

Abstract
Recovery of intracellular pH from cytosolic alkalosis has been attributed primarily to Cl– coupled acid loaders/base extruders such as Cl–/HCO3– or Cl–/OH– exchangers. We have studied this process in cortical astrocytes from wild-type and transgenic mouse models with gene deletion for the electrogenic sodium bicarbonate cotransporter 1 (NBCe1) and for carbonic anhydrase (CA) isoform II. An acute cytosolic alkalosis was induced by the removal of either CO2/HCO3– or butyric acid, and the subsequent acid loading was analysed by monitoring changes in cytosolic H+ or Na+ using ion-sensitive fluorescent dyes. We have identified that NBCe1 reverses during alkalosis and contributes more than 70% to the rate of recovery from alkalosis by extruding Na+ and HCO3–. After CA inhibition or in CAII-knockout (KO) cells, the rate of recovery was reduced by 40%, and even by 70% in the nominal absence of CO2/HCO3–. Increasing the extracellular K+ concentration modulated the rate of acid loading in wild-type cells, but not in NBCe1-KO cells. Removing chloride had only a minor effect on the recovery from alkalosis. Reversal of NBCe1 by reducing pH/[HCO3–] was demonstrated in astrocytes and in Xenopus oocytes, in which human NBCe1 was heterologously expressed. The results obtained suggest that reversed NBCe1, supported by CAII activity, plays a major role in acid-loading cortical astrocytes to support recovery from cytosolic alkalosis.
Key points
The regulation of H+i from cytosolic alkalosis has generally been attributed to the activity of Cl–-coupled acid loaders/base extruders in most cell types, including brain cells.
The present study demonstrates that outwardly-directed sodium bicarbonate cotransport via electrogenic sodium bicarbonate cotransporter 1 (NBCe1) mediates the major fraction of H+i regulation from cytosolic alkalosis in mouse cortical astrocytes.
Cl–-coupled acid-loading transporters play only a minor role in the regulation of H+i from alkalosis in mouse cortical astrocytes.
NBCe1-mediated H+i regulation from alkalosis was dominant, with the support of intracellular carbonic anhydrase II, even when the intra- and extracellular [HCO3–] was very low (<1mM), as in nominally CO2/HCO3– free condition.
A reversed NBCe1 in astrocytes may also be significant for stabilizing extracellular pH in brain tissue.
Introduction
A powerful control of intra- and extracellular pH is necessary for normal cell function because a plethora of molecular and biochemical processes are sensitive to protons (Srivastava et al. 2007). Cells regulate the concentration of protons by buffering H+ ions and mediating the transport of acid/base equivalents across the membrane. In the brain, a modest shift of intra- and extracellular H+ ion concentration can have significant effects on brain functions, such as neuronal excitability, synaptic transmission and metabolism (Deitmer & Rose, 1996; Chesler 2003; Deitmer & Chesler, 2009). This is the result of the proton sensitivity of a large number of processes, such as ion channel gating and conductance, synaptic transmission, cell-to-cell communication via gap junctions, and enzymatic activity in brain energy metabolism (Traynelis and Cull-Candy 1990; Deitmer and Rose 1996; Devries 2001; Ruminot et al. 2011). Astrocytes, the major glial cell type in the mammalian nervous system, are known to significantly modulate the acid/base status of the brain (Deitmer 1991, Rose and Deitmer 1994, 1995; Deitmer and Rose 1996; Ransom 2000; Chesler 2003). The major acid/base coupled transporters identified in mammalian astrocytes so far comprise sodium hydrogen exchanger 1, electrogenic sodium bicarbonate cotransporter 1 (NBCe1), sodium-dependent chloride/bicarbonate exchange, chloride/bicarbonate exchange, monocarboxylate transporters, glutamate transporters, plasma membrane Ca2+ pump and vacuolar-type H+ pump (Chesler 2003; McAlear and Bevensee 2003; Deitmer 2007).
Anion exchanger (AE)-mediated Cl–/HCO3– exchange is known as a common acid-loading mechanism in many tissues, including brain cells (Raley-Susman et al. 1993; Kobayashi et al. 1994; Havenga et al. 1994; Leem and Vaughan-Jones 1998; Brett et al. 2002; Casey et al. 2009; Ruffin et al. 2014). The equilibrium potential of NBCe1 with a stoichiometry of 2 HCO3-: 1 Na+, is close to the resting membrane potential of astrocytes (−70 mV to −80 mV). NBCe1 has been shown to operate both inwardly and outwardly (i.e. as acid extruder and acid loader), such as during de- and hyperpolarization, respectively, in leech glial cells (Munsch and Deitmer 1994; Deitmer 1992; Deitmer 1991; Deitmer and Schlue 1989) and mammalian astrocytes (O'Connor et al. 1994; Brooks and Turner 1994).
Therefore, the present study investigated the contribution of acid-loading transporters involved in the regulation of H+i from an acute intracellular alkalosis induced by the removal of either CO2/HCO3– or butyrate, with the latter occurring in the presence and absence of CO2/HCO3–, in mouse cortical astrocytes. The results obtained suggest that outwardly-directed transport of bicarbonate by NBCe1 is the major acid-loading process that re-acidifies the cytosol of cortical astrocytes after an acute alkali load. However, Cl–-coupled acid loading (e.g. by AE-mediated Cl–/HCO3– exchange) plays only a minor role in the regulation of H+i from alkalosis in cortical astrocytes.
Methods
Chemicals and reagents
Standard chemicals, tissue culture reagents, 6-ethoxy-1,3-benzothiazole-2-sulphonamide (EZA) and 4,4′-diisothio-cyanatostilbene-2,2′-disulphonic acid (DIDS) were purchased from Sigma (St Louis, MO, USA). 2',7'-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF)-acetoxymethyl (AM) ester and asante natrium green-2 (ANG-2) AM were obtained from Molecular Probes (Eugene, OR, USA) and TEF Labs (Austin, TX, USA), respectively. N-cyanosulphonamide (S0859) was obtained from Sanofi-Aventis (Frankfurt-Hoechst, Germany).
Animals, primary astrocyte culture from cerebral cortex
Cortical astrocyte culture was prepared from wild-type (WT) (C57BL/6, p0 to p3), NBCe1-KO (129S6/SvEv and Black Swiss background, p0 to p3; originally received from Dr Gary E. Shull, UC College of Medicine, University of Cincinnati, Cincinnati, OH, USA) and carbonic anhydrase (CA)II-KO (Jackson Laboratories, Bar Harbor, MI, USA) mice, as described previously (Stridh et al. 2012). Astrocytes were plated on poly-d-lysine-coated glass coverslips and maintained in Dulbecco's modified Eagle's medium containing 5% fetal calf serum and 5% horse serum. The medium was renewed after 24 h of plating and subsequently every 3 days. Cells were used for experiments when they were 10–20 days in vitro. Animals were maintained under a 12:12 h day/night cycle at constant room temperature with free access to water and standard mouse diet in the animal facility of the Technical University of Kaiserslautern. All procedures involving animals were approved by the Landesuntersuchungsamt Rheinland-Pfalz, Koblenz (23 177-07).
Intracellular H+ imaging
To measure the intracellular H+ concentration ([H+]i) changes in cultured cortical astrocytes, we used a confocal imaging system and the AM of a proton sensitive dye, BCECF. The dye was loaded into the cells by incubating them with 3 μM BCECF-AM in Hepes-buffered solution (pH 7.4) for 15 min at room temperature. Cells were then mounted into a chamber under a confocal laser scanning microscope (LSM 510; Zeiss, Oberkochen, Germany) and continuously superfused with solutions (pH 7.4) buffered with either Hepes (in mM): 140 NaCl, 5 KCl, 10 α-d-glucose, 0.5. NaH2PO4, 10 Hepes, 1 MgCl2 and 2 CaCl2 or with CO2/HCO3− (in mM): 114 NaCl, 5 KCl, 0.5 NaH2PO4, 10 α-d-glucose, 26 NaHCO3, 1 MgCl2 and 2 CaCl2. BCECF was excited consecutively at 488 nm (proton-sensitive wavelength) and 458 nm (close to isosbestic point) and the changes in fluorescence emission were monitored at > 505 nm (using a long pass filter; LP 505 filter). Images were obtained every 5 s (0.2 Hz) with a 40× water immersion objective. The fluorescence emission intensity of 488 nm excitation changes inversely with a change in [H+]i, whereas the fluorescence emission intensity of 458 nm excitation is largely pH-insensitive. The changes of [H+]i were monitored using the ratio F458/F488. The ratio was converted into pH and absolute intracellular proton concentrations ([H+]i) using the nigericin-based calibration technique. Cells were perfused with calibration solutions titrated to pH 6.0, pH 6.5, pH 7.0 and pH 7.5 containing (in mM): 0.01 nigericin, 15 NaCl, 130 KCl, 20 Hepes, 1 MgCl2 and 1 CaCl2. Mean ratio values (F458/F488) were plotted as a function of pH to create the calibration curve. The ratio was then converted to intracellular pH (pHi) and [H+]i (Theparambil et al. 2014).
Intracellular Na+ imaging
To measure intracellular Na+, cells were loaded with 10 μM acetoxymethyl ester of the single wavelength sodium-sensitive dye ANG-2 for 60 min in Hepes-buffered solution at room temperature (20–22°C). To improve dye uptake, pluronic acid-F127 (0.08%) was added. After dye loading, the cover slip was placed in the experimental chamber and mounted on the stage of the confocal laser scanning microscope (LSM 510). Cells were then continuously superfused with either Hepes or CO2/HCO3−-buffered solutions. ANG-2 was excited by 488 nm laser wavelength every 5 s (0.2 Hz) with a 40× water-immersion objective and fluorescence emission was collected at a photomultiplier tube with a long pass filter (LP 515). The fluorescence intensities were normalized (ΔF/F) and these relative fluorescence intensity changes were then converted to [Na+]i using a gramicidin, monensin and ouabain based calibration method.
