

Abstract
Lynch Syndrome (LS) is an inherited form of colorectal cancer caused by germline mutations in the Mismatch Repair (MMR) genes. It accounts for approximately 5% of all colorectal cancers. The prevalence of Lynch Syndrome among US Hispanics is unknown. The objective of this study was to describe the germline mutations of Lynch Syndrome in Caribbean Hispanics from Puerto Rico and Dominican Republic. A total of 89 subjects were recruited through the Puerto Rico Familial Colorectal Cancer Registry and were classified according to Amsterdam and Bethesda clinical guidelines. For those tumors with lack of expression of MMR protein, gene sequencing was ordered. A total of 35 individuals with deficient MMR system were identified: 22 had MMR mutations and 13 had tumors with absent MMR protein expression. Our results show that the mutation spectrum of Caribbean Hispanic LS patients was composed mostly of MSH2 (66.7 %) mutations, followed by MLH1 (25.0 %). One mutation was identified in MSH6 (8.3 %). A previously unidentified mutation in MLH1 gene c.2044_2045del was found in one Caribbean Hispanic family. MMR mutation-positive individuals were found to be more likely to have a prominent family history of CRC and tumors located at the proximal colon. Compared to MSH2 mutation carriers, MLH1 mutation-positive individuals were more likely to have a strong family history of CRC and LS associated cancers. Furthermore, insurance coverage for genetic testing was found to be limited in the study population with 65.1% of the individuals recruited were denied coverage. This report presents the first description of the mutation spectrum and clinicopathologic characteristics of LS Caribbean Hispanics patients.
Keywords: Lynch syndrome, Hispanics, Mismatch repair system, MLH1, MSH2, MSH6 and/or PMS2
INTRODUCTION
Colorectal cancer (CRC) is a common malignancy among US Hispanics [1]. During the last decade, the mortality rate of CRC in Puerto Rico (PR) has remained high despite advances in medical technology [2]. During the period of 2006–2010, CRC accounted for 13.1% of cancer deaths in males and 13.6% of cancer deaths in females[3]. Thus, efforts dedicated towards identifying individuals with high-risk of developing CRC should be guided towards early screening and/or surveillance in order to reduce the morbidity and mortality associated to CRC. Genetic testing for highly penetrant germline mutations is an excellent option for individuals who belong to high-risk families, helping to establish pre-symptomatic, molecular diagnosis that could affect a patient’s prognosis and treatment options [4].
Lynch Syndrome (LS), also known as hereditary non-polyposis colorectal cancer (HNPCC), is a subset of hereditary CRC that accounts for 2–4% of all CRC cases [5]. LS is inherited in an autosomal dominant pattern, has high penetrance and is characterized by an increased risk of CRC and endometrial cancers [5,6]. Moreover, it accounts for a smaller risk of several other associated malignancies including ovarian, skin, gastric, small bowel, and pancreatic cancer [6]. Previous studies have shown that as many as 1 in every 35 patients newly diagnosed with CRC may have LS [7,8]. Carriers of these mutations have approximately 70% lifetime risk for developing CRC [9,7]. However, regular surveillance can reduce this high cancer risk in LS patients and their relatives [10,11]. Thus, identifying patients with LS provides an opportunity to prevent cancer through intensified screening, surveillance and/or prophylactic surgery for these patients.
LS is associated with germline mutations in the DNA mismatch repair (MMR) pathway genes [12]. The MMR pathway genes, MLH1, MSH2, MSH6 or PMS2, produce fundamental proteins that are responsible for maintaining DNA integrity, correcting nucleotide mismatches that have been overlooked by the usual editing function of DNA polymerase [13]. Loss of MMR function results in an increased mutation rate through the accumulation of polymerase errors, thus increasing genomic microsatellite instability [14]. Strategies for identifying MMR gene mutation carriers include the evaluation of personal and family cancer history using family pedigrees, molecular diagnostic testing of tumors, clinical prediction models, and germline DNA mutational analysis. In this study, we describe the characteristics of a high-risk cancer Caribbean Hispanic population identified through the population-based Puerto Rico Central Cancer Registry (http://www.salud.gov.pr/PRCancer/Pages/default.aspx) and recruited in the Puerto Rico Familial Colorectal Cancer Registry (PURIFICAR).
The aims of this study were to describe the MMR genes mutation spectrum in Caribbean Hispanic families (Puerto Rico and Dominican Republic) with LS and to characterize MMR-deficient and proficient CRC patients with regards to clinicopathologic criteria. Furthermore, we examined the health insurance approval rate for genetic testing in Puerto Rico. Since the MMR mutation spectrum of LS has not been sufficiently studied in any Caribbean Hispanic population, our study will fill this knowledge gap. Studying and describing the results found among Caribbean Hispanics enables the comparison of LS characteristics and mutations across ethnicities and countries. Moreover, due to genetic ancestry background differences among Caribbean Hispanics, compared to other Hispanics groups [15], there is potential for identifying novel MMR mutations associated with LS.
RESEARCH METHODS
Probands for the study were Caribbean Hispanics from Puerto Rico and the Dominican Republic with suspected history of hereditary CRC identified. Caribbean Hispanic was defined as an individual born in Puerto Rico or Dominican Republic. Patients were recruited through the population-based Puerto Rico Central Cancer Registry and recruited in the Puerto Rico Familial Colorectal Cancer Registry (PURIFICAR http://purificar.rcm.upr.edu/index_eng.html). PURIFICAR is located at the University of Puerto Rico Medical Sciences Campus and receives direct or in-kind support by the National Institutes of Health (NCI R03 CA130034), the University of Puerto Rico Comprehensive Cancer Center, the Puerto Rico Colorectal Cancer Coalition (www.coloncancerpr.org), and the Puerto Rico Gastroenterology Association (www.gastropr.org). Recruitment was approved by the Institutional Review Board of the University of Puerto Rico Medical Sciences Campus.
Individuals recruited through PURIFICAR are Caribbean Hispanics with a genetic diagnosis of LS and a genetic and/or clinical diagnosis of Familial adenomatous polyposis (FAP), MYH-associated polyposis (MAP), attenuated FAP, and hamartomatous polyposis syndrome. Recruited probands sign an informed consent and complete a standard questionnaire that includes medical, environmental and detailed family history of cancer. After completion of the questionnaire, probands are referred to the University of Puerto Rico Cancer Genetics Clinic, (led by M.C.C.). Patients attending the clinic receive a variety of services including: genetic counseling and testing, endoscopic surveillance and diagnosis, radiological and surgical procedures, referral to specialists, in addition to direct communication with their medical insurance companies to assist in the approval of services. Blood samples for each participant are obtained and DNA isolated from each sample. For each proband, a family pedigree was completed using PROGENY software (http://www.progenygenetics.com). To confirm the cancer diagnosis of the proband, pathology reports were obtained, in addition to medical and surgical notes from the medical records. Affected family members living in the US, Dominican Republic and/or PR were likewise invited to enroll in PURIFICAR.
