

Abstract
Cancers detected at a late stage are often refractory to treatments and ultimately lethal. Early detection can significantly increase survival probability, but attempts to reduce mortality by early detection have frequently increased overdiagnosis of indolent conditions that do not progress over a lifetime. Study designs that incorporate biomarker trajectories in time and space are needed to distinguish patients who progress to an early cancer from those who follow an indolent course. Esophageal adenocarcinoma (EA) is characterized by evolution of punctuated and catastrophic somatic chromosomal alterations and high levels of overall mutations but few recurrently mutated genes aside from TP53. Endoscopic surveillance of Barrett’s esophagus (BE) for early cancer detection provides an opportunity for assessment of alterations for cancer risk in patients who progress to EA compared to nonprogressors. We investigated 1,272 longitudinally collected esophageal biopsies in a 248 Barrett’s patient case-cohort study with 20,425 person-months of follow-up, including 79 who progressed to early-stage EA. Cancer progression risk was assessed for total chromosomal alterations, diversity, and chromosomal region-specific alterations measured with single nucleotide polymorphism arrays in biopsies obtained over esophageal space and time. A model using 29 chromosomal features was developed for cancer risk prediction (Area under receiver operator curve=0.94). The model prediction performance was robust in two independent EA sets and outperformed TP53 mutation, flow cytometric DNA content and histopathologic diagnosis of dysplasia. This study offers a strategy to reduce overdiagnosis in BE and improve early detection of EA and potentially other cancers characterized by punctuated and catastrophic chromosomal evolution.
Keywords: Barrett’s esophagus, Overdiagnosis, Chromosome instability, Risk model, Evolution
Introduction
Vast amounts of time and resources are spent on reducing the burden cancer has on individuals and society. One approach to cancer control is to identify individuals at highest risk of progressing to cancer and interrupt the process with currently available interventions such as resection. Unfortunately, many attempts to reduce cancer mortality through early detection tend to selectively identify individuals with slowly progressing or indolent conditions such as nonprogressing lesions of the esophagus, breast, prostate, thyroid, lung, and skin (1), resulting in overdiagnosis and overtreatment, and concomitantly fail to detect rapidly progressing disease (2).
Overdiagnosis and overtreatment are particularly relevant for persons with Barrett’s esophagus (BE). BE develops in an estimated 2% to 10% of patients who have chronic heartburn (3). In response to an acid and bile reflux environment, BE appears to be a protective adaptation in which the normal, stratified squamous epithelium of the esophagus is replaced by a specialized intestinal metaplastic columnar epithelium (4) with properties that protect the esophagus from reflux injury (3, 5). While BE is the only known precursor to esophageal adenocarcinoma (EA), the absolute lifetime risk of a BE patient developing EA appears to be low with the estimated annual incidence of cancer in large population-based studies ranging from 0.12% to 0.43% (3, 6–8). The vast majority of patients (90–95%) in endoscopic biopsy surveillance programs for early cancer detection will neither be diagnosed with nor die of EA during their lifetime, resulting in overdiagnosis and overtreatment (3, 7). However, if EA is not detected until it is symptomatic, it is usually advanced and incurable with a 5-year survival of less than 15% (9).
The inherently dynamic, stochastic evolutionary processes that lead to cancer and the diversity of somatic genomic mutations and chromosome alterations in cancer make it difficult to identify specific alterations that may be used to predict risk of progression to cancer (10–12). The ideal study to identify robust markers of cancer risk would include a study design with sufficient sample size, spatial and temporal tissue sampling with prospective follow-up, a non-progressing control population with the same precursor condition, and a cancer outcome rather than non-valid surrogate endpoints. Cancer-only studies in EA have provided information about possible targets for treatment of advanced cancers (13–20), but lack non-progressing control populations required for early detection research. Cross-sectional studies have been frequently conducted in which tissue samples are compared across different patients that represent different stages of progression, but these may not be representative of steps in progression in an individual patient given the diversity of genomic alterations present in different EAs (21–24).
Most EAs have extensive chromosomal instability, high levels of chromosome copy number alterations, and frequent catastrophic chromosomal events including whole-genome doublings (13, 15–21, 23, 24). In addition, EA has a high overall mutation frequency and a distinct mutation spectrum, yet with the exception of TP53, no recurrently mutated genes have been identified at high frequency in high-risk BE or EA to be useful as predictors of EA risk (13, 14, 22, 23). BE is an excellent model in which to study the somatic genomic evolutionary process at a stage before the widespread genomic instability that characterizes advanced EA. The clinical practice of periodic endoscopic biopsy surveillance for early cancer detection has allowed the systematic collection of mapped surveillance biopsies sampled over space and time from Barrett’s and normal control tissue in patients who did or did not progress to EA (25, 26). We have recently reported that patients with BE who do not progress to EA typically have low levels of somatic chromosomal alterations (SCA) that remain stable over prolonged periods of time, whereas those who progress to EA develop high levels of SCA, increased diversity, and evolve punctuated chromosome instability and catastrophic whole genome doublings within four years of EA detection (26).