All calibration solutions contained (in mM): 140 (Na+ + K+), 30 Cl−, 113 gluconic acid, 10 Hepes, 10 glucose, 1 MgCl2 and 2 CaCl2. Gramicidin (3 μm), monensin (10 μM) and ouabain (1 mM) were added to the calibration solution to equilibrate intra- and extracellular Na+, K+ and H+. The ANG-2 fluorescence intensity increased with increasing [Na+]. The mean relative fluorescence intensities were plotted against the corres-ponding [Na+] (0, 5, 10, 20 and 50 mM), and the resulting calibration curve was fitted by a hyperbolic function.
Heterologous expression of NBCe1 and electrophysiology in Xenopus laevis oocytes
Xenopus laevis females were purchased from Xenopus Express (Vernassal, France). Frogs were anaesthetized with 1 g l−1 of 3-aminobenzoic acid ethylester (MS-222; Sigma-Aldrich, Taufkirchen, Germany), rendered hypothermic and segments of ovarian lobules were surgically removed under sterile conditions. The procedure was approved by the Landesuntersuchungsamt Rheinland-Pfalz (Koblenz, Germany; 23 177-07/A07-2-003 §6) and has been described in detail previously (Becker & Deitmer, 2007). In brief, oocytes were isolated and singularized by collagenase treatment (Collagenase A, Roche, Mannheim, Germany) in Ca2+-free oocyte solution at 28°C for 2 h. The singularized oocytes were left overnight in an incubator at 18°C in Ca2+-containing oocyte solution (pH 7.8) to recover. The human NBCe1 cDNA (hkNBCe1) was cloned in an oocyte expression vector pGH19. Plasmid DNA was linearized with NotI and transcribed in vitro with T7 RNA-polymerase in the presence of the cap analogon m7G(5')ppp(5')G (mMessage mMachine; Ambion, Austin, TX, USA) to produce a capped RNA transcript. The cRNA was purified with the Qiagen RNeasy MinElute Cleanup Kit (Qiagen GmbH, Hilden, Germany) and stored at −80°C in DEPC-H2O. The oocyte solution had the composition (in mM): 82.5 NaCl, 2.5 KCl 2.5, 1 CaCl2, 1 MgCl2, 1 Na2HPO4 and 5 Hepes, titrated to pH 7.4. The bicarbonate-containing solution contained (in mM): 58.5 NaCl, 2.5 KCl, 1 CaCl2, 1 MgCl2, 1 Na2HPO4 and 24 NaHCO3, aerated with 5% CO2 and Hepes (5 mM), to stabilize the pH. Oocytes of the stages V and VI were selected and injected with 13.8 ng of NBCe1-cRNA dissolved in diethyl dicarbonate-H2O using glass micropipettes and a microinjection device (Nanoliter 2000; World Precision Instruments, Berlin, Germany).
For measurement of intracellular H+ and membrane potential, double-barreled microelectrodes were used, as described in detail previously (Deitmer, 1991). pHi measurements were stored digitally using custom PC software based on LabView (National Instruments Germany GmbH, München, Germany) and were routinely converted into the intracellular H+ concentration [H+]i. Amplitude and the rate of change of the measured [H+]i were analysed. For voltage clamp, electrodes filled with 3 M KCl were connected to the head-stages of an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA, USA). The experimental bath was grounded with a chlorided silver wire coated with agar dissolved in oocyte solution. Oocytes were clamped to a holding potential of −40 mV, and all experiments were carried out at room temperature (22 °C). Further details are provided in Becker & Deitmer (2007).
Statistical analysis
All statistical analysis were performed using SigmaPlot, version 11.0 (Systat Software Inc., Chicago, IL, USA) and Clamp Fit, version 10.2 (Molecular Devices, Sunnyvale, CA, USA). Statistical values are reported as the mean ± SEM. For calculation of significance differences between two groups, Student's t test was used with significance levels as appropriate (P ≤ 0.05, P ≤ 0.01 and P ≤ 0.001). Multiple comparisons were performed by Kruskal–Wallis one-way ANOVA on ranks with Dunn's test as post hoc analysis.
Results
The regulation of H+i from CO2/HCO3− removal induced alkalosis
We have used the addition and removal of CO2/HCO3− as one method to induce an acute intracellular acidification and alkalinization, respectively, in cultured cortical astrocytes. Changing from Hepes to CO2/HCO3− -buffered solution is expected to result in a sharp rise of H+i, which is attributed to the free diffusion of CO2 into cells and its conversion to H+i and HCO3− (Fig. 1A). Subsequently, cells regulate the intracellular pH from this acidification, as attributed to the activity of acid-extruding transporters (Fig. 1A). Changing back to nominally CO2/HCO3− free, Hepes-buffered external solution, at a constant pH of 7.4, is expected to induce a sharp fall of H+i, as attributed to the free diffusion of CO2 out of cell and the conversion of intracellular H+ and HCO3− to CO2, which then continues to diffuse out of cell (Fig. 1B). Cells regulate H+i back from this alkalosis by re-acidifying the cytosol, presumably with the help of acid-loading transporters (Fig. 1B).
Figure 1. CO2/HCO3− induced pHi/[H+]i shifts in cortical astrocytes.

A and B, cartoons depicting the events of H+i changes upon the addition (A) and removal (B) of CO2/HCO3− at a constant extracellular pH value. During the exposure of CO2/HCO3−, the free diffusion of CO2 into the cell and the subsequent conversion to H+ and HCO3−, the rate of which is enhanced by intracellular CA, induces an acute intracellular acid load. Activation of acid-extruding transporters such as NHE (sodium/hydrogen exchanger) and NBC (sodium bicarbonate cotransporter) (A) regulates H+i from this CO2-induced acid load. Similarly, the removal of CO2/HCO3− from external saline (B) induces an acute cytosolic alkali load attributed to the free diffusion of CO2 out of the cell and the conversion of intracellular H+ and HCO3− to CO2, which then continues to leave the cell. The presence of intracellular CA enhances the rate of this alkali load, whereas the activation of acid-loading transporters during this alkalosis re-acidifies the cytosol and regulate H+i back to the normal steady-state. C, original recordings (superimposed) of H+i shifts induced by the addition and removal of 5% CO2/26 mM HCO3− in mouse cortical astrocytes from WT and NBCe1-KO mice. D, CO2/HCO3− induced H+i recordings (superimposed) in the presence and absence of external Cl− and the NBCe1 inhibitor S0859 (50 μM) in WT astrocytes. E and F, rate of H+i rise (E) and acid/base flux (F) during regulation from CO2/HCO3− removal-induced alkalosis in WT astrocytes in the presence and absence of external Cl− (open and grey bars), in the presence of the NBCe1 inhibitor S0859 (brown bars) and in NBCe1-KO astrocytes (red bars). All values are shown as the mean ± SEM. *P≤ 0.05; n.s., non-significant (Kruskal–Wallis one-way ANOVA on ranks with Dunn's test as post hoc analysis).
We have studied the contribution of acid-loading transporters for the regulation of H+i from alkalosis induced by changing the external solution buffered with 5% CO2/26 mm HCO3− to a nominally CO2/HCO3−-free, Hepes-buffered solution in WT and NBCe1-KO astrocytes (Fig. 1C). The functional activity of acid-loading transporters was studied from the initial rate of change of H+i and the concomitant acid/base flux rate (JA/B mM min–1) during recovery from this alkalosis (Fig. 1C and D, highlighted by the dotted lines). The acid/base flux rate was calculated by multiplying the rate of pHi changes from alkalosis (ΔpHi min–1) with the total physicochemical buffer strength (βt = βCO2 + βi). The physicochemical buffer strength of astrocytes (β), as measured from the amplitude of CO2-induced acidification in the absence of extracellular sodium to exclude transport-mediated contribution to cytosolic H+ buffering, was found to be 49.8 ± 4.1 mM, of which 20.6 ± 1.7 mM was CO2-dependent (βCO2) and 29.1 ± 2.9 was intrinsic (βi) buffer capacity (Theparambil and Deitmer, 2015). Because the cytoplasmic buffer capacity, β, is expected to decrease with a decrease in [HCO3−]i/pHi, it should be noted that using single β value may have caused an overestimation of the transporter flux because it does not account for the dynamic changes in pHi during the recovery from alkalosis.
A rapid fall of [H+]i, to ∼40–50 nM, was observed upon the removal of CO2/HCO3− in both WT and NBCe1-KO astrocytes (Fig. 1C). The rate of rise of H+i and the concomitant acid/base flux rate during the recovery from alkalosis, however, were greatly reduced in astrocytes from NBCe1-KO mice compared to WT astrocytes, revealing a substantial contribution of NBCe1-mediated export of bicarbonate in the regulation of H+i from alkalosis (Fig. 1E and F). Pharmacological inhibition of NBCe1 with 50 μM S0859 (Fig. 1D, brown bar) (Ch'en et al. 2008; Heidtmann et al. 2015) also reduced the rate of H+i rise (Fig. 1E, red bar) and the concomitant acid/base flux rate (Fig. 1F, red bar) during the recovery from CO2/HCO3− removal-induced alkalosis in WT astrocytes. The rate of H+i rise and the concomitant acid/base flux from CO2/HCO3− removal-induced alkalosis were partly dependent (∼30–35%) on extracellular Cl− in WT astrocytes, indicating a significant, but minor contribution of a Cl−-coupled acid loader (Fig. 1E and F), possibly Cl−/HCO3− exchange or Na+-dependent Cl−/HCO3− exchange. The rate of H+i rise and the acid/base flux rate in WT astrocytes under control conditions were 16.6 ± 0.88 nM min−1 and −6.16 ± 0.12 mM min−1, respectively (Fig. 1E and F, open bars), and were reduced to 11.5 ± 1.3 nM min−1 and −4.53 ± 0.14 mM min−1 in the absence of external Cl− (Fig. 1E and F, grey bars), to 8.7 ± 0.39 nM min−1 and −1.82 ± 0.2 mM min−1 when NBCe1 was pharmacologically inhibited by S0589 (Fig. 1E and F, brown bars), and to 2.6 ± 0.15 nM min−1 and −1.36 ± 0.05 mM min−1 when NBCe1 protein was genetically deleted (NBCe1-KO; Fig. 1E and F, red bars).