The analysis of this study was based on individuals who met Amsterdam Criteria (I or II) or Bethesda clinical guidelines for suspected LS. Amsterdam I criteria are: 3 relatives with CRC, one 1st degree of the other two, at least two consecutive generations affected one relative diagnosed before 50 years of age and FAP is excluded[16]. The Amsterdam II criteria are: 3 relatives affected with LS associated cancers (CRC, endometrial, small bowel), one 1st degree relative of the other two, 2 consecutive generations affected, one LS cancer diagnosed before age 50 years, and FAP excluded in CRC cases [16]. Bethesda guidelines include (only one has to be met): CRC diagnosed before 50 years of age; presence of synchronous, metachronous CRC or other LS associated cancers; CRC with high microsatellite instability (MSI) in a patient 60 years of age or younger; CRC diagnosed in a first degree relative before 50 years of age and CRC diagnosed at any age in two first degree family members [17]. Individuals that did not met Amsterdam I/II or Bethesda clinical guidelines were excluded from the study.
The algorithm used for the selection and molecular testing of the probands is shown in Figure 1, as universal testing is not available in our institution during the study period. In summary, patients with personal history of CRC who met the clinical criteria (Amsterdam I and II and Bethesda guidelines) for LS (probands) had clinical tumor molecular testing for microsatellite instability (MSI) and immunohistochemistry (IHC) for the expression of the MLH1, MSH2, MSH6 and PMS2 proteins when tumor block was available. Genetic testing for the MMR genes was ordered through Myriad Genetics in patients who showed absence of MMR protein expression or when tumor was not available for examination. Large rearrangement testing was performed as part of genetic testing using Multiplex Ligation-dependent Probe Amplification (MLPA) procedure. Furthermore, when genetic testing results showed positive MMR gene mutations, genetic counseling was offered to at-risk relatives.
Figure 1.

Protocol for genetic testing of patients with suspected Lynch syndrome
Statistical Analysis
Sociodemographic and clinical characteristics in cases were described using frequency distributions for categorical variables and summary measures for quantitative variables. To assess comparability of study groups, chi-square test or Fisher’s exact test was used for categorical variables and Student’s t test or Mann-Whitney to compare quantitative variables. Statistical analyses were performed using SPSS version 17.0 (SPSS Inc. Released 2008. SPSS Statistics for Windows, Version 17.0. Chicago: SPSS Inc.) and STATA 12 (StataCorp. 2011. Stata Statistical Software: Release 12. College Station, TX: StataCorp LP).
RESULTS
Between 2010 and 2014 there were 89 CRC Caribbean Hispanic patients, with suspected LS recruited in PURIFICAR and referred to the University of Puerto Rico Clinical Cancer Genetics Clinic. Pedigree information was obtained for each proband; family history of cancer was self-reported. These 89 individuals represented a total of 78 families. Of the recruited individuals, 19 (21.3%) met Amsterdam Criteria (I/II) and 70 (78.7%) met Bethesda guidelines. From the 89 patients recruited who received genetic counseling, 31 had direct MMR germline genetic testing performed at a commercial laboratory and 58 underwent MMR protein analysis by IHC. Among the 31 patients who underwent germline mutation analysis, 22 (71.0%) had a germline mutation in one of the MMR genes (Table 1) and 9 had negative MMR mutation analysis result (Table 2). Among the 58 patients who underwent MMR protein analysis by IHC; 13 (22.4%) had absence of at least 1 MMR protein; 8.6% and 13.7% had absence of MLH1 and MSH2 proteins, respectively (Table 1). Tumor molecular testing for MSI was obtained for 30 out of the 89 suspect LS individuals. Of the 30 individuals tested 73.4% were microsatellite stable and 26.6% were MSI high (data not shown). In summary, a total of 35 individuals with deficient MMR system were identified: 22 had MMR mutations and 13 had tumors with absent MMR protein expression.
Table 1.
Clinicopathologic characteristics of Caribbean Hispanic colorectal cancer patients according to MMR germline mutations and MMR protein-deficient status.
| Study ID | Mutation | Gender | Age at CRC | Tumor Location | Family History of CRC | Family History LS Associated Cancers | Amsterdam (A)/Bethesda (B) |
|---|---|---|---|---|---|---|---|
| MLH1 | |||||||
|
| |||||||
| 8343 | c.1024del16 | F | 43 | Sigmoid | Yes | Yes | A I |
| 9009 | c.2044_2045del | F | 58 | Transverse | Yes | Yes | A II |
| 9009.02 | c.2044_2045del | M | 42 | Transverse | Yes | Yes | A II |
| 9009.03 | c.2044_2045del | M | 42 | Transverse | Yes | Yes | A II |
| 9306 | c.1024del16 | M | 39 | Descending | Yes | Yes | A I |
| 9306.01 | c.1024del16 | F | 35 | Yes | Yes | B | |
| Pte. CUR | c.2044_2045del | M | Yes | Yes | A II | ||
| Pte. MMP | c.1855delG | M | 29 | Right Colon | Yes | No | B |
|
| |||||||
| MSH2 | |||||||
|
| |||||||
| 9106 | c.IVS5-1G>T | F | 47 | Ascending | Yes | Yes | B |
| Pte. DMM | c.IVS5-1G>T | F | Endometrial (44) | Yes | Yes | B | |
| 9109 | c.905T>A | F | 51 | Ascending | No | Yes | A II |
| 9162 | c.1705delGA | F | 42 | Skin | Yes | No | A II |
| 9162.01 | c.1705delGA | F | Yes | No | A II | ||
| 9162.02* | c.1705delGA | F | - | - | Yes | No | A II |
| 9162.03* | c.1705delGA | F | - | - | Yes | No | A II |
| 9249 | c.1457del4 | F | 39 | Rectosigmoid | No | No | B |
| 9258 | c. del exons 1–3 | F | 57 | Yes | Yes | B | |
| Pte. JCG | c. del exons 1–3 | F | 32 | Rectum | Yes | Yes | B |
| 9500 | c.IVS11+1G>A | M | 45 | Yes | No | B | |
| Pte. HFO | c.1308insT | F | 61 | Yes | No | A I | |
| 9560 | c.876insC | M | Yes | No | B | ||
|
| |||||||
| MSH6 | |||||||
|
| |||||||
| 9476 | c.3119delTT | F | 34 | Cecum | Yes | Yes | B |
|
| |||||||
| Immunohistochemistry Analysis for MMR proteins (Absent Protein) | |||||||
|
| |||||||
| MLH1 | |||||||
|
| |||||||
| 9124 | M | 46 | Yes | No | A II | ||
| 9253 | M | 22 | Colon | No | No | B | |
| 9413 | M | 57 | Ascending | Yes | Yes | B | |
| 9420 | M | 30 | Sigmoid | Yes | Yes | B | |
| 9473 | F | 34 | Cecum | Yes | No | B | |
|
| |||||||
| MSH2 | |||||||
|
| |||||||
| 8313 | F | 75 | Rectum | No | Yes | A II | |
| 8397 | F | 59 | Sigmoid | Yes | Yes | A I | |
| 8160 | M | 59 | Ascending | No | Yes | B | |
| 9091.01 | M | 58 | Right Colon | Yes | Yes | B | |
| 9423 | M | 43 | Right Colon | No | Yes | B | |
| 17017 | M | 75 | Rectum | No | Yes | B | |
| 9611 | F | 40 | No | Yes | B | ||
| Pte. ESO | F | 40 | Rectosigmoid | No | Yes | B | |
Mutation carriers.