We hypothesized that an EA risk prediction model based on SCA will improve risk stratification of BE patients. We developed cancer risk prediction models using genome-wide SCA assessed over time and space in a large study with longitudinal follow-up and tested whether the risk models improved cancer prediction relative to current risk stratification approaches. These models were derived from a 248 person case-cohort study designed with a cancer endpoint and used longitudinal SCA data from 1,272 biopsies obtained by unbiased sampling at 2 cm intervals throughout the BE segment measured at two time points. The resulting SCA-based risk models were compared to histopathologic assessment of dysplasia, DNA content flow cytometry and TP53 mutations for cancer risk prediction. We show how the model can be applied in practice to deal with stochastic chromosome evolutionary processes during neoplastic progression. These results provide a path forward to identifying BE patients at highest risk for progression to EA who will benefit most from intervention.
Materials and Methods
Detailed methods are presented in Supplementary methods. The cohort study has been approved by the University of Washington Human Subjects Review Committee since 1983. A case-cohort study design (27, 28) was adopted and patients were drawn from a cohort of 516 research participants with histopathologically documented BE at baseline and were followed based on a standard protocol (25). The case-cohort study included 248 individuals followed for 20,425 person-months, including all (n=79) individuals in the cohort who progressed to an endpoint of EA (progressors) and 169 who did not progress to EA during follow-up (nonprogressors) (Supplementary Table S1). Human Omni1-Quad v1.0 SNP arrays were used to assess genome-wide SCA in epithelial isolated cell populations every 2 cm in the BE segment at two time points (T1=baseline, T2=penultimate) per individual (26). Genomes were divided into 3,064 1Mb segments and the frequencies of five SCA types in each 1Mb segment (genome build hg19) and their risk relative (Hazard Ratio) for future cancer were quantified. Three methods were applied independently to identify SCA features for EA risk prediction including bootstrap and ranking, a combination of bootstrap and ranking with Lasso, and Lasso only. Eighty-six regions were selected for EA risk prediction. A 29-feature model (27 specific 1Mb regions representing the 86 regions, and two summation features from these 86 specific 1Mb segments) was built for risk prediction. The 29 features were trained either with T1 data only or T1+T2 data and cross-validated to obtain two EA risk prediction models. The 29 SCA features were also measured using SNP array data from six independent EA surgical specimens and from an independent set of 47 esophageal cancers from TCGA Research Network (29). The performance of the SCA based risk prediction models was compared with TP53 mutations and with dysplasia and DNA content flow cytometry using Receiver operating characteristic (ROC) prediction performance.
Results
Total SCA and SCA diversity
We have shown previously that total SCA and diversity increase in BE progressors at times closer to EA diagnosis (26). Therefore, we assessed the amount of five types of SCA (chromosome copy loss, copy gain, copy neutral loss of heterozygosity “cnLOH”, copy gain with balanced allele ratio “balanced gain”, and homozygous deletion) and SCA diversity throughout the genome as predictors of EA progression in BE at baseline (T1) and at the last endoscopy just prior to or at EA diagnosis in progressors or the penultimate endoscopy in nonprogressors (T2). Increasing amount of total SCA at T1 was associated with increased risk of progression (Fig. 1A). ROC curves were used to assess total SCA performance for EA prediction using biopsies from T1 (Area under ROC curve (AUC)=0.78) or T1 combined with T2 (T1+T2) (AUC=0.80, Fig. 1B). Increasing genome-wide SCA diversity between T1 biopsies in an individual was also associated with increased risk of progression (Fig. 1C) and had similar ROC curves to total SCA (T1 AUC=0.79, T1+T2 AUC=0.80) (Fig. 1D). Thus, total SCA and SCA diversity are overall measures of chromosomal instability that confer an increased risk of progression to EA.
Fig. 1. Total SCA, SCA diversity and EA risk.

(A) Kaplan-Meier survival curves showing cumulative probability to develop EA over time per risk group stratified by total SCA at T1. SCA amount per biopsy was calculated as the sum of all base pairs of SCA from start to end of each alteration. For each patient, the T1 biopsy with the maximum SCA was used. (B) ROC curves showing performance of maximum SCA for EA risk prediction using T1 data only (grey line, AUC=0.78) and T1+T2 combined data (black line, AUC=0.80). (C) Kaplan-Meier survival curves showing cumulative probability to develop EA over time per risk group stratified by maximum SCA diversity across 3,064 1Mb genomic segments at T1. (D) ROC curves showing performance of maximum SCA diversity for EA risk prediction using T1 data only (grey line AUC=0.79), and T1+T2 combined data (black line AUC=0.80). Kaplan-Meier plots were adjusted for case-cohort study (see Supplementary methods).