The regulation of H+i from butyrate removal-induced alkalosis in the presence of CO2/HCO3−
Because the complete removal of CO2/HCO3− from external buffer would form a large outwardly-directed concentration gradient for both CO2 and HCO3−, we further studied the contribution of bicarbonate-dependent acid loaders at a constant extracellular CO2/HCO3− concentration. Therefore, we used the addition and removal of butyric acid (40 mM butyrate) as another method to induce an acute acidification and alkalinization, respectively, in cultured cortical astrocytes. Cytosolic alkalosis induced by the removal of butyrate is attributed to the free diffusion of butyric acid (membrane-permeable as undissociated acid) out of the cell and the subsequent association of intracellular butyrate and H+ ions to butyric acid, which then continues to leave the cell. The diffusion of butyric acid carries acid equivalents out of the cell and lasts until all butyrate is extruded. Because, during the exposure of butyrate, regulatory transport processes result in a recovery from the induced acidosis, the removal of butyrate causes a transient cytosolic alkalosis. The regulation of H+i from this alkalosis is attributed to the activity of acid-loading transporters, and was investigated in more detail in the present study in the presence of 5% CO2/26 mm HCO3−.
The removal of 40 mM butyrate in the presence of 5% CO2/26 mM HCO3− induced a sharp fall of [H+]i, which was associated with a rise in intracellular [HCO3−], estimated at the low peak of H+i, to be 26.5 mm, as calculated from the Henderson–Hasselbalch equation (Fig. 2A). The rate of subsequent [H+]i rise and the concomitant acid/base flux after this alkalosis were 15.8 ± 0.6 nM min−1 and −5.4 ± 0.23 mM min−1, respectively, in WT astrocytes, although this was reduced by 75% in NBCe1-KO cells to 4.76 ± 0.35 nM min−1 and −1.3 ± 0.07 mM min−1 (Fig. 2A, C and D). This suggests a substantial contribution of NBCe1-mediated bicarbonate export to the regulation of H+i from alkalosis in astrocytes. The absence of Cl− in the external solution buffered with CO2/HO3− did not significantly change the rate of H+i rise (Fig. 2B, grey trace) and the concomitant flux rate during the recovery from alkalosis, with rates of 14.2 ± 0.82 nM min−1 and −5.02 ± 0.32 mM min−1 (Fig. 2C and D, grey bars), suggesting that Cl−-dependent acid loading mechanisms do not contribute to the regulation from alkalosis under these conditions. Inhibition of CA activity with 30 μM EZA in the absence (as well as in the presence; not shown here) of external Cl− (Fig. 2B, orange trace), however, significantly reduced the rate of change of H+i and the acid/base flux rate during recovery from alkalosis by ∼30%, with rates of 9.4 ± 0.45 nM min−1 and −3.5 ± 0.11 mM min−1 (Fig. 2C and D, orange bars), suggesting that intracellular CA supports NBCe1-mediated outward transport of bicarbonate.
Figure 2. Butyrate removal-induced [H+]i shifts in the presence of CO2/HCO3− in cortical astrocytes.
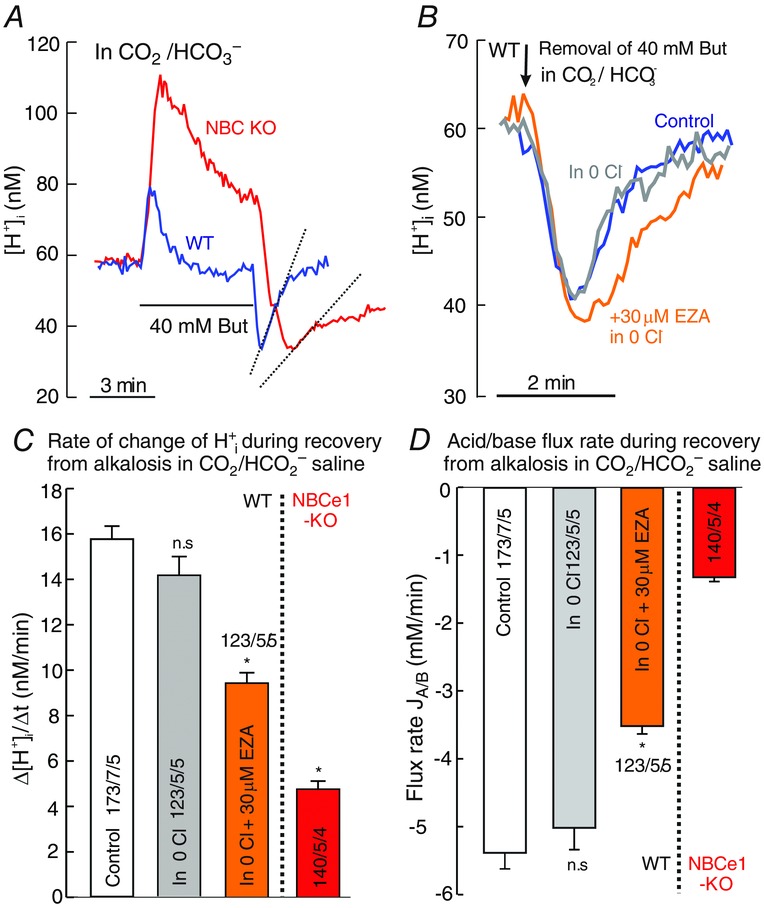
A, original recordings (superimposed) of H+i shifts induced by the addition and removal of 40 mM butyrate from extracellular solution containing 5% CO2/26 mm HCO3− in WT and NBCe1-KO astrocytes. B, superimposed traces of H+i shifts in WT astrocytes upon the removal of butyrate in CO2/HCO3− containing solution with and without Cl− (blue and grey traces, respectively) and in the presence of CA inhibitor EZA (ethoxyzolamide, 30 μM) in Cl− -free solution. C and D, bar plots of the rate of H+i rise from butyrate removal-induced alkalosis in CO2/HCO3−-buffered saline (C) and the concomitant acid/base flux rates (D) in WT astrocytes in the presence and absence of external Cl− (open and grey bars respectively), in the presence of CA inhibitor EZA in Cl− free saline (orange bars) and in NBCe1-KO astrocytes (red bars). All values are shown as the mean ± SEM. *P ≤ 0.05; n.s., non-significant (Kruskal–Wallis one-way ANOVA on ranks with Dunn's test as post hoc analysis).
The regulation of H+i from butyrate removal-induced alkalosis in the nominal absence of CO2/HCO3−
In some cell types, a bicarbonate independent, Cl−-dependent acid-loader, chloride/hydroxyl exchanger (CHE), was shown to be the major regulator of H+i from alkalosis in the nominal absence of CO2/HCO3− (Leem and Vaughan-Jones 1997, 1998; Sun et al. 1996). Therefore, the regulation of [H+]i from butyrate removal-induced alkalosis was also investigated in nominally CO2/HCO3−-free, Hepes-buffered solution, aiming to determine whether CHE contributed to the regulation of H+i from alkalosis. To calculate the acid/base flux rate (JA/B), we used the intrinsic buffer capacity (βi) of astrocytes. We found a substantial reduction in the rate of [H+]i rise and the concomitant acid/base flux rate during the recovery from alkalosis from 15.8 ± 0.58 nM min−1 and −5.4 ± 0.23 mM min−1 in a solution buffered with 5% CO2/26 mM HCO3−, to 8.3 ± 36 nM min−1 −1.9 ± 0.09 mM min−1, respectively, in Hepes-buffered solution under control conditions (Fig. 3A and B; open bars in Fig. 3D and E). The rate of H+i rise and the corresponding acid/base flux rate during regulation from alkalosis in Hepes-buffered solution did not change in the absence of external Cl−, with rates of 7.9 ± 0.9 nM min−1 and −1.95 ± 0.05 mM min−1, respectively, (Fig. 3A grey trace; grey bars in Fig. 3D and E), although they were significantly reduced to 3.7 ± 0.19 nM min−1 and −0.67 ± 0.02 mM min−1, respectively, in the presence of the anion transport inhibitor DIDS (0.5 mM), suggesting that CHE-mediated acid loading does not contribute to the regulation of H+i from alkalosis in cortical astrocytes (brown trace in Fig. 3A; brown bars in Fig. 3D and E).
Figure 3. Butyrate removal-induced H+i shifts in the nominal absence of CO2/HCO3− in cortical astrocytes.