Table 2.
Clinicopathological characteristics of MMR mutation-negative Caribbean Hispanic colorectal cancer patients.
| Study ID | Gender | Age at CRC | Tumor Location | Family History of CRC | Family History of LS Associated Cancers | Amsterdam (A)/Bethesda *(B) |
|---|---|---|---|---|---|---|
| Mutation Negative | ||||||
|
| ||||||
| 9013 | F | 62 | Rectosigmoid | No | No | B |
| 9105 | F | 41 | Rectosigmoid | No | Yes | B |
| 9108 | F | 35 | Descending | No | No | B |
| 9127 | M | 42 | Splenic flexure | Yes | No | B |
| 9151 | M | 44 | Transverse | No | No | B |
| 9166 | F | 54 | Transverse | Yes | No | A I |
| 9280 | F | 50 | Descending | Yes | No | A I |
| 9285 | F | 74 | Descending | Yes | Yes | B |
| 9292 | F | 47 | Cecum | No | Yes | B |
Lynch Syndrome Cohort
MLH1 Germline Mutations
Mutations on MLH1 accounted for 25.0% of the MMR mutations detected by germline sequence analysis (Table 1). A total of 3 mutations (c.1024del16, c.2044_2045del, c.1855delG) were identified (Table 1). The first mutation, c.1024del16, was previously identified by Pino et al., in the kindred of 3 individuals [18]. The c.1024del16 mutation, found in two unrelated individuals, is located in exon 11 of MLH1. Even though this mutation does not cause a splice site change (same as WT), 16 base pairs are deleted in exon 11 causing a premature truncation (creation of stop codon) of the MLH1 protein at amino acid 361. MLH1 mutation c.2044_2045del, present in exon 18 of the gene, causes the deletion of two nucleotides (AT). The deletion of the AT nucleotides causes a frameshift of the MLH1 protein (p.Met682ValfsX11), by producing a premature stop of the protein 11 amino acids away from the frameshift site. This MLH1 c.2044_2045del mutation was recently described by our group as a novel mutation, unique to Puerto Rico, since no reports are available in international databases [19]. The c.1855delG mutation causes a frameshift at codon 619 in the MLH1 protein (p.Ala619Leufs*18).
MSH2 Germline Mutations
A total of 8 mutations in 8 different families (13 individuals) were found in MSH2 (Table 1).(66.7% of mutations detected). The c.905T>A SNP of MSH2 exon 5, that was described in one individual, results in a premature stop codon in the protein (p.Leu302X). This SNP was described previously by Tomita et al., and was classified as pathogenic due to the changes that occur at the protein level [20]. Another mutation, c.1705delGA, was found in a proband (9162) and 3 additional relatives of the proband. The third mutation identified in MSH2, c.1457del4, was identified in one proband and results in the truncation of the MSH2 protein. Two MSH2 mutations were identified in coding intervening sequences (IVS), which were c.IVS11+1G>A and c.IVS5-1G>T. The variant c.IVS11+1G>A causes a nucleotide substitution adjacent to exon 11 of MSH2, which can affect mRNA processing. MSH2 variant c.IVS5-1G>T causes a nucleotide change at the beginning of exon 6 of MSH2. The MSH2 mutation c. del exons 1–3, was previously identified in a Japanese proband and was associated with anticipation effects in LS [21]. The last two mutations identified were c.1308insT and c.876insC.
MSH6 germline mutations
Only one proband (8.3% of mutations detected) showed a mutation on MSH6 (Table 1). The mutation, c.3119delTT, causes a deletion of two nucleotides TT in exon 7 producing a frameshift of the MSH6 protein (p. Phe1040Terfs).
Clinico-pathological characteristics of MMR-mutation positive LS individuals are described in Table 1. Most MMR mutation-positive individuals were females (75%); there was a high prevalence of family history of CRC (90.5%) and LS associated cancers (61.9%). MMR mutation-positive individuals were diagnosed with CRC between the ages of 29 to 61 and were more likely to meet Amsterdam criteria (54.5%) compared to Bethesda (45.5%). The most significant family history of CRC was for individual 8343, with 8 relatives with CRC including the father, 2 siblings, 3 paternal uncles, 1 paternal aunt, and 1 paternal grandfather (Figure 2). Overall, the most common LS associated cancer was endometrial cancer accounting for 29.2% of the affected relatives. Other LS associated tumors included: 5 patients with stomach cancers, 2 with ovarian cancers, and 1 with small intestine cancer.
Figure 2.
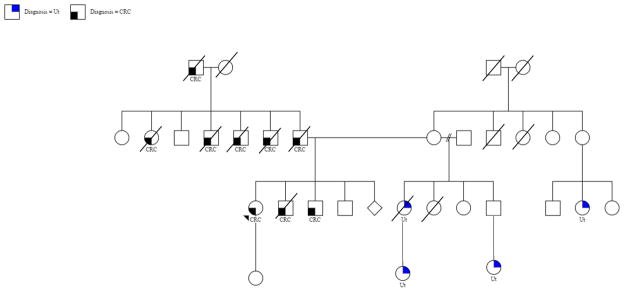
Pedigree with information of family history of cancer for proband 8343.