SCA frequency and hazard ratio
We hypothesized that prediction accuracy could be further improved by identifying only those SCA features selected during development of EA. The case- cohort study was designed to determine the temporal relationship between SCA and patient outcomes while preserving the characteristics of the entire cohort, allowing a cost-effective approach for genomic investigations (26–28). This study design allowed quantification of genome-wide SCA hazard ratios (HR) for risk of progression to EA to distinguish genomic alterations that occurred primarily during progression to cancer from those detected at similar frequencies in nonprogressors (Fig. 2). After SCA calls were made throughout the genome for each biopsy (Supplementary methods), the genome of each sample was divided into 3,064 one megabase (1Mb) segments and each of the five SCA types were called as a binary variable, either present or absent. HRs were calculated for all five SCA types independently at each 1Mb segment. Some high frequency SCA such as frequent loss and homozygous deletion spanning CDKN2A, FHIT, and WWOX had a similar frequency in both nonprogressors and progressors and therefore conferred no or low risk (low HR) of progression to EA (Fig. 2A). In contrast, progressors were characterized by many low to moderate frequency SCA segments with high HRs that were infrequent in nonprogressors such as loss and cnLOH on chromosome 17p linked to TP53 and amplification spanning ERBB2 (Fig. 2B). Large-scale chromosome alterations, coupled with intra- or inter-individual heterogeneity, resulted in a large portion of the genome having significant HRs for one or multiple SCA types in the same chromosomal regions (Fig. 2C).
Fig. 2. SCA frequency and hazard ratio (HR) for EA risk.

SCA types are colored for cnLOH (blue), loss (green), balanced gain (yellow), allele specific copy gain (orange). SCA data were combined from baseline (T1) and last endoscopy before EA diagnosis or final endoscopy (T2) for hazard ratio (HR) estimation. Each data point in the scatter plots represents a 1Mb segment SCA HR for EA (x-axis) vs. its SCA frequency (y-axis) in nonprogressors (A) and progressors (B). The plots show all 1Mb segments in which the frequency in nonprogressors and/or progressors is significantly larger than zero (statistical significance adjusted for multiple comparisons). Many of the 1Mb segments with SCA were correlated due to whole chromosome arm alterations or large chromosomal events spanning many Mb. SCA HR (x-axis) and genomic location (y-axis) are shown in panel C. Regions of significant homozygous deletions with significant HR were small and few in number and therefore not plotted.
SCA feature selection for EA risk prediction
A stepwise feature selection was performed within each of the five SCA types using univariate hazard ratios to identify 1Mb genome segments that were significantly associated (p<0.1) with development of EA. Out of 15,320 possible 1Mb segments representing all five types of SCA throughout the 3,064 Mb genomic segments, 9,391 were significantly associated with progression to EA (Fig. 3). Further feature selection to reduce correlated events resulted in 86 SCA regions for cancer risk prediction (Supplementary Table S2, and Supplementary methods). To reduce the number of variables for EA prediction and minimize overtraining, bootstrapping and feature construction and ranking were used to identify a smaller set of predictors from the 86 SCA regions, resulting in 29 SCA features (Table 1, Fig. 3, and Supplementary methods). These 29 SCA features capture regions indicative of an overall process of genomic instability and take into account large genomic regions that are correlated (co-occur). Thus, any one of the 86 1Mb segments may involve many megabases and may not in and of itself be causative for progression. The robustness of this feature selection approach was supported by two independent feature selection methods (30) (Supplementary Fig. S1 and Supplementary methods).
Fig. 3. Procedures for prediction model feature selection.

Schematic diagraming multi-step approach to SCA data dimension reduction, and feature selection resulting in 29 SCA features to be used for risk prediction models.
Table 1.
Selected SCA features for EA risk prediction models
| Chromosome: (1Mb segment) of SCA location selected in model | SCA type | HR | Frequency of SCA by patient (T1 + T2) in progressors (%)1 | Frequency of SCA by patient (T1 + T2) in nonprogressors (%)1 | Average size contiguous SCA (Mb) spanning selected location in progressors2 (SD) | Average size contiguous SCA (Mb) spanning selected location in nonprogressors2 (SD) |
|---|---|---|---|---|---|---|
| 1:36–37† | loss | >30 | 2.5 | 0.6 | 25.5(31.8) | 14¶ |