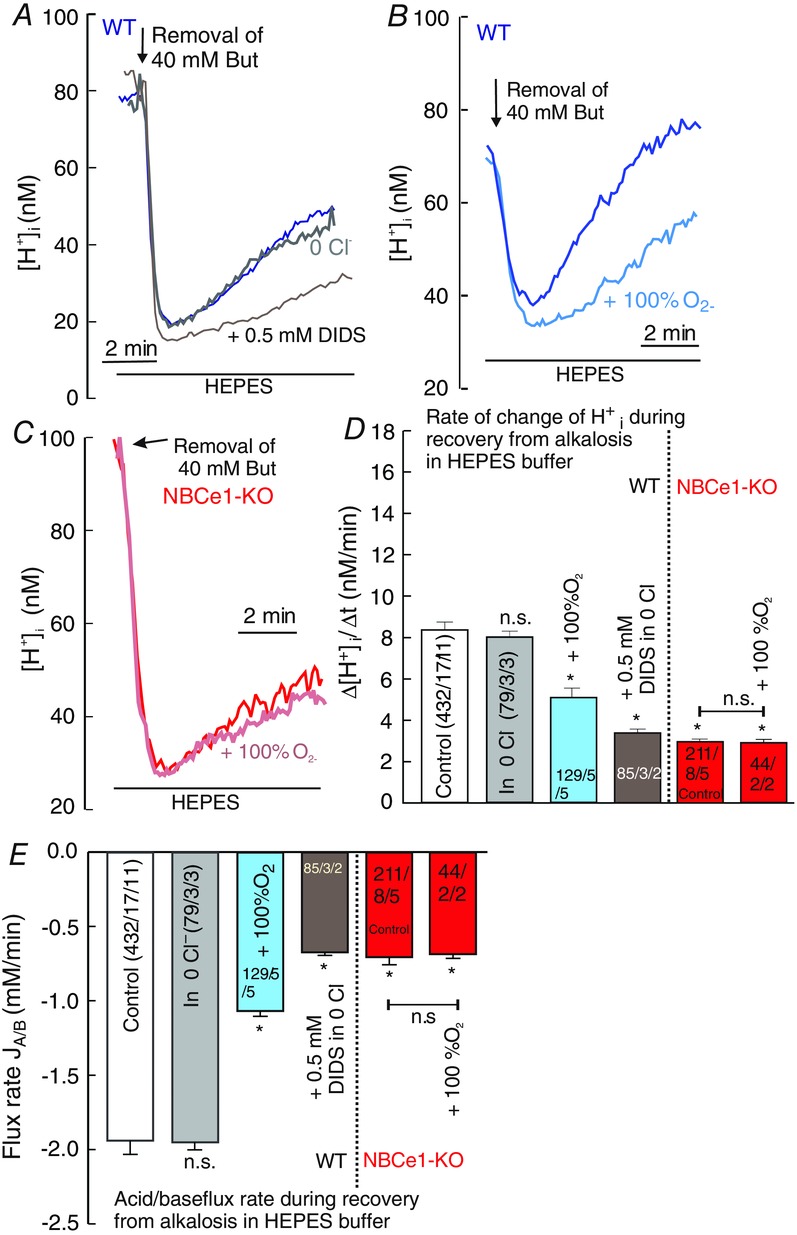
A, original recordings (superimposed) of H+i fall and subsequent regulation induced by the removal of butyrate in nominally CO2/HCO3−-free, Hepes-buffered, solution with and without Cl− (blue and grey traces, respectively) and in the presence of anion transporter inhibitor DIDS (0.5 mM) in Cl− free saline (brown trace) in WT astrocytes. B and C, H+i recordings in 100% O2-aerateded and non-aerated solution in WT (B) and in NBCe1-KO astrocytes (C). D and E, bar plots of the rate of H+i rise (D) and the concomitant acid/base flux rates (E) during recovery from alkalosis in Hepes-buffered solution in WT astrocytes with and without Cl− (open and grey bars), 100% O2- aerated solution (light blue bars), in the presence of 0.5 mM DIDS (brown bars) and in NBCe1-KO astrocytes in 100% O2-aerated and non-aerated solutions (red bars). All values are shown as the mean ± SEM. *P ≤ 0.05; n.s., non-significant (Kruskal–Wallis one-way ANOVA on ranks with Dunn's test as post hoc analysis).
Because Hepes-buffered solutions still contain micromolar concentrations of bicarbonate (180 μM at pH 7.4), derived from equilibrated air CO2 in the solution, we hypothesized that NBCe1 may still contribute to the bicarbonate-dependent acid-loading process during the regulation of [H+]i from alkalosis even without additionally added bicarbonate as a result of the high bicarbonate sensitivity of NBCe1 reported previously (Theparambil et al. 2014). We tested this hypothesis by reducing residual CO2/HCO3− from Hepes-buffered solution by aerating the solution with 100% O2, and then compared the rate of H+i rise and corresponding acid/base flux rate during the recovery from alkalosis with the non-aerated solution, in WT, as well as NBCe1-KO astrocytes. The reduction of residual CO2/HCO3− from the Hepes buffered solution decreased the rate of H+i rise and the concomitant acid/base flux rate during the regulation from alkalosis significantly in WT astrocytes from 8.3 ± 0.4 nM min−1 and −1.9 ± 0.09 mM min−1 to 5.1 ± 0.5 nM min−1 and −1.07 ± 0.03 mM min−1, respectively (Fig. 3B, D and E, light blue trace and bars). NBCe1-KO astrocytes showed a much slower rate of [H+]i rise and acid/base flux rate during recovery from alkalosis, with rates of 2.9 ± 0.1 nM min−1 and −0.71 ± 0.05 mM min−1, respectively, compared to the WT cells, whereas aerating the solution with 100% O2 had no additional effect on the regulation of H+i from alkalosis in these NBCe1-KO cells (Fig. 3C, D and E, red traces and bars). These results suggest that, as attributed to its high bicarbonate sensitivity, NBCe1-mediated outward transport of bicarbonate significantly contributes to the regulation of H+i from alkalosis in cortical astrocytes even under conditions of very low intracellular bicarbonate concentrations (of up to 250 μM at the low peak during alkalosis of 30 nM [H+]i).
The contribution of CAII to NBCe1-mediated regulation from alkalosis in the nominal absence of CO2/HCO3−
We have shown that the inhibition of CA with 30 μM EZA reduces NBCe1-mediated H+i regulation from alkalosis in the presence of 5% CO2/26 mM HCO3− (Fig. 2B–D) and therefore we hypothesized that the rapid production of intracellular HCO3− by CA activity provides a steady supply of substrate for the transporter. Here, we have compared the rate of [H+]i rise and the concomitant acid/base flux rate during the regulation from alkalosis induced by removal of butyrate in the absence and presence of the CA inhibitor EZA (30 μM) in a Cl−-free Hepes-buffered solution. The rate of H+i rise and the acid/base flux rate were significantly reduced in the presence of 30 μM EZA in WT astrocytes by ∼63% from 7.9 ± 0.9 nM min−1 and −1.95 ± 0.05 mM min−1, respectively, to 3.0 ± 0.1 nM min−1 and −0.71 ± 0.02 mM min−1 (blue and orange traces in Fig. 4A; open bars in Fig. 4D and E; 0 Cl− control taken from Fig. 3D and E), whereas inhibition of CA in NBCe1-KO astrocytes had no effect on the rate of H+i regulation from alkalosis (Fig. 4B, D and E, red and brown traces and red bars). This was confirmed in astrocytes with the CAII gene deleted, where the rate of H+i rise and the acid/base flux rate during regulation from alkalosis were reduced to 2.79 ± 0.19 nM min−1 and −0.77 ± 0.03 mM min−1, respectively (green trace in Fig.4C; green bars in Fig.4D and E) and hence were similarly low as in the presence of EZA and in NBCe1-KO cells (Fig.4D and E). This suggests that the rapid production of intracellular bicarbonate catalysed by CA activity significantly supports the NBCe1-mediated outward transport of bicarbonate during the recovery from intracellular alkalosis in nominally CO2/HCO3−-free solution.
Figure 4. The effect of suppressing CA activity on the rate of H+i regulation from butyrate removal induced alkalosis in the nominal absence of CO2/HCO3−.

A and B, original recordings showing the fall of H+i and subsequent regulation induced by the removal of butyrate in a Cl−-free, Hepes-buffered solution without and with CA inhibitor EZA (30 μM) in WT (A, blue and orange trace) and in NBCe1-KO astrocyte (B, red and brown trace). C, H+i recordings of butyrate removal induced alkalosis and subsequent regulation in WT (blue trace) and in CAII-KO astrocyte (green trace). D and E, bar plots of the rate of H+i rise (D) and the concomitant acid/base flux rate (E) during recovery from alkalosis in Hepes-buffered solutions in the presence and absence of EZA in WT astrocytes (open bars) in CAII KO astrocytes (green bars) and in NBCe1-KO astrocytes with and without EZA in Cl− free solution (red bars). All values are shown as the mean ± SEM. *P ≤ 0.05; n.s., non-significant (Kruskal–Wallis one-way ANOVA on ranks with Dunn's test as post hoc analysis).
The electrochemical gradient governs the direction of bicarbonate transport via NBCe1 in cortical astrocytes and in Xenopus oocytes
The outward transport mode of NBCe1 was further studied by lowering the extracellular [HCO3−]/pH at 5% CO2 under different membrane potentials in cortical astrocytes and in Xenopus oocytes where hNBCe1 was heterologously expressed. It was hypothesized that, when astrocytes are depolarized by raising external [K+], the electrical gradient necessary to overcome for extruding bicarbonate via the electrogenic NBCe1 would be much larger, and hence the rate of acidification would be reduced, after lowering the external [HCO3−] from 26 mM to 10 mM (Fig. 5A). A robust increase in [H+]i, with a rate of rise of 26.7 ± 1.7 nM min−1 and acid/base flux rate of −9.29 ± 0.28 mM min−1, was monitored upon lowering the extracellular [HCO3−] from 26 to 10 mM at an external [K+] of 5 mM, and this was substantially reduced to 6.9 ± 0.53 nM min−1 and −3.7 ± 0.18 mM min−1, respectively, when the extracellular [K+] was raised to 15 mM (Fig. 5C and D, open bars). By contrast, in astrocytes from NBCe1-KO mice, lowering the extracellular [HCO3−] from 26 to 10 mM in the presence of 5 mM K+ induced only a slow rise of H+i of 5.4 ± 0.93 nM min−1 and an acid/base flux rate of −2.08 ± 0.15 mM min−1 (Fig. 5B–D, red trace and red bars). The kinetics of H+i rise and acid/base flux did not change significantly when the extracellular [K+] was elevated to 15 mM in NBCe1-KO cells (Fig. 5C and D). These results suggest that the rapid acidification observed in astrocytes upon lowering the extracellular bicarbonate concentration was largely attributed to the outwardly-directed transport of bicarbonate mediated by NBCe1.