Characteristics of MMR Mutation-Negative Caribbean Hispanic Patients
Normal germline MMR expression was identified in 9 individuals that underwent genetic testing (Table 2). These individuals where evaluated for all MMR genes (MLH1, MSH2, MSH6, PMS2) and in addition, 5 of the patients were tested for EPCAM mutations demonstrating normal gene sequencing and/or rearrangements. Age at CRC diagnosis for MMR mutation negative individuals ranged from 35 to 74 years of age. Furthermore, mutation negative individuals had less relatives with CRC and LS associated cancers (Table 2). The two individuals (9285 and 9292) that had a family history of LS associated cancers had relatives with pancreatic cancer history (data not shown). MMR mutation negative probands were more likely to meet Bethesda guidelines (77.8%) for LS rather than Amsterdam criteria (Table 2). The decision to undergo direct MMR genetic testing as a first-step instead of tumor molecular screening with MMR IHC was based on patient’s preference and/or lack of tumor availability.
Clinicopathological characterization according to MMR-status
Differences in clinicopathological characteristics according to MMR-status (proficient vs. deficient) for patients who meet Amsterdam Criteria or Bethesda guidelines are described in Table 3. Regardless of their MMR proficiency status, patients had similar ages at CRC diagnosis and gender. Tumor location was identified as significantly predominant in the proximal colon in MMR-deficient individuals, when compared to MMR-proficient that were predominantly distal (57.1% vs. 25.6%; p=0.016). Moreover, presence of family history of CRC was increased in MMR-deficient patients compared to patients with MMR proficient tumors (74.3% vs. 36.5%; p=0.001). Family history of LS associated cancers was reported more frequently among MMR-deficient patients (68.6%) compared to MMR-proficient patients (49.0%), although this difference was not statistically significant (p=0.074) (Table 3).
Table 3.
Comparison of MMR-deficient versus MMR-proficient Caribbean Hispanic patients.
| Clinical Characteristics | MMR Status
|
p-value | |
|---|---|---|---|
| Deficient N = 35 | Proficient N = 54 | ||
| Age at CRC Diagnosis (Mean ± SD) | 46.3± 13.4 | 44.6 ± 12.1 | 0.782a |
| Gender (n(%)) | |||
| Male | 14 (40.0) | 23 (42.6) | 0.808b |
| Female | 21 (60.0) | 31 (57.4) | |
| Tumor Location (n(%)) | |||
| Proximal/Right Colon | 12 (57.1) | 10 (25.6) | 0.016b |
| Distal/Rectum | 9 (42.9) | 29 (74.4) | |
| Family History of CRC (n(%)) | |||
| Yes | 26 (74.3) | 19 (36.5) | 0.001b |
| No | 9 (25.7) | 33 (63.5) | |
| Family History LS Associated Cancers (n(%)) | |||
| Yes | 24 (68.6) | 24 (49.0) | 0.074b |
| No | 11 (31.4) | 25 (51.0) | |
| Amsterdam/ Bethesda Criteria (n(%)) | |||
| Amsterdam I | 4 (11.4) | 3 (5.6) | <0.001c |
| Amsterdam II | 11 (31.4) | 1 (1.9) | |
| Bethesda | 20 (57.1) | 50 (92.6) | |
Mann-Whitney U test;
Pearsons Chi-Square Test;
Fisher’s Exact Test
Differences between LS patients according to affected MMR gene (MLH1 vs. MSH2)
MMR-deficient individuals were grouped according to MLH1 and MSH2 mutation/absent protein status (Table 4). The clinicopathological characteristics evaluated were similar between MLH1 and MSH2 mutation carriers. For MLH1 carriers, we observed that tumor location was predominantly proximal (62.5%), most probands had family history of CRC (92.3%) and LS associated cancers (76.9%) (Table 4). LS patients with deficient MSH2 were mostly females (71.4%) and had less LS associated cancers in their families (58.3%). Age at CRC diagnosis was similar between the two groups; the mean age at diagnosis was 39.7 ± 11.2 and 52.0 ± 12.8 for MLH1 and MSH2 affected individuals, respectively.
Table 4.
Clinicopathologic characterization of Lynch Syndrome patients according to specific MMR-deficient gene.
| Clinical Characteristics | MMR
|
p-value | |
|---|---|---|---|
| MLH1 N = 13 | MSH2 N = 21 | ||
| Age at CRC Diagnosis (Mean ± SD) | 39.7 ±11.2 | 52.0 ± 12.8 | 0.17a |
| Gender (n(%)) | |||
| Male | 8 (61.5) | 6 (28.6) | 0.058b |
| Female | 5 (38.5) | 15 (71.4) | |
| Tumor Location (n(%)) | |||
| Proximal/Right Colon | 5 (62.5) | 6 (50.0) | 0.582b |
| Distal/Rectum | 3 (37.5) | 6 (50.0) | |
| Family History of CRC (n(%)) | |||
| Yes | 12 (92.3) | 13 (61.9) | 0.051b |
| No | 1 (7.7) | 8 (38.1) | |
| Family History LS Associated Cancers (n(%)) | |||
| Yes | 10 (76.9) | 13 (61.9) | 0.074b |
| No | 3 (23.1) | 8 (38.1) | |
| Amsterdam/ Bethesda Criteria (n(%)) | |||
| Amsterdam I | 2 (15.4) | 2 (9.5) | 0.690c |
| Amsterdam II | 5 (38.5) | 6 (28.6) | |
| Bethesda | 6 (46.2) | 13 (61.9) | |
Mann-Whitney U test;
Pearson’s Chi Square Test;
Fisher’s Exact Test
Muir-Torre LS subtype individuals
Muir-Torre (MT) is a rare CRC syndrome considered to be a subtype of LS [22]. In this study, a total of 6 individuals representing 5 families were identified as MT patients, all of which showed skin manifestations (sebaceous carcinoma). All the families had the MSH2 gene affected. A total of 4 families (5 individuals) had mutations in the MSH2 gene that included the following mutations: c.1705delGA, c.IVS5-1G>T, c.1457del4 and c. del exons1–3 (Table 1). One patient (9423) was identified as having absent MSH2 protein as determined by IHC (Table 1).