| 2: 226–227 | cnLOH | >30 | 8.6 | 0.0 | 48.7 (46.7) | § |
| 5: 93–94 | cnLOH | 7.9 | 11.9 | 0.6 | 102.7 (41.1) | 132¶ |
| 6: 1–2 | gain | 8.2 | 12.7 | 0.6 | 30.1 (26.0) | 32¶ |
| 6: 5–6 | gain | 5.9 | 15.2 | 0.6 | 35.3 (22.7) | 32¶ |
| 6: 29–30 | cnLOH | 5.2 | 14.9 | 3.0 | 24.1 (19.3) | 34.6 (12.3) |
| 6: 146–147 | cnLOH | 8.5 | 6.5 | 0.6 | 91.8 (35.2) | 67.3 (1.2) |
| 7: 77–78 | loss | 23.1 | 6.3 | 0.0 | 74.0 (43.5) | § |
| 7: 78–79 | cnLOH | >30 | 6.5 | 0.0 | 67.6 (38) | § |
| 8:138–139 | cnLOH | >30 | 5.9 | 0.0 | 35.8 (40.1) | § |
| 9: 0–1 | loss | 2.0 | 59.5 | 33.1 | 25.8 (16.1) | 10.2 (12.2) |
| 9: 33–34 | loss | 4.8 | 38.0 | 12.4 | 27.5 (18.1) | 18.5 (13.8) |
| 9: 65–66 | loss | 2.6 | 12.7 | 3.6 | 23.9 (27.4) | 5.1 (1.7) |
| 11: 38–39 | cnLOH | 6.9 | 4.6 | 0.6 | 34.9 (17.8) | 50.5 (0.7) |
| 11: 50–51 | cnLOH | 3.8 | 3.3 | 1.2 | 39.2 (17.7) | 28 (32.5) |
| 11: 110–111 | cnLOH | 4.3 | 6.0 | 1.8 | 55.3 (20.4) | 72.3 (11.3) |
| 12: 45–46 | loss | 4.0 | 11.4 | 2.4 | 39.9 (44.1) | 57.2 (44.2) |
| 13: 42–43 | cnLOH | 18.0 | 17.7 | 0.6 | 81.9 (27.2) | 51¶ |
| 15: 70–71 | gain | 14.4 | 40.5 | 1.8 | 58.6 (22.2) | 68 (19.7) |
| 17: 8–9 | loss | 8.9 | 46.8 | 7.1 | 21.0 (3.5) | 22 (4.2) |
| 17: 9–10 | cnLOH | 8.5 | 24.2 | 2.4 | 20.7 (3.0) | 16 (6.7) |
| 17: 12–13 | cnLOH | 10.8 | 24.2 | 1.8 | 20.4 (4.0) | 22 (1) |
| 17: 37–38 | gain | 12.3 | 34.2 | 2.4 | 23.2 (22.3) | 20.5 (23.9) |
| 18: 19–20 | gain | 4.6 | 60.8 | 21.3 | 16.2 (19.9) | 26.5 (26.0) |
| 19: 48–49 | cnLOH | >30 | 5.8 | 0.0 | 25.0 (9.7) | § |
| X: 42–43 | loss | >30 | 13.9 | 0.0 | 46.5 (19.2) | § |
| Y: 13–14 | loss | 4.6 | 54.4 | 16.6 | 15.3 (3.0) | 13.2 (5.8) |
| Sum of copy loss events in the 86 regions | ||||||
| Sum of all SCA events in the 86 regions | ||||||
The frequency was calculated from all biopsies from both time points within each patient.
The average of the sizes of SCA in a chromosome arm surrounding the SCA locations used in prediction models.
Boundaries for each 1 Mb segment follow the standard as in chromosome 1: 36–37 which includes the nucleotides on chromosome 1 from 36,000,001 to 37,000,000 base pairs on human genome reference hg19.
Only one sample with SCA in this region so no variance in average SCA size can be calculated.
No samples had SCA spanning this region.
Loss = allele specific copy loss; Gain = allele specific copy gain.
The performance of these 29 features for EA risk prediction in BE was evaluated by multiple model training and cross-validation methods (Supplementary Fig. S2 and Supplementary methods). First, the 29 SCA features from T1 were used with one individual patient’s SCA data omitted during each round to train the prediction models. Parameters of the models were averaged to obtain the T1 risk prediction model (T1-model), and the model then was tested for predicting EA outcome using SCA data from T1 (AUC=0.94, Fig. 4A). The performance of predictions using the leave-one-out-sample approach (the Jackknife cross-validation of T1 SCA) showed similar results with slightly lower AUC (AUC=0.86, Supplementary Fig. S3). Next, the 29 SCA features from T2 were used to test the T1-model. T2 biopsies were collected from independent locations in the esophagus on average 64.7 months after T1 biopsies. The T1-model was robust for EA risk prediction when it was tested using 29 SCA features from T2 (AUC=0.84, Fig. 4B).
Fig. 4. ROC curves of EA risk prediction models, histopathology, and DNA content flow cytometry.

ROC curve of the T1-model applied to SCA data from T1 (A) or T2 (B) biopsies to predict EA risk. (C) ROC curve using the T1T2-model to predict EA risk based on maximum SCA calls by patient (T1+T2 data), where the maximum SCA at each of the 29 SCA features was used from any biopsy, regardless of whether it came from T1 or T2,. AUC=area under the ROC curve. (D–F) Histopathology and DNA content flow abnormalities were treated as binary variables to generate ROC curves, with each curve consisting of three points; one with sensitivity equal to 0 (perfect specificity), one with specificity equal to 0 (perfect sensitivity) and the third being the sensitivity and specificity for EA risk prediction of the binary variable. (D) ROC curves of histopathologic diagnosis of high-grade dysplasia (HGD) alone (blue), and low-grade dysplasia (LCD) and/or HGD (red) from 1, 2, 3, or 4 biopsies per 2 cm sampled in BE along the esophagus from T1 data. (E) ROC curves of histopathologic diagnosis of high-grade dysplasia (HGD) alone (blue), and low-grade dysplasia (LGD) and/or HGD (red) from 1, 2, 3, or 4 biopsies per 2 cm sampled in BE along the esophagus from combined T1+T2 data. (F) ROC curves of DNA content flow cytometric assessment of tetraploidy and/or aneuploidy from 1 biopsy per 2 cm in the esophagus from T1 (grey curve) and from combined T1+T2 data (black curve).