Figure 5. NBCe1 transport can be reversed in cortical astrocytes and in X. laevis oocytes.
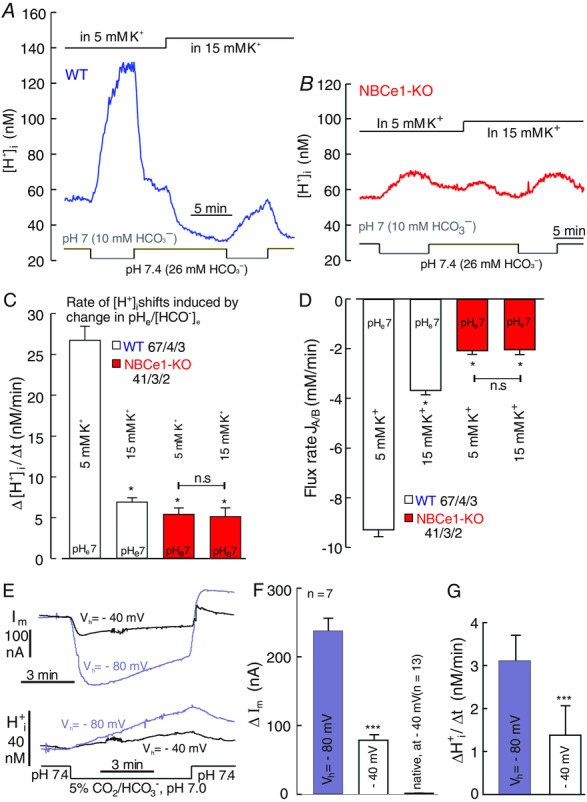
A and B, original recording of H+i shifts induced by lowering the extracellular [HCO3−] from 26 to 10 mM/pH 7.4 to 7 at 5% [CO2] in 5 and 15 mM extracellular [K+] in WT (A) and in NBCe1-KO (B) astrocytes. C and D, bar plots of the rate of H+i rise (C) and acid/base flux rates (D) upon lowering the extracellular [HCO3−] in 5 and 15 mM extracellular [K+] in WT and NBCe1-KO astrocytes (open and red bars). All values are shown as the mean ± SEM. *P ≤ 0.05; n.s., non-significant (Kruskal–Wallis one-way ANOVA on ranks with Dunn's test as post hoc analysis). E, original recordings from Xenopus oocytes in which hNBCe1 protein was heterologously expressed, showing NBCe1-mediated inward current and intracellular H+ rise upon lowering [HCO3−] at two different holding potentials (−40 mV and −80 mV). F and G, bar plot of currents obtained from NBCe1-cRNA-injected and native oocytes (F) and rate of intracellular H+ rise (G), which was substantially reduced at −40 mV compared to −80 mV. All values are shown as the mean ± SEM. ***P ≤ 0.001; n.s., non-significant (Student's t test).
When hNBCe1 protein was heterologously expressed in Xenopus oocytes, we observed a larger inward current and rate of acidification at a holding potential of −80 mV compared to the more positive holding potential of −40 mV, being 238 ± 18 nA vs. 79 ± 8 nA and 3.1 ± 0.6 nM min−1 vs. 1.4 ± 0.7 nM min−1, respectively, when the extracellular [HCO3−] was reduced from 24 mM (pH 7.4) to 10 mM (pH 7.0), respectively (Fig. 5E–G). Native oocytes showed only small, negligible, steady-state current changes of 4.11 ± 0.9 nA at – 40 mV (Fig. 5F) and 6.22 ± 2.9 nA at −80 mV, as attributed to their resting conductance, and no change in [H+]i (Becker & Deitmer, 2007). These results strongly suggest that NBCe1-mediated bicarbonate transport can be inwardly or outwardly-directed depending on the chemical and electrical gradient across the cell membrane, consistent with a HCO3−:Na+ stoichiometry of 2:1 (Becker & Deitmer, 2004).
The cytosolic changes of Na+i during the recovery from alkalosis
The transport activity of NBCe1 is also expected to induce changes in intracellular [Na+] ([Na+]i). Because reversed NBCe1 would have to extrude Na+ against a large electrochemical gradient, we also monitored cytosolic changes of [Na+]i in WT and in NBCe1-KO astrocytes during the addition and removal of CO2/HCO3− and 40 mM butyrate (in the presence of CO2/HCO3−). The addition of CO2/HCO3− or 40 mM butyrate evoked a sharp rise in [Na+]i, with subsequent regulation in WT astrocytes (Fig. 6A, C, D, F), whereas cytosolic Na+ in NBCe1-KO astrocytes decreased upon the addition of either CO2/HCO3− or butyrate (Fig. 6B, C, E, F). We conclude that the rise of Na+i in WT astrocytes was attributed to NBCe1 being inwardly activated (Figs 1C and 2A) and assume that the amplitude and rates of changes in both WT and NBCe1-KO cells were considerably affected by the activity of Na+/K+ ATPase.
Figure 6. Intracellular sodium changes induced by NBCe1 in cortical astrocytes.
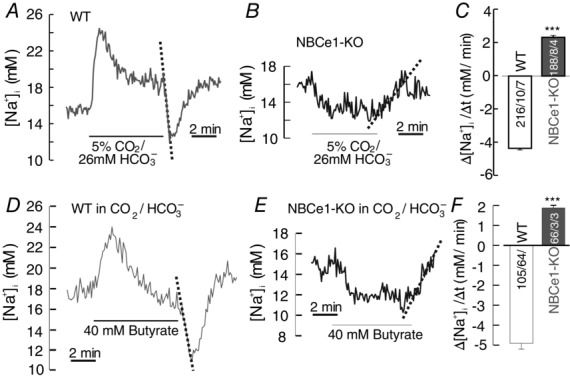
A and B, original recordings of cytosolic Na+ change upon the addition and removal of 5% CO2/26 mM HCO3− in WT (A) and NBCe1-KO (B) astrocytes. The dotted lines highlight the cytosolic Na+ changes upon the removal of CO2/HCO3−. C, bar plot of the rates of cytosolic Na+ changes in WT astrocytes (open bar) and in NBCe1-KO astrocytes (Gray bar) upon removal of CO2/HCO3−. D and E, original recordings of cytosolic Na+ changes in WT (D) and NBCe1-KO (E) astrocytes upon the addition and removal of 40 mM butyrate in the presence of 5% CO2/26 mM HCO3−. The dotted lines highlight the cytosolic Na+ changes upon the removal of butyrate. F, bar plot of the rates of cytosolic Na+ in WT astrocytes (open bar) and in NBCe1-KO astrocytes (Gray bar) upon the removal of 40 mM butyrate. All values are shown as the mean ± SEM. ***P ≤ 0.001; n.s., non-significant (Student's t test).
During the removal of CO2/HCO3− or 40 mM butyrate, we observed a rapid fall of Na+i of −4.3 ± 0.1 mM min−1 and −4.9 ± 0.29 mM min−1, respectively, with subsequent recovery in WT astrocytes (Fig. 6A, C, D, F), whereas [Na+]i in NBCe1-KO astrocytes rose back to its original baseline (Fig. 6B, C, E, F). These results show that Na+i increased upon the addition of CO2/HCO3− or butyrate (reflecting NBCe1-mediated recovery from acidosis) and was extruded after CO2/HCO3− or butyrate removal (reflecting NBCe1-mediated recovery from alkalosis), in line with the finding that NBCe1 is an acid extruder during intracellular acidification during the addition, and an acid loader during the removal, of CO2/HCO3− or butyrate. The rapid fall of Na+i is attributed to the NBCe1 activity starting without delay after the removal of CO2/HCO3− or butyrate, even at the time when the H+i is still falling as a result of CO2 or butyric acid leaving and dominating the change in H+i. This fall in Na+i upon removal of CO2/HCO3− and butyrate was only transient, and Na+i subsequently increased again, presumably attributed to Na+-coupled membrane transporters, activated by the increased Na+ gradient across the membrane but not identified in the present study. The decrease of Na+i in NBCe1-KO cells observed during the addition of either CO2/HCO3− or butyrate may be a result of some moderate activation of the Na+/K+ ATPase, although this was not studied in any more detail in the present study.
Discussion
In the present study, we could identify NBCe1 as the acid/base transporter which is, to a large extent, responsible for the recovery from cytosolic alkalosis, as induced by the removal of either CO2/HCO3− or butyrate from the external solution, in mouse cortical astrocytes. We conclude that NBCe1 is challenged by the cytosolic alkalosis to reverse and carry sodium and bicarbonate out of the cells. Thus, NBCe1 in these cells is not only an acid extruder/base loader as reported previously (Brune et al. 1994; Brookes & Turner, 1994; Bevensee et al. 1997; Theparambil et al. 2014), but also an acid loader/base extruder. For most cells, chloride/base -coupled transporters were suggested to be responsible for regulating pHi from alkalosis; however, in cortical astrocytes, Cl−-coupled transport made only a minor contribution only when the HCO3− gradient induced was large (e.g. during removal of external CO2/HCO3−). At low, submillimolar [HCO3−]i/o, as in the nominal absence of CO2/HCO3−, reversed NBCe1 was still responsible for 65–70% of the rate of acidosis, with the support of CA activity rapidly delivering the substrate HCO3−. Up to 30% of the rate of acidosis could not be assigned to NBCe1 activity and ∼20% of this rate of acidosis was neither Cl−-, nor HCO3−-dependent, and has remained unidentified.