Genetic Testing Insurance Coverage
After screening, tumor molecular testing was performed (IHC). Genetic testing was recommended for those individuals recruited through PURIFICAR who had CRC and showed absence of MMR proteins through IHC (Figure 1). Of the 89 individuals who qualified based on selection algorithm, insurance coverage for genetic testing was denied to 65.1% (58) individuals. The percentage of denial of insurance coverage was greater among state insurance compared to private insurance (93.3% State insurance, 58.1% private insurance, 13.5% Medicare did not covered genetic test). Health insurance companies did not state reasons for genetic testing coverage denial other than the insurance policy for the patient did not cover genetic testing. Genetic testing was performed through commercial laboratories (including Myriad Genetics Laboratories (http://www.myriadpro.com), Quest Laboratories and University of Puerto Rico Medical Sciences Campus).
DISCUSSION
LS is a hereditary colorectal cancer syndrome that affects approximately 5% of CRC cases [23], with a penetrance of approximately 80% [24]. The characteristics of the LS Caribbean Hispanic cancer patients were detailed in the current study. 35 individuals with deficient MMR system were identified. Of these 35 individuals, 22 had MMR mutations and 13 had tumors with absent MMR protein expression. Furthermore, 9 individuals who satisfied clinical criteria for genetic testing were found to be negative for MMR mutations. Furthermore, we showed that even though MMR-proficient and deficient individuals had similar ages at diagnosis (p=0.782), MMR-deficient individuals present with stronger family history of CRC than their MMR-proficient counterparts. Additionally, most MMR-deficient individuals met Amsterdam criteria I and II as compared to MMR-proficient individuals, which most met Bethesda criteria. As previously described in other non-Hispanic populations, LS Caribbean Hispanic patients had proximal tumors [25], had more relatives with CRC [25–27] and LS associated cancers [26,28,25]. Moreover, the most common MMR gene affected in non-Hispanic populations was MLH1, while among our population of Caribbean Hispanics MSH2 was the most commonly affected gene [21,29–31,25,26].
Dominguez-Valentín et al. published a review pertaining to the mutation spectrum of LS cases in South America [26]. A total of 267 families were included with 99 pathogenic or disease-causing MMR mutations identified [26] The review included LS families from Argentina, Brazil, Uruguay and Colombia, which we will discuss individually [26] Giraldo et al 2005 described MLH1 and MSH2 mutations in a cohort 23 unrelated Colombian families [32]. Similar to Caribbean Hispanics, LS suspect Colombian Hispanics met Bethesda guidelines more frequently (78.7% and 52.2%, respectively) [32]. However, mutations in Colombian families were present more frequently in MLH1 than MSH2 as compared to our cohort [32]. Furthermore, the studies from Uruguay (461 patients) [33] and Brazil (25 families) [34] showed similar results with MLH1 as the most frequently mutated gene in their studies. A study of South American LS families from Brazil (101 families), Argentina (16 families) and Uruguay (6 families), demonstrated that 56.3% of the MMR mutations were found on the MLH1 gene [28]. In contrast, a different report of 43 unrelated Argentinian families that fulfill Amsterdam criteria, for which 5 mutations were encountered, most of these were found at the MSH2 gene, similar to our Caribbean Hispanics [35]. Differences in the spectrum of MMR mutations between Hispanic populations could be due to differences in the, sample size, selection bias, as well as, genetic ancestry of the individual populations. For instance, Caribbean Hispanics have higher percentage of African ancestry compared to Argentinians who have higher European ancestry [36]. Further studies are needed to elucidate the ancestral origin of MMR genes in the populations mentioned above, which can increase the knowledge on the inheritance of LS among affected Hispanic individuals.
The MLH1 mutations described in the study were present in exons 11 and 18, and most caused frameshift mutations of the proteins (Table 1). Interestingly, deletion of exons 1–3 of MSH2 was identified in a Muir-Torre patient (Table 1) and was previously identified in a Japanese patient with LS [21]. One variant of uncertain significance (VUS) was identified in MLH1 in one individual (data not shown). The VUS identified was a nucleotide change in exon 8 of MLH1, which substituted Alanine for a Guanine at nucleotide 655 and causes an amino acid change p.Ile219Val (data not shown). This VUS have been previously described in the literature, however, no clinical significance has been found [37–40]. Recently, our group described a previously unidentified MLH1 mutation, c.2044–2045del, found only in a Caribbean Hispanic family (Table 1) [19]. This novel mutation opens the possibility of founder MMR mutations in Puerto Rico. The novel MLH1 mutation is believed to be transmitted through the maternal line of the proband, however, confirmatory tests could not be performed since most of the maternal family members are deceased [19]. Puerto Ricans are an admixed population of three ancestral populations: European, Africans and Taínos. As evidence by both the historical admixture occurring in the island and mitochondrial DNA analysis, most of the maternal lineage of Puerto Ricans is of Taíno ancestry [41]. Due to genetic ancestry and geographical isolation of Puerto Rico, founder MMR mutations could be present in the island. Additional studies with large sample sizes and improve access to genetic testing will aid in identifying founder MMR mutations in Puerto Rico and understand their role in the carcinogenesis of CRC in the island.
Insurance coverage for genetic testing in hereditary cancer risk assessment has been included in the Affordable Care Act (ACA) [42]. According to ACA, BRCA genetic testing is mandatory for women with breast cancer that meet criteria (early age of onset and strong family history). However, genetic testing for LS is not directly mandated by the Act [42]. In contrast, Medicare covers LS genetic testing for individuals that meet Amsterdam or Bethesda criteria per the National Comprehensive Cancer Network (NCCN) [43]. Recently, the NCCN also published recommendations for universal LS screening for any individual diagnosed with CRC at age 70 or younger using tumor-based molecular genetic testing (IHC for MMR proteins or MSI analysis). In PR, two clinical laboratories are performing all tumor-based molecular genetic testing for LS patients. Even though ACA and Medicare recommend testing for individuals at high-risk, genetic testing in PR is still limited. Our study showed that medical insurance companies did not approve 41.2% of individuals who qualified for genetic testing based on NCCN guidelines. One of the most common reasons for genetic testing denial was the lack of this benefit in the insurance purchased by the companies for their employees. Legislation for information about genetic testing coverage and inclusion of this benefit in accordance to the NCCN guidelines by major health insurance carriers would increase insurance approval rates. Limitation on genetic testing approval has a direct negative impact in the evaluation of patients at risk of hereditary cancer and their relatives, and ultimately increases the burden of cancer for this minority population. Furthermore, due to the high-risk of development for other non-colorectal cancers, suspect LS patients not approved for genetic testing might not receive adequate screening and preventive strategies. Additional studies are needed to understand the negative impact of limited genetic testing on PR.
