor and protein kinase A, in SH-SY5Y cells and hippocampal neurones. J Neurochem. 2002;80:520–530. doi: 10.1046/j.0022-3042.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- Dubin AE, Rathouz MM, Mapp KS, Berg DK. Cyclic AMP and the nicotinic response of bovine adrenal chromaffin cells. Brain Res. 1992;586:344–347. doi: 10.1016/0006-8993(92)91646-v. [DOI] [PubMed] [Google Scholar]
- Dunn TA, Wang C-T, Colicos MA, Zaccolo M, DiPilato LM, Zhang J, Tsien RY, Feller MB. Imaging of cAMP levels and protein kinase A activity reveals that retinal waves drive oscillations in second-messenger cascades. J Neurosci. 2006;26:12807–12815. doi: 10.1523/JNEUROSCI.3238-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi S-H, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Flores-Hernandez J, Hernandez S, Snyder GL, Yan Z, Fienberg AA, Moss SJ, Greengard P, Surmeier DJ. D1 dopamine receptor activation reduces GABAA receptor currents in neostriatal neurons through a PKA/DARPP-32/PP1 signaling cascade. J Neurophysiol. 2000;83:2996–3004. doi: 10.1152/jn.2000.83.5.2996. [DOI] [PubMed] [Google Scholar]
- Frazier CJ, Buhler AV, Weiner JL, Dunwiddie TV. Synaptic potentials mediated via α-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J Neurosci. 1998;18:8228–8235. doi: 10.1523/JNEUROSCI.18-20-08228.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Hall M, Adler LE, Leonard S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry. 1995;38:22–33. doi: 10.1016/0006-3223(94)00252-X. [DOI] [PubMed] [Google Scholar]
- Gill CH, Peters JA, Lambert JJ. An electrophysiological investigation of the properties of a murine recombinant 5-HT3 receptor stably expressed in HEK 293 cells. Br J Pharmacol. 1995;114:1211–1221. doi: 10.1111/j.1476-5381.1995.tb13335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–491. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Green WN, Ross AF, Claudio T. Acetylcholine receptor assembly is stimulated by phosphorylation of its γsubunit. Neuron. 1991;7:659–666. doi: 10.1016/0896-6273(91)90378-d. [DOI] [PubMed] [Google Scholar]
- Hales TG, Dunlop JI, Deeb TZ, Carland JE, Kelley SP, Lambert JJ, Peters JA. Common determinants of single channel conductance within the large cytoplasmic loop of 5-hydroxytryptamine type 3 and α4β2 nicotinic acetylcholine receptors. J Biol Chem. 2006;281:8062–8071. doi: 10.1074/jbc.M513222200. [DOI] [PubMed] [Google Scholar]
- Hoyle E, Genn RF, Fernandes C, Stolerman IP. Impaired performance of alpha7 nicotinic receptor knockout mice in the five-choice serial reaction time task. Psychopharmacology (Berl) 2006;189:211–223. doi: 10.1007/s00213-006-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Wang G, Lee AJ, Stornetta RL, Zhu JJ. The organization of two new cortical interneuronal circuits. Nat Neurosci. 2013;16:210–218. doi: 10.1038/nn.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones IW, Wonnacott S. Precise localization of α7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci. 2004;24:11244–11252. doi: 10.1523/JNEUROSCI.3009-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komal P, Evans G, Nashmi R. A rapid agonist application system for fast activation of ligand-gated ion channels. J Neurosci Methods. 2011;198:246–254. doi: 10.1016/j.jneumeth.2011.04.024. [DOI] [PubMed] [Google Scholar]
- Komal P, Gudavicius G, Nelson CJ, Nashmi R. T-cell receptor activation decreases excitability of cortical interneurons by inhibiting α7 nicotinic receptors. J Neurosci. 2014;34:22–35. doi: 10.1523/JNEUROSCI.2093-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell SJ, Gee VJ, Harkness PC, Doward AI, Baker ER, Gibb AJ, Millar NS. RIC-3 enhances functional expression of multiple nicotinic acetylcholine receptor subtypes in mammalian cells. Mol Pharmacol. 2005;68:1431–1438. doi: 10.1124/mol.105.017459. [DOI] [PubMed] [Google Scholar]
- Levin ED. Nicotinic receptor subtypes and cognitive function. J Neurobiol. 2002;53:633–640. doi: 10.1002/neu.10151. [DOI] [PubMed] [Google Scholar]
- Lewis BL, O'Donnell P. Ventral tegmental area afferents to the prefrontal cortex maintain membrane potential ‘up’ states in pyramidal neurons via D1 dopamine receptors. Cereb Cortex. 2000;10:1168–1175. doi: 10.1093/cercor/10.12.1168. [DOI] [PubMed] [Google Scholar]
- Liu X-B, Murray KD. Neuronal excitability and calcium/calmodulin-dependent protein kinase type II: Location, location, location. Epilepsia. 2012;53:45–52. doi: 10.1111/j.1528-1167.2012.03474.x. [DOI] [PubMed] [Google Scholar]