NBCe1 can be an acid loader
From the stoichiometry of NBCe1, the direction of net sodium bicarbonate cotransport at resting state can be predicted (Sciortino & Romero 1999). When NBCe1 was first identified in the basolaterlal membrane of the proximal tubule cells of salamander kidney, the stoichiometry was suggested to be 3 HCO3−:1 Na+ (Boron & Boulpeap 1983). The larger the HCO3−:Na+ coupling ratio, the more effectively the negative membrane potential can push bicarbonate against the inward sodium gradient. Therefore, in kidney epithelial cells, NBCe1 is outwardly-directed at steady-state and functioning as an acid loader.
In the mammalian brain, where NBCe1 is predominantly expressed in astrocytes, NBCe1 operates with a stoichiometry of 2 HCO3−:1 Na+ (Bevensee et al. 1997; Brune et al. 1994, Theparambil et al. 2014), similar to the NBCe1 characterized in the leech giant glial cell (Deitmer & Schlue, 1989; Deitmer & Szatkowski, 1990) but different from Müller cells, a special type of radial glial cell in the retina, where NBCe1 was shown to operate with a stoichiometry 3 HCO3−:1 Na+ (Newman, 1991). The equilibrium potential of NBCe1 (ENBC) as calculated from the 2:1 stoichiometry was found to lie close to the resting membrane potential of cells, (Chesler, 2003; Deitmer & Schlue, 1989; Munsch & Deitmer, 1994; O'Connor et al. 1994), whereas, with the 3:1 stoichiometry, it was near to 0 mV (Newman, 1991).
The present study suggests that NBCe1 in cortical astrocytes mediates an outwardly-directed bicarbonate transport, when [HCO3−]i rises during alkalosis in astrocytes. NBCe1 thus functions as the major acid loader, which regulates H+i from alkalosis in cortical astrocytes. ENBC was calculated from the experimentally measured concentrations of intracellular Na+ and H+/HCO3− at the peak of alkalosis, using the equation (Deitmer & Schlue, 1989):
where R is the universal gas constant, T is the temperature in Kelvin, F is the Faraday constant, z is the total valence of ions, [Na+]o is the extracellular concentration of sodium used (140 mM), [Na+]i is the intracellular concentration of sodium (22.0 ± 0.57 mM) during the removal of butyric acid in CO2/HCO3−-containing solution, [HCO3−]o is the extracellular concentration of bicarbonate (26 mM), [HCO3−]i is the intracellular concentration of bicarbonate (at the peak alkalosis 26.5 mM, as calculated from the peak alkaline pHi after the removal of butyrate) and n is the stoichiometry of NBCe1 (i.e. 2).
At the peak alkalosis, the reversal potential of NBCe1 in astrocytes with the stoichiometry of 2 HCO3−:1 Na+ was calculated to be −46 mV, whereas the membrane resting potential of astrocytes is expected to be more negative (−65 mV to −80 mV) (Kimelberg et al. 1979; Walz & Herz, 1983; Chesler & Kraig, 1989) than the above calculated reversal potential of NBCe1. Therefore, at more negative potentials than ENBC, sodium bicarbonate cotransport via NBCe1 is thermodynamically posed to extrude sodium and bicarbonate. When the membrane potential is more positive than ENBC (e.g. by elevating extracellular K+ from 5 to 15 mM), inwardly-directed bicarbonate transport of NBCe1 would be favoured. In agreement with this, we could show that the rate of acidification and acid/base flux attributed to the outwardly-directed bicarbonate transport of NBCe1 upon lowering external [HCO3−] from 26 to 10 mM was significantly reduced when astrocytes were depolarized by elevating external [K+] from 5 to 15 mM. Similarly, NBCe1-mediated inward current and the rate of H+i rise, in line with electrogenic outward transport of HCO3−, were also reduced at more positive holding membrane potentials in X. laevis oocytes, in which hNBCe1 (with a stoichiometry of 1 Na+ and 2 HCO3−, Becker & Deitmer, 2004) was heterologously expressed. Furthermore, removal of either CO2/HCO3− or butyrate resulted in a transient decrease of Na+i in astrocytes, in line with an outwardly directed NBCe1. The [Na+]i changes and rates of [Na+]i changes measured presumably do not reflect the precise quantity of transported Na+ via the NBCe1 because other Na+-transport processes, in particular the Na+-K+-ATPase, are expected to be active in these cells.
The recovery from intracellular alkalosis was only partly dependent on Cl− in cortical astrocytes
The regulation of H+i from alkalosis in the brain has often been attributed to Cl−-dependent acid-loading transporters such as AEs (Alper, 2009). Neurons have been shown to express AE3, and this carrier was identified as the sole acid-loading transporter in these cells (Hentschke et al. 2006; Kopito et al. 1989; Raley-Susman et al. 1993; Svichar et al. 2009). In astrocytes, functional evidence for Cl−/HCO3− exchange was based on the intracellular alkalosis observed upon the removal of extracellular chloride in bicarbonate-containing solution (Mellergård et al. 1994, Shrode and Putnam, 1994). In the present study, we observed a partial dependency of extracellular Cl− for the H+i regulation from alkalosis (∼30%) only when there was a large outwardly-directed bicarbonate gradient (e.g. during the alkalosis induced by the removal of CO2/HCO3−). It should be noted in the present study that removing CO2/HCO3− initially forms a large outwardly-directed concentration gradient for both CO2 and HCO3−. In line with the low functional activity of Cl−/HCO3− exchange reported in cortical astrocytes, such a large bicarbonate concentration gradient might be required for its activation during alkalosis (Theparambil & Deitmer 2015). However, when alkalosis was induced by the removal of butyrate in the presence or absence of CO2/HCO3−, the regulation of H+i from alkalosis was independent of extracellular Cl−, suggesting a minor contribution of Cl−/HCO3− and/or Cl−/OH− exchange to the regulation of H+i in cortical astrocytes under physiological conditions. These results are in contrast to the generally proposed mechanisms of H+i regulation from alkalosis for many cell types, where acid loading had been proposed to be mediated primarily by Cl−-dependent transporters such as Cl−/HCO3− exchangers or Cl−/OH− exchanger (Brett et al. 2002; Leem and Vaughan Jones 1997, 1998; Ruffin et al. 2014; Casey et al. 2009).
The role of CA activity for the extrusion of bicarbonate at low [HCO3−]
We could show that NBCe1-mediated regulation of H+i from alkalosis was dominant even when the intra- and extracellular concentrations of bicarbonate were very low, as in the nominally CO2/HCO3−-free, Hepes-buffered solution. Although Hepes-buffered solution is generally considered to be CO2/HCO3−-free, it still contains micromolar concentration of bicarbonate (∼180 μM at pH 7.4, as calculated by Henderson–Hasselbalch equation), which is derived from the equilibrated atmospheric CO2 in the solution. Therefore, a rise of [HCO3−]i up to 300 μm was calculated during alkalosis after the removal of butyrate. The rate of regulation from alkalosis in Hepes-buffered solution was significantly reduced in NBCe1-KO astrocytes by ∼70% compared to WT astrocytes. Therefore, we hypothesized that NBCe1 may export bicarbonate even from this very low intracellular concentration and regulate H+i from alkalosis, as attributed to its high bicarbonate sensitivity as previously reported (Theparambil et al. 2014). In agreement with the above hypothesis, we could show a significant reduction in the rate of H+i regulation from alkalosis in WT astrocytes, but not in NBCe1-KO astrocytes, when residual CO2/HCO3− was reduced by continuous aeration of Hepes-buffered solution with 100% O2. Indeed, the acid/base flux rate decreased to approximately half in Hepes-buffered solutions aerated with O2 (Fig. 3E). This corresponds to the finding obtained in NBCe1-expressing Xenopus oocytes, where the NBCe1 transport current was reduced to 50% by reducing residual HCO3− by O2 aeration of the solution (Theparambil et al. 2014). It should be noted that the present study does not indicate any preference for the transported species of NBCe1, bicarbonate (HCO3−) or carbonate (CO32−), as has been discussed repeatedly (Romero et al. 2013). The concentration of carbonate in nominally CO2/HCO3− free saline of pH 7.4 is, however, extremely low.
The presence of intra- and extracellular isoforms of CA appears to be crucial for enhancing the efficacy of CO2/HCO3− buffer system because the catalytic activity of CA accelerates the rate of reversible hydration of CO2 many fold (Maren 1967; Hassan et al. 2013). In addition, CAs are also known to facilitate the transport activity of many acid/base-coupled transporters, including NBCe1 (Becker and Deitmer 2007; Schüler et al. 2011; Becker et al. 2014). In the present study, we have observed that the main intracellular CA isoform, CAII (Hassan et al. 2013), enhances NBCe1-mediated export of bicarbonate in astrocytes, as evaluated from the reduced rate of NBCe1-mediated H+i regulation from alkalosis and the concomitant acid flux rate, when CA activity was either pharmacologically inhibited by 30 μM EZA or when cells from CAII-KO mice were used. It is hypothesized that the rapid production of bicarbonate by CAII activity provides a fast, steady supply of substrate for NBCe1, and thereby enhances its transport activity. Such support of CAII is crucial when the intracellular concentration of bicarbonate is low, especially because the transporter has to carry bicarbonate together with sodium against the large inwardly-directed sodium gradient. In agreement with this hypothesis, inhibition of CAII reduced the rate of NBCe1-mediated H+i regulation from alkalosis in Hepes-buffered, nominally CO2/HCO3−-free solution by 65–70%, and even by 40% in a solution containing 5% CO2/26 mM HCO3−.