This study has several limitations. First, although most individuals recruited through PURIFICAR are population-based, the Registry also receives direct referral by physicians and surgeons from the community, which may not adequately represent the general population. Thus, the mutation spectrum of MMR genes in Caribbean Hispanics may be wider and more varied than the one presented in this publication. Second, selection of individuals that meet Amsterdam I/II or Bethesda guidelines versus universal testing can introduce selection bias. NCCN guidelines rely on detailed cancer family history in order for a patient to be classified as Amsterdam I/II or Bethesda. This detailed information on family history is not always available, since clinicians depend on the patient’s memory in order to obtain it. Thus, a portion of LS individuals that do not meet criteria are not identifiable through these guidelines [44]. Studies have indicated that a universal testing approach for LS in newly diagnosed CRC patients is sensitive [44], feasible [8] and cost effective [45,46]. Currently, there are no universal testing protocols being implemented in Puerto Rico and clinicians use the NCCN guidelines to refer patients for IHC/MSI and/or genetic testing. Studies for the adequate implementation of universal testing in Puerto Rico are needed to increase early detection of LS patients in the island. Lastly, genetic testing for LS in PR is inadequate, with significant denials of insurance coverage for individuals meeting NCCN guidelines. Lack of approval not only affects the mutation spectrum seen in PR, but also the patient’s clinical management.
Implementation of protocols for the approval of genetic testing and/or IHC, or protocols for universal testing for LS is necessary to capture the majority of LS cases in PR. In conclusion, we report the mutation spectrum of LS in Caribbean Hispanics, with MSH2 being the most commonly mutated gene in our study population. Moreover, we observed that MMR-deficient individuals had significant family history of CRC, when compared to MMR-proficient patients. Furthermore, we did not observe phenotype differences between MLH1-deficient and MSH2-deficient individuals. Future studies should be directed towards understanding the differences in the mutation spectrum of Caribbean Hispanic LS patients and implementation of widespread clinical guidelines for LS diagnosis in PR, including NCCN guidelines, IHC and genetic testing.
Acknowledgments
This work was partially supported by the National Institute on Minority Health Disparities Award Number 8U54MD 007587-03 and U54MD007587; NCI Award Number 5K22CA115913-03, R21CA167220-01, 5R03CA130034-02, and U54CA096297; Center for Collaborative Research in Health Disparities RCMI Award Number G12MD007600. In addition, the authors will like to thank the patients involved in the study for their treasured contribution.
Footnotes
Compliance with Ethical Standards
None of the authors have any commercial conflicts to disclose. The procedures performed during the research described here were in accordance with ethical standards of responsible human experimentation both institutional and national. Institutional Review Board of the University of Puerto Rico Medical Sciences Campus approved this study. Informed consent was obtained for patients included in the study.
References
- 1.Varela A, Jandorf L, Duhamel K. Understanding factors related to Colorectal Cancer (CRC) screening among urban Hispanics: use of focus group methodology. Journal of cancer education : the official journal of the American Association for Cancer Education. 2010;25 (1):70–75. doi: 10.1007/s13187-009-0015-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soto-Salgado M, Suarez E, Calo WA, Cruz-Correa M, Figueroa-Valles N, Ortiz AP. Incidence and mortality rates for colorectal cancer in Puerto Rico and among Hispanics, non-Hispanic whites and non-Hispanic blacks in the United Staes, 1998–2002. Cancer. 2009;115 (13):3016–3023. doi: 10.1002/cncr.24340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tortolero-Luna G, Zavala-Zegarra D, Perez-Rios N, Torres-Cintron CR, Ortiz-Ortiz KJ, Traverso-Ortiz M, Roman-Ruiz Y, Veguilla-Rosario I, Vazquez-Cubano N, Merced-Velez MF, Ojeda-Reyes G, Hayes-Velez FJ, Ramos-Cordero M, Lopez-Rodriguez A, Perez-Rosa N. Cancer in Puerto Rico, 2006–2013. Puerto Rico Central Cancer Registry 2013 [Google Scholar]
- 4.Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis) Genet Med. 2014;16 (1):101–116. doi: 10.1038/gim.2013.166. [DOI] [PubMed] [Google Scholar]
- 5.Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138 (6):2044–2058. doi: 10.1053/j.gastro.2010.01.054. S0016-5085(10)00168-X [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aarnio M, Mecklin JP, Aaltonen LA, Nystrom-Lahti M, Jarvinen HJ. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer. 1995;64 (6):430–433. doi: 10.1002/ijc.2910640613. [DOI] [PubMed] [Google Scholar]
- 7.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Clendenning M, Sotamaa K, Prior T, Westman JA, Panescu J, Fix D, Lockman J, LaJeunesse J, Comeras I, de la Chapelle A. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26 (35):5783–5788. doi: 10.1200/JCO.2008.17.5950. JCO.2008.17.5950 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, Fix D, Lockman J, Comeras I, de la Chapelle A. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352 (18):1851–1860. doi: 10.1056/NEJMoa043146. 352/18/1851 [pii] [DOI] [PubMed] [Google Scholar]
- 9.Hampel H, Stephens JA, Pukkala E, Sankila R, Aaltonen LA, Mecklin JP, de la Chapelle A. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology. 2005;129 (2):415–421. doi: 10.1016/j.gastro.2005.05.011. S0016-5085(05)00881-4 [pii] [DOI] [PubMed] [Google Scholar]