Application of longitudinal SCA data for EA risk prediction
We sought to develop a risk model that could be applied to SCA data for risk prediction regardless of whether it was collected from one or multiple time points from an individual patient. The same 29 features used in the T1-model were used to train a model with SCA data from both endoscopic time points (T1 and T2) treated independently to test whether this combined dataset, with roughly twice the number of endoscopies, would improve the accuracy and robustness of EA risk prediction in BE (Supplementary Fig. S2, Supplementary methods). To minimize over-training, a random set of 2/3 of the combined T1 and T2 data were used for model training and the remaining 1/3 were used as a set to test prediction performance (AUC=0.86, Supplementary Fig. S4, Supplementary methods). This procedure was repeated 10,000 times, and the model parameters from these iterations were averaged, resulting in a single “T1T2-model”. This model was applied to a composite (maximum by patient T1+T2) SCA call at each of the 29 SCA features for EA risk prediction (AUC=0.94, Fig. 4C, Supplementary Fig. S2, Supplementary methods). A bootstrap method showed the T1T2-model had consistently higher AUC than the T1-model (bootstrap ranking test p=0.0136, see Supplementary methods). The T1T2-model was trained with data from two time points treated independently and therefore provides flexibility to be used for data collected from either one or multiple time points. The T1T2-model, which generates an EA risk score (predicted probability of EA) ranging from low to high EA risk (0 to 1, Supplementary methods), was used in subsequent analyses.
Testing the prediction models with independent sets of EA samples
Validation of a prediction model would ideally be performed on a large, independent sample set from a separate prospective BE cohort. This is challenging due to the prolonged nature of the study, including some nonprogressors followed for more than two decades, and the relative rarity of EA outcomes without use of surrogate endpoints. However, the T1-model and T1T2-models generated high EA risk scores of ≥0.96 in six advanced EAs from esophagectomy specimens (Supplementary Table S2) and risk scores of >0.99 in 39 of 47 EA samples from TCGA (downloaded September, 2014) (29) from individuals who were not part of the case-cohort study (Supplementary methods).
Comparison of SCA with histopathology, DNA content flow cytometry, and TP53 mutation for EA risk prediction in BE
Histopathological evaluation of surveillance biopsies is the current clinical standard to identify patients at high risk of developing EA. In this study, histopathology was assessed using a standard protocol of four biopsies every two centimeters, whereas SCA was assessed in one biopsy every two cm in the esophagus. To compare the performance of the T1T2-model with histopathological diagnosis of dysplasia, the prediction performance was evaluated for histopathology using 1, 2, 3 or 4 biopsies per two cm intervals for EA risk prediction using only T1 histopathology data (Fig. 4D), and combined T1+T2 histopathology data (Fig. 4E). High-grade dysplasia (HGD) or any dysplasia (either HGD or low-grade (LGD)) were separately evaluated for EA risk prediction. The highest AUC was obtained using 4 biopsies every 2 cm in combined T1+T2 data for HGD only (AUC=0.81), but dropped to a maximum AUC=0.75 with 1 biopsy per 2 cm (from LGD+HGD in combined T1+T2).
DNA content flow cytometry had been previously performed at the same time points in 239 of the 248 participants in this case-cohort study in separate biopsies every two centimeters (31, 32). The performance of flow cytometry for EA risk prediction was evaluated using results from T1 only (AUC=0.75), and combined T1+T2 (AUC=0.79) (Fig. 4F). The T1T2-model utilizing only one biopsy every 2 cm (AUC=0.94) outperformed both dysplasia and DNA content flow cytometry, even when evaluating the histopathology in up to four times the number of biopsies.
TP53 mutations status in exons 5–9 had been previously assessed in a longitudinal study in separate biopsies every two cm in the esophagus at a single time point before last endoscopy or EA diagnosis in 122 participants in this study, including 33 who subsequently progressed to EA (33). In these 122 patients TP53 mutations had 42% sensitivity and 92% specificity for future progression to EA. In comparison, for these 122 patients, the 29-SCA feature model with a 0.6 risk score threshold (Supplementary methods) resulted in 82% sensitivity and 92% specificity for future progression to EA using the T1-model, and 91% sensitivity and 90% specificity for future progression to EA using the T1T2-model
Dynamic stratification of EA risk: illustration of risk management during stochastic evolution to cancer
The stochastic nature of the development of cancer means that a cancer risk assessment at a single point in time may be insufficient (34). To demonstrate how the prediction model might be used for dynamic EA risk stratification over time, the T1T2-model was applied to T1 SCA data to stratify patients into low, intermediate and high risk scores (Fig. 5A, Supplemental methods). The patients with intermediate risk were then further stratified by using T2 SCA data (Fig. 5B). An additional 13 patients, eight of whom ultimately developed cancer, were identified as high risk based on the T2 data. This demonstrated that applying this risk prediction model to SCA data from a second time point increased the number of BE patients who could be stratified into low and high risk groups.