- Livingstone PD, Dickinson JA, Srinivasan J, Kew JNC, Wonnacott S. Glutamate-dopamine crosstalk in the rat prefrontal cortex is modulated by alpha7 nicotinic receptors and potentiated by PNU-120596. J Mol Neurosci. 2010;40:172–176. doi: 10.1007/s12031-009-9232-5. [DOI] [PubMed] [Google Scholar]
- Maller JL, Kemp BE, Krebs EG. In vivo phosphorylation of a synthetic peptide substrate of cyclic AMP-dependent protein kinase. Proc Natl Acad Sci USA. 1978;75:248–251. doi: 10.1073/pnas.75.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man H-Y, Sekine-Aizawa Y, Huganir RL. Regulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci USA. 2007;104:3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Margiotta JF, Berg DK, Dionne VE. Cyclic AMP regulates the proportion of functional acetylcholine receptors on chicken ciliary ganglion neurons. Proc Natl Acad Sci USA. 1987;84:8155–8159. doi: 10.1073/pnas.84.22.8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Matsumoto A, Enomoto T, Nishizaki T. Activation of nicotinic acetylcholine receptors induces long-term potentiation in vivo in the intact mouse dentate gyrus. Eur J Neurosci. 2000;12:3741–3747. doi: 10.1046/j.1460-9568.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- Meinkoth JL, Ji Y, Taylor SS, Feramisco JR. Dynamics of the distribution of cyclic AMP-dependent protein kinase in living cells. Proc Natl Acad Sci USA. 1990;87:9595–9599. doi: 10.1073/pnas.87.24.9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Shen K. In and out of the postsynaptic region: signalling proteins on the move. Trends Cell Biol. 2000;10:238–244. doi: 10.1016/s0962-8924(00)01764-5. [DOI] [PubMed] [Google Scholar]
- Moss SJ, McDonald BJ, Rudhard Y, Schoepfer R. Phosphorylation of the predicted major intracellular domains of the rat and chick neuronal nicotinic acetylcholine receptor α7 subunit by cAMP-dependent protein kinase. Neuropharmacology. 1996;35:1023–1028. doi: 10.1016/s0028-3908(96)00083-4. [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Naira AC, Hemmings HC, Jr, Greengard P. Protein kinases in the brain. Annu Rev Biochem. 1985;54:931–976. doi: 10.1146/annurev.bi.54.070185.004435. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Lester HA. CNS localization of neuronal nicotinic receptors. J Mol Neurosci. 2006;30:181–184. doi: 10.1385/JMN:30:1:181. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Kandel ER. A macromolecular synthesis-dependent late phase of long-term potentiation requiring cAMP in the medial perforant pathway of rat hippocampal slices. J Neurosci. 1996;16:3189–3198. doi: 10.1523/JNEUROSCI.16-10-03189.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondrejcak T, Wang Q, Kew JNC, Virley DJ, Upton N, Anwyl R, Rowan MJ. Activation of α7 nicotinic acetylcholine receptors persistently enhances hippocampal synaptic transmission and prevents Aβ-mediated inhibition of LTP in the rat hippocampus. Eur J Pharmacol. 2012;677:63–70. doi: 10.1016/j.ejphar.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Perry DC, Xiao Y, Nguyen HN, Musachio JL, Dávila-García MI, Kellar KJ. Measuring nicotinic receptors with characteristics of α4β2, α3β2 and α3β4 subtypes in rat tissues by autoradiography. J Neurochem. 2002;82:468–481. doi: 10.1046/j.1471-4159.2002.00951.x. [DOI] [PubMed] [Google Scholar]
- Phillips AG, Vacca G, Ahn S. A top-down perspective on dopamine, motivation and memory. Pharmacol Biochem Behav. 2008;90:236–249. doi: 10.1016/j.pbb.2007.10.014. [DOI] [PubMed] [Google Scholar]
- Porter NM, Twyman RE, Uhler MD, Macdonald RL. Cyclic AMP-dependent protein kinase decreases GABAA receptor current in mouse spinal neurons. Neuron. 1990;5:789–796. doi: 10.1016/0896-6273(90)90338-g. [DOI] [PubMed] [Google Scholar]
- Sala F, Nistri A, Criado M. Nicotinic acetylcholine receptors of adrenal chromaffin cells. Acta Physiol (Oxf) 2008;192:203–212. doi: 10.1111/j.1748-1716.2007.01804.x. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Gorelova N, Durstewitz D, Yang CR. Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J Neurosci. 2001;21:3628–3638. doi: 10.1523/JNEUROSCI.21-10-03628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ. The variance of sodium current fluctuations at the node of Ranvier. J Physiol. 1980;307:97–129. doi: 10.1113/jphysiol.1980.sp013426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ. Interpreting power spectra from nonstationary membrane current fluctuations. Biophys J. 1981;35:289–300. doi: 10.1016/S0006-3495(81)84790-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydserff S, Sutton EJ, Song D, Quirk MC, Maciag C, Li C, Jonak G, Gurley D, Gordon JC, Christian EP, Doherty JJ, Hudzik T, Johnson E, Mrzljak L, Piser T, Smagin GN, Wang Y, Widzowski D, Smith JS. Selective α7 nicotinic receptor activation by AZD0328 enhances cortical dopamine release and improves learning and attentional processes. Biochem Pharmacol. 2009;78:880–888. doi: 10.1016/j.bcp.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Thomsen MS, Hansen HH, Timmerman DB, Mikkelsen JD. Cognitive improvement by activation of α7 nicotinic acetylcholine receptors: from animal models to human pathophysiology. Curr Pharm Des. 2010;16:323–343. doi: 10.2174/138161210790170094. [DOI] [PubMed] [Google Scholar]