- 10.Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, Lynch P, Burke W, Press N. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA. 2006;296 (12):1507–1517. doi: 10.1001/jama.296.12.1507. 296/12/1507 [pii] [DOI] [PubMed] [Google Scholar]
- 11.Vasen HF, Blanco I, Aktan-Collan K. Revised guidelines for the clinical management of Lynch Syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013 doi: 10.1136/gutjnl-2012-304356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pritchard CC, Grady WM. Colorectal cancer molecular biology moves into clinical practice. Gut. 2011;60 (1):116–129. doi: 10.1136/gut.2009.206250. gut.2009.206250 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pena-Diaz J, Jiricny J. Mammalian mismatch repair: error-free or error-prone? Trends in biochemical sciences. 2012;37 (5):206–214. doi: 10.1016/j.tibs.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Medina-Arana V, Delgado L, Bravo A, Martín J, Fernández-Peralta AM, González-Aguilera JJ. Tumor spectrum in lynch syndrome, DNA mismatch repair system and endogenous carcinogens. Journal of Surgical Oncology. 2012;106 (1):10–16. doi: 10.1002/jso.23054. [DOI] [PubMed] [Google Scholar]
- 15.Avena S, Via M, Ziv E, Perez-Stable EJ, Gignoux CR, Dejean C, Huntsman S, Torres-Mejia G, Dutil J, Matta JL, Beckman K, Burchard EG, Parolin ML, Goicoechea A, Acreche N, Boquet M, Rios Part M, del C, Fernandez V, Rey J, Stern MC, Carnese RF, Fejerman L. Heterogeneity in genetic admixture across different regions of Argentina. PLoS One. 2012;7(4):e34695. doi: 10.1371/journal.pone.0034695. PONE-D-11-17545 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116(6):1453–1456. doi: 10.1016/s0016-5085(99)70510-x. S0016508599005715 [pii] [DOI] [PubMed] [Google Scholar]
- 17.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96 (4):261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pino MS, Mino-Kenudson M, Wildemore BM, Ganguly A, Batten J, Sperduti I, Iafrate AJ, Chung DC. Deficient DNA mismatch repair is common in Lynch syndrome-associated colorectal adenomas. J Mol Diagn. 2009;11 (3):238–247. doi: 10.2353/jmoldx.2009.080142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marques-Lespier JM, Diaz-Algorri Y, Gonzalez-Pons M, Cruz-Correa M. Report of a Novel Mutation in MLH1 Gene in a Hispanic Family from Puerto Rico Fulfilling Classic Amsterdam Criteria for Lynch Syndrome. Gastroenterology research and practice. 2014;2014:527946. doi: 10.1155/2014/527946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomita N, Fukunaga M, Ohzato H, Tamura S, Sugimoto K, Aihara T, Miki H, Takatsuka Y, Matsuura N, Iwanaga T, Fukayama N, Sugano K. The novel germline mutation of hMSH2 gene in a case of a hereditary non-polyposis colorectal cancer (HNPCC) patient who meets the revised Amsterdam criteria. Japanese journal of clinical oncology. 2003;33 (9):486–489. doi: 10.1093/jjco/hyg082. [DOI] [PubMed] [Google Scholar]
- 21.Stella A, Surdo NC, Lastella P, Barana D, Oliani C, Tibiletti MG, Viel A, Natale C, Piepoli A, Marra G, Guanti G. Germline novel MSH2 deletions and a founder MSH2 deletion associated with anticipation effects in HNPCC. Clin Genet. 2007;71 (2):130–139. doi: 10.1111/j.1399-0004.2007.00745.x. [DOI] [PubMed] [Google Scholar]
- 22.South CD, Hampel H, Comeras I, Westman J, Frankel WL, de La Chapelle A. The frequency of Muir-Torre syndrome among Lynch Syndrome families. J Natl Cancer Inst. 2008;100 (4):277–281. doi: 10.1093/jnci/djm291. [DOI] [PubMed] [Google Scholar]
- 23.De Jesus-Monge WE, Gonzalez-Keelan C, Zhao R, Hamilton SR, Rodriguez-Bigas M, Cruz-Correa M. Mismatch repair protein expression and colorectal cancer in Hispanics from Puerto Rico. Fam Cancer. 2010;9 (2):155–166. doi: 10.1007/s10689-009-9310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lynch H, de La Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet. 1999;36:801–818. [PMC free article] [PubMed] [Google Scholar]
- 25.Alvarez K, Hurtado C, Hevia MA, Wiedlandt AM, de la Fuente M, Church J, Carvallo P, Lopez-Kostner F. Spectrum of MLH1 and MSH2 Mutations in Chilean Families with Suspect Lynch Syndrom. Dis Colon Rectum. 2010;53:450–459. doi: 10.1007/DCR.0b013e3181d0c114. [DOI] [PubMed] [Google Scholar]
- 26.Dominguez-Valentin M, Nilbert M, Wernhoff P, Lopez-Kostner F, Vaccaro C, Sarroca C, Palmero EI, Giraldo A, Ashton-Prolla P, Alvarez K, Ferro A, Neffa F, Caris J, Carraro DM, Rossi BM. Mutation spectrum in South American Lynch syndrome families. Hered Cancer Clin Pract. 11(1):18. doi: 10.1186/1897-4287-11-18. 1897-4287-11-18 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thiffault I, Foulkes WD, Marcus VA, Farber D, Kasprzak L, MacNamara E, Wong N, Hutter P, Radice P, Bertario L, Chong G. Putative common origin of two MLH1 mutations in Italian-Quebec hereditary non-polyposis colorectal cancer families. Clin Genet. 2004;66:137–143. doi: 10.1111/j.1399-0004.2004.00274.x. [DOI] [PubMed] [Google Scholar]
- 28.Valentin MD, da Silva FC, dos Santos EM, Lisboa BG, de Oliveira LP, de Ferreira FO, Gomy I, Nakagawa WT, Aguiar S, Junior, Redal M, Vaccaro C, Valle AD, Sarroca C, Carraro DM, Rossi BM. Characterization of germline mutations of MLH1 and MSH2 in unrelated south American suspected Lynch syndrome individuals. Fam Cancer. 2011;10 (4):641–647. doi: 10.1007/s10689-011-9461-y. [DOI] [PubMed] [Google Scholar]