Fig. 5. SCA based risk stratification of BE patients over time.
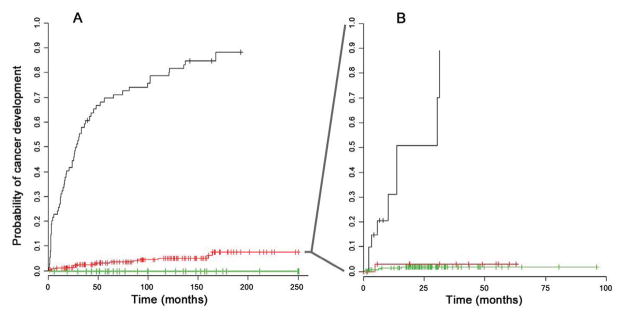
(A) The Kaplan-Meier (KM) curves of BE patients at baseline stratified into three EA risk groups based on the T1T2-model SCA data from T1 biopsies only. Risk groups included 35 patients with low EA risk (green, progressors=0, nonprogressors=35), 147 patients with medium EA risk (red, progressors=17, nonprogressors=130) and 66 with high EA risk (black, progressors=62, nonprogressors=4) (B) The KM curve of the 147 medium risk patients identified at baseline were further stratified into low, medium, and high risk group using the SCA data from T2 biopsies. This resulted in 13 patients changing to low EA risk (green, progressors=1, nonprogressors=12, 117 patients remaining at medium EA risk (red, progressors=5, nonprogressors=112), and 13 patients changing to high EA risk (black, progressors=8, nonprogressors=5). Four patients in the medium risk group from 6A had T1 data only and are plotted in 6A but not plotted in 6B. The proportion of patients stratified in each risk group reflects the makeup of the case-cohort study design which is enriched with progressors; different proportions would be expected when examining the entire cohort. The KM plots were adjusted for case-cohort study (see Supplementary methods).
Discussion
While progress has been made in characterizing cancer genomes, other strategies beyond this catalog are needed to identify markers of future progression for early cancer detection. Our results provide an EA risk prediction model that achieved a 0. 94 AUC using 29 SCA features representative of chromosomal instability from individuals who progress to EA compared to those who remain cancer free during follow-up. Comparing the somatic genomes of progressors before cancer to the genomes of nonprogressors allowed us to identify SCA features that capture high-risk somatic genomic characteristics for accurate EA risk prediction. To our knowledge, this is the first cancer risk prediction model based on longitudinal investigation of genome-wide SCA with consideration of temporal and spatial heterogeneity to account for the dynamic, stochastic evolution in neoplastic progression.
There is a strong rationale for using the process of chromosome instability in BE as a biologically significant measure of risk for future progression to EA, rather than focusing on specific gene mutations. EA has been shown to have a high overall frequency of point mutations, yet with the exception of TP53, few individual genes are recurrently mutated in more than 15% of EAs (14, 20, 22). Most of the genes that are recurrently mutated in EA are also mutated at similar frequency in non-dysplastic BE, HGD, and EA (23). In contrast, the vast majority of EAs have high levels of chromosomal instability (13, 18) and punctuated events in which large regions of the genome are altered and detectable by SNP arrays. Catastrophic events are also common with half of EAs having evidence of genome doublings (19) and nearly a third developing chromothripsis (20). In our study, chromothripsis was detected using SNP arrays (20, 35) (see Supplementary methods) in 13 of 79 progressors (16.5%, CI: 9.4%–26.9%) before detection of EA (data not shown). All 13 patients with chromothripsis had risk scores of 1, indicating the SNP array-based SCA features used in our risk models identify individuals who have undergone punctuated or catastrophic chromosomal events. Our study design allowed us to discount low-risk chromosomal lesions arising in nonprogressors and capture complex chromosomal alterations arising from punctuated and catastrophic events in cancer (19, 20, 36, 37). Therefore, measuring chromosome instability with SNP arrays provided a cost-effective tool for robust assessment of cancer progression risk in individuals with BE.
Whole genome sequencing technology is rapidly becoming accessible for discovery research, but it is currently cost-prohibitive compared to using SNP array technology to assess chromosomal alterations for clinical cancer risk prediction. Our study required measuring SCA in 1,272 biopsies, and SNP arrays provided a relatively inexpensive tool to assess SCA including cnLOH. Previous studies have used fluorescence in situ hybridization to assess copy number alterations, but this technology does not scale-up sufficiently to encompass the 86 regions captured by the 29 SCA features. Additionally, 39 of the 86 regions are cnLOH, which FISH cannot measure. We used a method to enrich for epithelial cells that does not require flow cytometric cell sorting and can readily be performed to reduce normal cell contamination. Our approach should be adaptable to SNP array platforms that have been validated for use in formalin fixed paraffin embedded samples routinely processed in clinical laboratories (38). The 29-feature model measures genomic segments representative of larger or correlated SCA events in somatic genome evolution, thus creating an opportunity for translating progression-associated chromosome instability measures to technology platforms applicable to clinical settings for screening for high-risk BE (23, 39).