- Trantham-Davidson H, Neely LC, Lavin A, Seamans JK. Mechanisms underlying differential D1 versus D2 dopamine receptor regulation of inhibition in prefrontal cortex. J Neurosci. 2004;24:10652–10659. doi: 10.1523/JNEUROSCI.3179-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhler MD, McKnight GS. Expression of cDNAs for two isoforms of the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem. 1987;262:15202–15207. [PubMed] [Google Scholar]
- Ungar AR, Moon RT. Inhibition of protein kinase A phenocopies ectopic expression of hedgehog in the CNS of wild-type and cyclops mutant embryos. Dev Biol. 1996;178:186–191. doi: 10.1006/dbio.1996.0209. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Vijayaraghavan S, Schmid HA, Halvorsen SW, Berg DK. Cyclic AMP-dependent phosphorylation of a neuronal acetylcholine receptor α-type subunit. J Neurosci. 1990;10:3255–3262. doi: 10.1523/JNEUROSCI.10-10-03255.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, O'Donnell P. D1 dopamine receptors potentiate NMDA-mediated excitability increase in layer V prefrontal cortical pyramidal neurons. Cereb Cortex. 2001;11:452–462. doi: 10.1093/cercor/11.5.452. [DOI] [PubMed] [Google Scholar]
- Wevers A, Monteggia L, Nowacki S, Bloch W, Schütz U, Lindstrom J, Pereira EF, Eisenberg H, Giacobini E, de Vos RA, Steur EN, Maelicke A, Albuquerque EX, Schröder H. Expression of nicotinic acetylcholine receptor subunits in the cerebral cortex in Alzheimer's disease: histotopographical correlation with amyloid plaques and hyperphosphorylated-tau protein. Eur J Neurosci. 1999;11:2551–2565. doi: 10.1046/j.1460-9568.1999.00676.x. [DOI] [PubMed] [Google Scholar]
- Whiting PJ, Liu R, Morley BJ, Lindstrom JM. Structurally different neuronal nicotinic acetylcholine receptor subtypes purified and characterized using monoclonal antibodies. J Neurosci. 1987;7:4005–4016. doi: 10.1523/JNEUROSCI.07-12-04005.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Rowan MJ, Anwyl R. Long-term potentiation is mediated by multiple kinase cascades involving CaMKII or either PKA or p42/44 MAPK in the adult rat dentate gyrus in vitro. J Neurophysiol. 2006;95:3519–3527. doi: 10.1152/jn.01235.2005. [DOI] [PubMed] [Google Scholar]
- Yan Z. Regulation of GABAergic inhibition by serotonin signaling in prefrontal cortex: molecular mechanisms and functional implications. Mol Neurobiol. 2002;26:203–216. doi: 10.1385/MN:26:2-3:203. [DOI] [PubMed] [Google Scholar]
- Yang K, Dani JA. Dopamine D1 and D5 receptors modulate spike timing-dependent plasticity at medial perforant path to dentate granule cell synapses. J Neurosci. 2014;34:15888–15897. doi: 10.1523/JNEUROSCI.2400-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Paspalas CD, Jin LE, Picciotto MR, Arnsten AFT, Wang M. Nicotinic α7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex. Proc Natl Acad Sci USA. 2013;110:12078–12083. doi: 10.1073/pnas.1307849110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, Crawford N, Kelly JS, Kerr LE, Marston HM, Spratt C, Finlayson K, Sharkey J. Impaired attention is central to the cognitive deficits observed in alpha 7 deficient mice. Eur Neuropsychopharmacol. 2007;17:145–155. doi: 10.1016/j.euroneuro.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Young JW, Finlayson K, Spratt C, Marston HM, Crawford N, Kelly JS, Sharkey J. Nicotine improves sustained attention in mice: evidence for involvement of the α7 nicotinic acetylcholine receptor. Neuropsychopharmacology. 2004;29:891–900. doi: 10.1038/sj.npp.1300393. [DOI] [PubMed] [Google Scholar]
- Zhong H, Sia G-M, Sato TR, Gray NW, Mao T, Khuchua Z, Huganir RL, Svoboda K. Subcellular dynamics of type II PKA in neurons. Neuron. 2009;62:363–374. doi: 10.1016/j.neuron.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Hablitz JJ. Dopamine modulation of membrane and synaptic properties of interneurons in rat cerebral cortex. J Neurophysiol. 1999;81:967–976. doi: 10.1152/jn.1999.81.3.967. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]