- 29.Hu F, Li D, Wang Y, Yao X, Zhang W, Liang J, Lin C, Ren J, Zhu L, Wu Z, Li S, Li Y, Zhao X, Cui B, Dong X, Tian S, Zhao Y. Novel DNA variants and mutation frequencies of hMLH1 and hMSH2 genes in colorectal cancer in the Northeast China population. PLoS One. 2013;8(4):e60233. doi: 10.1371/journal.pone.0060233. PONE-D-13-00890 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berzina D, Irmejs A, Kalniete D, Borosenko V, Nakazawa-Miklasevica M, Ribenieks K, Trofimovics G, Gardovskis J, Miklasevics E. Novel germline MLH1 and MSH2 mutations in Latvian Lynch syndrome families. Exp Oncol. 2012;34(1):49–52. 2946 [pii] [PubMed] [Google Scholar]
- 31.Tang R, Hsiung C, Wang JY, Lai CH, Chien HT, Chiu LL, Liu CT, Chen HH, Wang HM, Chen SX, Hsieh LL. Germ line MLH1 and MSH2 mutations in Taiwanese Lynch syndrome families: characterization of a founder genomic mutation in the MLH1 gene. Clin Genet. 2009;75 (4):334–345. doi: 10.1111/j.1399-0004.2009.01162.x. CGE1162 [pii] [DOI] [PubMed] [Google Scholar]
- 32.Giraldo A, Gomez A, Salguero G, Garcia H, Aristizabal F, Gutierrez O, Angel LA, Padron J, Martinez C, Martinez H, Malaver O, Florez L, Barvo R. MLH1 and MSH2 mutations in Colombian families with hereditary nonpolyposis colorectal cancer (Lynch syndrome)--description of four novel mutations. Fam Cancer. 2005;4 (4):285–290. doi: 10.1007/s10689-005-4523-7. [DOI] [PubMed] [Google Scholar]
- 33.Sarroca C, Valle AD, Fresco R, Renkonen E, Peltomaki P, Lynch H. Frequency of hereditary non-polyposis colorectal cancer among Uruguayan patients with colorectal cancer. Clin Genet. 2005;68 (1):80–87. doi: 10.1111/j.1399-0004.2005.00458.x. [DOI] [PubMed] [Google Scholar]
- 34.Rossi BM, Lopes A, Oliveira Ferreira F, Nakagawa WT, Napoli Ferreira CC, Casali Da Rocha JC, Simpson CC, Simpson AJ. hMLH1 and hMSH2 gene mutation in Brazilian families with suspected hereditary nonpolyposis colorectal cancer. Annals of surgical oncology. 2002;9 (6):555–561. doi: 10.1007/BF02573891. [DOI] [PubMed] [Google Scholar]
- 35.Vaccaro CA, Bonadeo F, Roverano AV, Peltomaki P, Bala S, Renkonen E, Redal MA, Mocetti E, Mullen E, Ojea-Quintana G, Benati ML, Rivello HG, Clark MB, Lynch JF, Lynch HT. Hereditary nonpolyposis colorectal cancer (Lynch Syndrome) in Argentina: report from a referral hospital register. Dis Colon Rectum. 2007;50 (10):1604–1611. doi: 10.1007/s10350-007-9037-y. [DOI] [PubMed] [Google Scholar]
- 36.Salari K, Choudhry S, Tang H, Naqvi M, Lind D, Avila PC, Coyle NE, Ung N, Nazario S, Casal J, Torres-Palacios A, Clark S, Phong A, Gomez I, Matallana H, Perez-Stable EJ, Shriver MD, Kwok PY, Sheppard D, Rodriguez-Cintron W, Risch NJ, Burchard EG, Ziv E. Genetic admixture and asthma-related phenotypes in Mexican American and Puerto Rican asthmatics. Genet Epidemiol. 2005;29 (1):76–86. doi: 10.1002/gepi.20079. [DOI] [PubMed] [Google Scholar]
- 37.Marcos I, Borrego S, Urioste M, Garcia-Valles C, Antinolo G. Mutations in the DNA mismatch repair gene MLH1 associated with early-onset colon cancer. The Journal of pediatrics. 2006;148 (6):837–839. doi: 10.1016/j.jpeds.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 38.Sheng JQ, Chan TL, Chan YW, Huang JS, Chen JG, Zhang MZ, Guo XL, Mu H, Chan AS, Li SR, Yuen ST, Leung SY. Microsatellite instability and novel mismatch repair gene mutations in northern Chinese population with hereditary non-polyposis colorectal cancer. Chinese journal of digestive diseases. 2006;7 (4):197–205. doi: 10.1111/j.1443-9573.2006.00269.x. [DOI] [PubMed] [Google Scholar]
- 39.Rahner N, Friedrichs N, Wehner M, Steinke V, Aretz S, Friedl W, Buettner R, Mangold E, Propping P, Walldorf C. Nine novel pathogenic germline mutations in MLH1, MSH2, MSH6 and PMS2 in families with Lynch syndrome. Acta oncologica. 2007;46 (6):763–769. doi: 10.1080/02841860701230217. [DOI] [PubMed] [Google Scholar]
- 40.Campbell PT, Curtin K, Ulrich CM, Samowitz WS, Bigler J, Velicer CM, Caan B, Potter JD, Slattery ML. Mismatch repair polymorphisms and risk of colon cancer, tumour microsatellite instability and interactions with lifestyle factors. Gut. 2009;58 (5):661–667. doi: 10.1136/gut.2007.144220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez-Cruzado JC, Toro-Labrador G, Viera-Vera J, Rivera-Vega MY, Startek J, Latorre-Esteves M, Roman-Colon A, Rivera-Torres R, Navarro-Millan IY, Gomez-Sanchez E, Caro-Gonzalez HY, Valencia-Rivera P. Reconstructing the population history of Puerto Rico by means of mtDNA phylogeographic analysis. Am J Phys Anthropol. 2005;128 (1):131–155. doi: 10.1002/ajpa.20108. [DOI] [PubMed] [Google Scholar]
- 42.Patient Protection and Affordable Care Act (2010). vol 42 U.S.C. USA
- 43.Secretary's Advisory Committee on Genetic Health and Society. Coverage and Reimbursment of Genetic Tests and Services. 2006 http://oba.od.nih.gov/oba/sacghs/reports/CRreport.pdf.
- 44.Pérez-Carbonell L, Ruiz-Ponte C, Guarinos C, Alenda C, Payá A, Brea A, Egoavil CM, Castillejo A, Barberá VM, Bessa X, Xicola RM, Rodríguez-Soler M, Sánchez-Fortún C, Acame N, Castellví-Bel S, Piñol V, Balaguer F, Bujanda L, De-Castro M-L, Llor X, Andreu M, Carracedo A, Soto J-L, Castells A, Jover R. Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut. 2011 doi: 10.1136/gutjnl-2011-300041. [DOI] [PubMed] [Google Scholar]
- 45.Mvundura M, Grosse SD, Hampel H, Palomaki GE. The cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancer. Genet Med. 2010;12 (2):93–104. doi: 10.1097/GIM.0b013e3181cd666c. [DOI] [PubMed] [Google Scholar]
- 46.Ladabaum U, Wang G, Terdiman J, Blanco A, Kuppermann M, Boland CR, Ford J, Elkin E, Phillips KA. Strategies to identify the Lynch syndrome among patients with colorectal cancer: a cost-effectiveness analysis. Annals of internal medicine. 2011;155 (2):69–79. doi: 10.7326/0003-4819-155-2-201107190-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]