Physiological significance of the reversibility of NBCe1 in astrocytes
Astrocytes, the most abundant glial cells in the mammalian CNS, are closely packed with neurons. Indeed, astrocytic processes tightly ensheath many synapses in the brain (Grosche et al. 1999; Theodosis et al. 2008). Such close physical proximity of cells results in narrow extracellular spaces in the brain, which form a platform for an active bidirectional dialogue between glial cells and neurons (Haydon & Carmingnoto, 2006; Halassa & Haydon, 2010). It has been shown that neuronal activity leads to a significant rise in the extracellular concentration of K+ (up to 12 mM) (Kofuji & Newman, 2004),whereas the extracellular [H+] is also known to rise transiently at high frequency stimulation in some brain regions (Kristhal et al.1987; Du et al. 2014). This extracellular K+ rise would depolarize astrocytes, which in turn stimulates NBCe1-mediated uptake of bicarbonate, leading to intracellular alkalosis (Chesler & Craig, 1987; Deitmer & Szatkowski, 1990). The rise of extracellular [H+]/acidification after neuronal activity was suggested to be a result of this glial uptake of sodium bicarbonate and the release of H+ from exocytosed acidic neurotransmitter vesicles (Rose & Deitmer 1994; Ransom, 2000; Miesenbock et al. 1998). In the present study, we hypothesize that, when neuronal activity subsides, and extracellular [K+] recovers to its basal value, the net electrochemical gradient might support the reversal of NBCe1 in astrocytes, as stimulated by a K+-induced rise in [HCO3−]i, a rise in [Na+]i after uptake of the neurotransmitter glutamate, and by extracellular acidification. Reversed NBCe1 activity may regulate pHi from neuronal activity-induced glial alkalosis, and the export of bicarbonate would help to stabilize the pH and enhance H+ buffer capacity in the extracellular spaces. Furthermore, reversed NBCe1 may even cause extracellular alkalosis, which could result in enhanced neuronal excitability and promote epileptiform activity. Astrocytic re-acidification may also be significant for promoting the extrusion of H+-coupled substrates, such as lactate, to the extracellular space from astrocytes, because the enhanced extracellular H+ buffer strength would neutralize the acid equivalents coupled with these substrates. Facilitation of monocarboxylate transporter-mediated lactate transport by NBCe1 was indeed demonstrated when these two proteins were co-expressed in X. laevis oocytes (Becker et al. 2004). Therefore, we propose that the reversed NBCe1 in astrocytes may also link glial and neuronal metabolism in the brain.
Acknowledgments
We thank Dr Gary E. Shull (University of Cincinnati) for providing the transgenic mice (NBCe1-KO).
Glossary
- AE
anion exchanger
- AM ester
acetoxymethyl ester
- ANG-2
asante natrium green-2
- BCECF
2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein
- CA
carbonic anhydrase
- CHE
chloride hydroxyl exchanger
- DIDS
4,4′-diisothiocyanostilbene 2,2′-disulphonic acid
- EZA
6-ethoxy-2-benzothiazolesulphonamide
- KO
knockout
- LSM-510
laser scanning microscope-510
- NBCe1
electrogenic sodium bicarbonate cotransporter 1
- NHE
sodium hydrogen exchanger; S0859, N-cyanosulphonamide
- WT
wild-type
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
SMT, ZN and JWD conceived and designed the experiments. SMT, ZN, AT and JWD collected, assembled, analysed and interpreted the data. SMT and JWD drafted the article.
Funding
This study was supported by a grant from the Deutsche Forschungsgemeinschaft (DE 231/24-2) and by the IRTG 1830 of the DFG to JWD.
References
- Becker HM, Bröer S, Deitmer JW. Facilitated lactate transport by MCT1 when coexpressed with the sodium bicarbonate cotransporter (NBC) in Xenopus oocytes. Biophys J. 2004;86:235–247. doi: 10.1016/S0006-3495(04)74099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker HM, Deitmer JW. Carbonic anhydrase II increases the activity of the human electrogenic Na+/ HCO3− cotransporter. J Biol Chem. 2007;282:13508–13521. doi: 10.1074/jbc.M700066200. [DOI] [PubMed] [Google Scholar]
- Becker HM, Klier M. Carbonic anhydrase and their interplay with acid/base-coupled membrane transporters. In: Frost S, Robert McKenna R, Deitmer JW, editors. Carbonic Anhydrase: Mechanism, Regulation, Links to Disease and Industrial Applications. Vol. 75. 2014. pp. 105–134. Subcell Biochem. [Google Scholar]
- Bevensee MO, Apkon M, Boron WF. Intracellular pH regulation in cultured astrocytes from rat hippocampus. II. Electrogenic Na/HCO3 cotransport. J. Gen. Physiol. 1997;110:467–483. doi: 10.1085/jgp.110.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boron WF, Boulpaep EL. Intracellular pH regulation in the renal proximal tubule of the salamander. Basolateral HCO3− transport. J Gen Physiol. 1983;81:53–94. doi: 10.1085/jgp.81.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett CL, Kelly T, Sheldon C, Church J. Regulation of Cl−/HCO3− exchangers by cAMP-dependent protein kinase in adult rat hippocampal CA1 neurons. J Physiol. 2002;545:837–853. doi: 10.1113/jphysiol.2002.027235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes N, Turner RJ. K+-induced alkalinization in mouse cerebral astrocytes mediated by reversal of electrogenic Na+- HCO3− cotransport. Am J Physiol Cell Physiol. 1994;267:C1633–C1640. doi: 10.1152/ajpcell.1994.267.6.C1633. [DOI] [PubMed] [Google Scholar]
- Brune T, Fetzer S, Backus KH, Deitmer JW. Evidence for electrogenic Na/HCO3 cotransport in cultured rat cerebellar astrocytes. Pflügers Arch. 1994;429:64–71. doi: 10.1007/BF02584031. [DOI] [PubMed] [Google Scholar]
- Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2009;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- Ch'en FF, Villafuerte FC, Swietach P, Cobden PM, Vaughan-Jones RD. S0859, an N-cyanosulphonamide inhibitor of sodium-bicarbonate cotransport in the heart. Br J Pharmacol. 2008;153:972–982. doi: 10.1038/sj.bjp.0707667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M, Kraig RP. Intracellular pH of astrocytes increases rapidly with cortical stimulation. Am J Physiol Regul Integr Comp Physiol. 1987;253:R666–R670. doi: 10.1152/ajpregu.1987.253.4.R666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M, Kraig RP. Intracellular pH transients of mammalian astrocytes. J Neurosci. 1989;9:2011–2019. doi: 10.1523/JNEUROSCI.09-06-02011.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the Brain. Physiol Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Deitmer JW. Electrogenic sodium-dependent bicarbonate secretion by glial cells of the leech central nervous system. J Gen Physiol. 1991;98:637–655. doi: 10.1085/jgp.98.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitmer JW. Evidence for glial control of extracellular pH in the leech central nervous system. Glia. 1992;5:43–47. doi: 10.1002/glia.440050107. [DOI] [PubMed] [Google Scholar]
- Deitmer JW. Acid-base transport and pH regulation. In: Gibson GE, Dienel GA, editors. Handbook of Neurochemistry and Molecular Neurobiology, Vol.5: Brain Energetics. Integration of Molecular and Cellular Processes. Berlin Heidelberg: Springer-Verlag; 2007. pp. 469–486. [Google Scholar]
- Deitmer JW, Rose CR. pH regulation and proton signalling by glial cells. Progr Neurobiol. 1996;48:73–103. doi: 10.1016/0301-0082(95)00039-9. [DOI] [PubMed] [Google Scholar]
- Deitmer JW. Neuron-glia pH regulation. In: Squire LR, Chesler M, editors. New Encyclopedia of Neuroscience, Vol. 6. Oxford: Academic Press; 2009. pp. 739–747. [Google Scholar]
- Deitmer JW, Rose CR. pH regulation and proton signalling by glial cells. Progr Neurobiol. 1996;48:73–103. doi: 10.1016/0301-0082(95)00039-9. [DOI] [PubMed] [Google Scholar]
- Deitmer JW, Schneider H-P. Acid-base transport across the leech giant glial cell membrane at low external bicarbonate concentrations. J Physiol. 1998;512.2:459–469. doi: 10.1111/j.1469-7793.1998.459be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitmer JW, Szatkowski MS. Membrane potential dependence of intracellular pH regulation by identified glial cells in the leech central nervous system. J Physiol. 1990;421:617–631. doi: 10.1113/jphysiol.1990.sp017965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitmer JW, Schlue WR. An inwardly directed, electrogenic sodium-bicarbonate co-transport in glial cells of the leech central nervous system. J Physiol. 1989;411:179–194. doi: 10.1113/jphysiol.1989.sp017567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries SH. Exocytosed protons feedback to suppress the Ca2+ current in mammalina cone photoreceptors. Neuron. 2001;32:1107–1117. doi: 10.1016/s0896-6273(01)00535-9. [DOI] [PubMed] [Google Scholar]