Formal criteria for evaluating surrogate biomarkers for disease outcome were developed nearly two decades ago (40). Surrogate markers such as high-grade dysplasia do not meet these criteria (40, 41); they cannot be objectively and reproducibly measured, do not accurately represent the true endpoint (EA) and are variably predictive of cancer with misclassification relative to risk of EA in the published literature ranging from 42% to 84% (3). Despite the poor reproducibility of histopathological evaluation of BE biopsies, histopathology has remained the standard for evaluating risk of progression to EA. Our SCA-based models improved EA risk prediction compared to traditional histopathological assessment of dysplasia using a diagnosis of HGD or combined LGD or HGD as predictors of future EA, even when the histopathological assessment was made in four times as many biopsies as the SCA assessment.
TP53 is the most commonly mutated gene in EA with mutation frequency of 72–81% (13, 14, 20, 22, 23). TP53 mutations can be detected before development of EA in BE with TP53 mutations having 44.1% sensitivity and a 91.4% specificity for future progression to EA base on a previously published longitudinal study (33). Weaver et al. sequenced commonly mutated genes in EA and reported that only TP53 mutations could discriminate HGD from non-dysplastic BE and non-BE controls in a cross-sectional study (23). A recent study found TP53 mutations in 81% of EAs and an additional 9% of the remaining EAs harbored structural alterations inactivating TP53 or amplifying MDM2 (20). We show that using specific features of SCA improves EA risk prediction over general measures of chromosomal instability such as total SCA and flow cytometry. Our results suggest assessing the outcome of disruptions of the TP53 signaling pathway and resultant high-risk chromosomal instability using measures of cancer risk associated chromosomal alterations improves EA risk prediction over using TP53 mutation alone.
Spatial diversity among cell populations within an individual’s Barrett’s segment and stochastic temporal evolutionary dynamics during neoplastic progression are challenges for early detection (10, 26, 34, 42). The majority of progressors (62 of 79) in this study were categorized by the SCA prediction model as high risk for cancer based on results from their T1 endoscopy alone. However, using samples from a second time point identified additional patients whose high risk SCA would have been missed if only a single time point were evaluated. For a new patient entering into the clinic, whether they will progress to EA and their actual time to cancer is unknown. In our study, measurements of SCA at follow-up time points improved assessment of EA risk in BE, especially for individuals with intermediate EA risk at baseline. Therefore, we suggest that models of cancer risk will be improved by incorporating both spatial and temporal dynamics of SCA and somatic genomic heterogeneity within individuals. Additional studies will be required to determine the optimal frequency and timing of sampling required for risk assessment.
There is a paucity of BE cohorts followed to a cancer endpoint available for validation because patients are generally managed using dysplasia with intervention before a cancer diagnosis, precluding longitudinal follow-up. We evaluated the 29-SCA features in six independent cancers and 47 EAs available at the time of manuscript preparation from TCGA (downloaded September, 2014) (29). While not an EA risk prediction validation, it is reassuring that 83% of EAs had risk scores of >0.99 using genotyping and chromosome copy number calls made with TCGA algorithms applied directly in our prediction model. Given that EA is a relatively rare cancer with low population incidence, and biorepositories of fresh frozen specimens collected in a longitudinal cohort are lacking, independent validation in BE will be difficult because many patients with dysplasia are treated, which may alter their disease trajectory. Future validation may be feasible using FFPE optimized SNP arrays such as OncoScan® FFPE or Infinium FFPE DNA Restore Kit with the HumanOmniExpress-FFPE array in formalin-fixed, paraffin embedded samples collected in a longitudinal cohort, or as part of the control arm of a randomized trial with a cancer outcome. Additional validation studies could be performed based on endoscopic “mapping” of EAs and surrounding BE prior to treatment similar to Gu et al (21), but also incorporating controls in endoscopic surveillance who do not progress to EA and have not undergone intervention, to assess the extent to which our EA risk prediction model can reduce over-diagnosis and over-treatment while more accurately defining the patients who will benefit most from therapy.
There are potential limitations to using SNP arrays to assess genomic instability in BE. In this study four progressors had low total SCA (<100 Mb) with low EA risk scores. These individuals may have had biopsies taken before the onset of large-scale chromosomal alterations, had a small, focal, chromosomally unstable cell population that was unsampled, or progressed to EA through alternate pathways such as microsatellite instability (14, 22). Whole genome sequencing has revealed somatic DNA structural changes and combinations of gene mutations that SNP array technologies do not measure (20). Characterization of these events could feasibly be incorporated into our SCA-based risk prediction model. However, the timing of structural alterations before cancer is unknown (14, 22, 23). Future studies measuring structural alterations, punctuated or catastrophic chromosomal events, and/or whole genome mutation rate or mutation spectrum, in addition to the 29-SCA features may improve our EA risk model, thereby extending the early detection window. Univariate analysis at a 1Mb resolution did not identify any genomic segments significantly associated with decreased EA risk. Whole genome sequencing or combinatorial analyses may allow identification of SCA events or breakpoints associated with protection from EA in nonprogressors. Successful translation of these additional measures into a clinically relevant model for cancer risk prediction will require well-designed longitudinal cohort studies with sufficient sample size and a cancer endpoint.