- Du J, Reznikov LR, Price MP, Zha XM, Lu Y, Moninger TO, Wemmie JA, Welsh MJ. Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala. Proc Natl Acad Sci U S A. 2014;111:8961–8966. doi: 10.1073/pnas.1407018111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosche J, Matyash V, Möller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron–glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci. 1999;2:139–143. doi: 10.1038/5692. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan SF, Cornish JL, Goodchild AK. Respiratory, metabolic and cardiac functions are altered by disinhibition of subregions of the medial prefrontal cortex. J Physiol. 2013;591:6069–6088. doi: 10.1113/jphysiol.2013.262071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havenga MJ, Bosman GJ, Appelhans H, De Grip WJ. Expression of the anion exchanger (AE) gene family in human brain. Identification of a new AE protein: AE0. Brain Res Mol Brain Res. 1994;25:97–104. doi: 10.1016/0169-328x(94)90283-6. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Hentschke M, Wiemann M, Hentschke S, Kurth I, Hermans-Borgmeyer I, Seidenbecher T, Jentsch TJ, Gal A, Hübner CA. Mice with a targeted disruption of the Cl−/HCO3− exchanger AE3 display a reduced seizure threshold. Mol Cell Biol. 2006;26:182–191. doi: 10.1128/MCB.26.1.182-191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidtmann H, Ruminot I, Becker HM, Deitmer JW. Inhibiton of monocarboxylate transporter by N-cyanosulphonamide S0859. Eur J Pharmacol. 2015 doi: 10.1016/j.ejphar.2015.05.049. doi: 10.1016/j.ejphar.2015. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Bowman C, Biddlecome S, Bourke RS. Cation transport and membrane potential properties of primary astroglial cultures from neonatal rat brains. Brain Res. 1979;177:533–550. doi: 10.1016/0006-8993(79)90470-0. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Morgans CW, Casey JR, Kopito RR. AE3 anion exchanger isoforms in the vertebrate retina: developmental regulation and differential expression in neurons and glia. J Neurosci. 1994;14:6266–79. doi: 10.1523/JNEUROSCI.14-10-06266.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR, Lee BS, Simmons DM, Lindsey AE, Morgans CW, Schneider K. Regulation of intracellular pH by a neuronal homolog of the erythrocyte anion exchanger. Cell. 1989;59:927–937. doi: 10.1016/0092-8674(89)90615-6. [DOI] [PubMed] [Google Scholar]
- Krishtal OA, Osipchuk YV, Shelest TN, Smirnoff SV. Rapid extracellular pH transients related to synaptic transmission in rat hippocampal slices. Brain Res. 1987;436:352–356. doi: 10.1016/0006-8993(87)91678-7. [DOI] [PubMed] [Google Scholar]
- Leem CH, Vaughan-Jones RD. Chloride-hydroxyl exchange in the guinea-pig ventricular myocyte: no role for bicarbonate. J Mol Cell Cardiol. 1997;29:2483–2489. doi: 10.1006/jmcc.1997.0485. [DOI] [PubMed] [Google Scholar]
- Leem CH, Vaughan-Jones RD. Sarcolemmal mechanisms for pHi recovery from alkalosis in the guinea-pig ventricular myocyte. J Physiol. 1998;509:487–496. doi: 10.1111/j.1469-7793.1998.487bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren TH. Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev. 1967;47:595–781. doi: 10.1152/physrev.1967.47.4.595. [DOI] [PubMed] [Google Scholar]
- McAlear SD, Bevensee MO. pH regulation in non-neuronal brain cells and interstitial fluid. Adv Mol Cell Biol. 2003;31:707–745. [Google Scholar]
- Mellergård P, Ouyang YB, Siesjö BK. Intracellular pH regulation in cultured rat astrocytes in CO2/ HCO3−containing media. Exp Brain Res. 1994;95:371–380. doi: 10.1007/BF00227129. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Munsch T, Deitmer JW. Sodium-bicarbonate cotransport current identified leech glial cells. J Physiol. 1994;474:43–55. doi: 10.1113/jphysiol.1994.sp020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Sodium-bicarbonate cotransport in retinal Müller (glial) cells of the salamander. J Neurosci. 1991;11:3972–3983. doi: 10.1523/JNEUROSCI.11-12-03972.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor ER, Sontheimer H, Ransom BR. Rat hippocampal astrocytes exhibit electrogenic sodium-bicarbonate co-transport. J Neurophysiol. 1994;72:2580–2589. doi: 10.1152/jn.1994.72.6.2580. [DOI] [PubMed] [Google Scholar]
- Raley-Susman KM, Sapolsky RM, Kopito RR. Cl–/ HCO3− exchange function differs in adult and fetal rat hippocampal neurons. Brain Res. 1993;614:308–314. doi: 10.1016/0006-8993(93)91049-x. [DOI] [PubMed] [Google Scholar]
- Ransom BR. Glial modulation of neural excitability mediated by extracellular pH: a hypothesis revisited. Prog Brain Res. 2000;125:217–228. doi: 10.1016/S0079-6123(00)25012-7. [DOI] [PubMed] [Google Scholar]
- Romero MF, Chen AP, Parker MD, Boron WF. The SLC4 family of bicarbonate (HCO3-) transporters. Mol Aspects Med. 2013;34:159–182. doi: 10.1016/j.mam.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Deitmer JW. Evidence that glial cells modulate extracellular pH transients induced by neuronal activity in the leech central nervous system. J Physiol. 1994;481.1:1–5. doi: 10.1113/jphysiol.1994.sp020413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Deitmer JW. Stimulus-evoked changes of extra- and intracellular pH in the leech central nervous system. I. Bicarbonate dependence. J Neurophysiol. 1995a;73:125–131. doi: 10.1152/jn.1995.73.1.125. [DOI] [PubMed] [Google Scholar]
- Rose CR, Deitmer JW. Stimulus-evoked changes of extra- and intracellular pH in the leech central nervous system. II. Mechanisms and maintenance of pH homeostasis. J Neurophysiol. 1995b;73:132–140. doi: 10.1152/jn.1995.73.1.132. [DOI] [PubMed] [Google Scholar]
- Ruffin VA, Salameh AI, Boron WF, Parker MD. Intracellular pH regulation by acid-base transporters in mammalian neurons. Front Physiol. 2014;5:43. doi: 10.3389/fphys.2014.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruminot I, Gutiérrez R, Peña-Münzenmayer G, Añazco C, Sotelo-Hitschfeld T, Lerchundi R, Niemeyer MI, Shull GE, Barros LF. NBCe1 mediates the acute stimulation of astrocytic glycolysis by extracellular K+ J Neurosci. 2011;31:14264–14271. doi: 10.1523/JNEUROSCI.2310-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüler C, Becker HM, McKenna R, Deitmer JW. Transport activity of the sodium bicarbonate cotransporter NBCe1 is enhanced by different isoforms of carbonic anhydrase. PLoS ONE. 2011;6:e27167. doi: 10.1371/journal.pone.0027167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciortino CM, Romero MF. Cation and voltage dependence of rat kidney lectrogenic Na+-HCO3− cotransporter, rkNBC, expressed in oocytes. Am J Physiol. 1999;277:F611–F623. doi: 10.1152/ajprenal.1999.277.4.F611. Renal Physiol. [DOI] [PubMed] [Google Scholar]
- Shrode LD, Putnam RW. Intracellular pH regulation in primary rat astrocytes and C6 glioma cells. Glia. 1994;12:196–210. doi: 10.1002/glia.440120305. [DOI] [PubMed] [Google Scholar]
- Srivastava J, Barber DL, Jacobson MP. Intracellular pH sensors: design principles and functional significance. Physiology. 2007;22:30–39. doi: 10.1152/physiol.00035.2006. [DOI] [PubMed] [Google Scholar]
- Stridh M, Alt M, Wittmann S, Heidtmann H, Aggarwal A, Riederer B, Seidler U, Wennemuth G, McKenna R, Deitmer JW, Becker HM. Lactate fluxes in astrocytes enhanced by a non-catalytic action of carbonic anhydrase II. J Physiol. 2012;590.10:2333–2351. doi: 10.1113/jphysiol.2011.220152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Leem CH, Vaughan-Jones RD. Novel chloride-dependent acid loader in the guinea-pig ventricular myocyte: part of a dual acid-loading mechanism. J Physiol. 1996;495:65–82. doi: 10.1113/jphysiol.1996.sp021574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svichar N, Waheed A, Sly WS, Hennings JC, Hübner CA, Chesler M. Carbonic anhydrases CA4 and CA14 both enhance AE3-mediated Cl−-HCO3− exchange in hippocampal neurons. J Neurosci. 2009;29:3252–3258. doi: 10.1523/JNEUROSCI.0036-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodosis DT, Poulain DA, Oliet SH. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol Rev. 2008;88:983–1008. doi: 10.1152/physrev.00036.2007. [DOI] [PubMed] [Google Scholar]
- Theparambil SM, Deitmer JW. High effective cytosolic H+ buffering in mouse cortical astrocytes attributable to fast bicarbonate transport. Glia. 2015 doi: 10.1002/glia.22829. doi: 10.1002/glia.22829 (in epub) [DOI] [PubMed] [Google Scholar]
- Theparambil SM, Ruminot I, Schneider HP, Shull GE, Deitmer JW. The electrogenic sodium-bicarbonate cotransporter NBCe1 is a high-affinity bicarbonate carrier in mouse cortical astrocytes. J Neurosci. 2014;34:1148–1157. doi: 10.1523/JNEUROSCI.2377-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature. 1990;345:347–350. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- Walz W, Hertz L. Functional interactions between neurons and astrocytes. II. Potassium homeostasis at the cellular level. Prog Neurobiol. 1983;20:133–183. doi: 10.1016/0301-0082(83)90013-8. [DOI] [PubMed] [Google Scholar]