Our approach to measure the process of chromosomal instability may also be successful in common cancer types such as breast, ovary, colon, and lung in which over half are characterized by chromosome instability, genome doublings, and catastrophic chromosomal alterations and for which over- and/or underdiagnosis are also challenges (12, 19, 43, 44). Cancer prevention and control models have been proposed for comprehensive EA incidence and mortality reduction strategies, beginning with general population models and moving toward more specific EA risk stratification tools (3, 45). The importance of any single mutation depends on the underlying inherited genotype, the environment when the mutation arose, and the current tissue architecture (46). An extension of our study will be to develop a comprehensive EA risk management plan that includes EA prevention strategies, host and environmental factors (3, 45, 47–49) and EA risk assessment based on our SCA model, which could then be applied to at risk populations (50). This will be achieved by using either quantitative methods or computer simulations to optimize an objective function that considers risk and benefits of patients (10, 51) and cost at the population level to determine the optimal number of risk groups and the timing of follow-up endoscopies for each risk group, and ultimately translating a measure of chromosomal instability into EA risk management in clinical practice.
Supplementary Material
Acknowledgments
Financial support: National Cancer Institute (NCI) P01CA091955 supported Xiaohong Li, Thomas G. Paulson, Carissa A. Sanchez, Patricia C. Galipeau, Karen Liu, Carlo C. Maley, Mary K. Kuhner, Thomas L. Vaughan, Brian J. Reid and Patricia L. Blount. NCI RC1 CA 146973 supported Xiaohong Li and Brian J. Reid. NCI K05CA124911 and R01 CA136725 supported Thomas L. Vaughan NCI R01 CA140657, R01 CA170595, R01 CA149566, R01 CA185138, CDMRP Breast Cancer Research Program Award BC132057 also supported Carlo C. Maley. NCI P30 CA015704 supported Steven G. Self. Fred Hutchinson Cancer Research Center Institutional Funds also supported Brian J. Reid. University of Washington, Department of Genome Sciences supported Mary K. Kuhner. Funding agencies were not involved in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank all the research participants who have made this study possible and Dave. Cowan for database support and figure preparation.
Footnotes
Conflict of interest statement: The authors disclose no conflicts of interest.
References
- 1.Esserman LJ, Thompson IM, Reid B, Nelson P, Ransohoff DF, Welch HG, et al. Addressing overdiagnosis and overtreatment in cancer: a prescription for change. Lancet Oncol. 2014;15:e234–42. doi: 10.1016/S1470-2045(13)70598-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Welch HG, Black WC. Overdiagnosis in cancer. J Natl Cancer Inst. 2010;102:605–13. doi: 10.1093/jnci/djq099. [DOI] [PubMed] [Google Scholar]
- 3.Reid BJ, Li X, Galipeau PC, Vaughan TL. Barrett’s oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer. 2010;10:87–101. doi: 10.1038/nrc2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang KK, Sampliner RE. Updated guidelines 2008 for the diagnosis, surveillance and therapy of Barrett’s esophagus. Am J Gastroenterol. 2008;103:788–97. doi: 10.1111/j.1572-0241.2008.01835.x. [DOI] [PubMed] [Google Scholar]
- 5.Orlando RC. Mucosal Defense in Barrett’s Esophagus. In: Sharma PSR, editor. Barrett’s Esophagus and Esophageal Adenocarcinoma. 2. Oxford, UK: Blackwell Publishing, Ltd; 2006. pp. 60–72. [Google Scholar]
- 6.Bhat S, Coleman HG, Yousef F, Johnston BT, McManus DT, Gavin AT, et al. Risk of malignant progression in Barrett’s esophagus patients: results from a large population-based study. J Natl Cancer Inst. 2011;103:1049–57. doi: 10.1093/jnci/djr203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hvid-Jensen F, Pedersen L, Drewes AM, Sorensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N Engl J Med. 2011;365:1375–83. doi: 10.1056/NEJMoa1103042. [DOI] [PubMed] [Google Scholar]
- 8.de Jonge PJ, van Blankenstein M, Looman CW, Casparie MK, Meijer GA, Kuipers EJ. Risk of malignant progression in patients with Barrett’s oesophagus: a Dutch nationwide cohort study. Gut. 2010;59:1030–6. doi: 10.1136/gut.2009.176701. [DOI] [PubMed] [Google Scholar]
- 9.Holmes RS, Vaughan TL. Epidemiology and pathogenesis of esophageal cancer. Sem Rad Oncol. 2007;17:2–9. doi: 10.1016/j.semradonc.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Li X, Blount PL, Vaughan TL, Reid BJ. Application of biomarkers in cancer risk management: evaluation from stochastic clonal evolutionary and dynamic system optimization points of view. PLoS Comput Biol. 2011;7:e1001087. doi: 10.1371/journal.pcbi.1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–24. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–33. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dulak AM, Schumacher SE, van Lieshout J, Imamura Y, Fox C, Shim B, et al. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res. 2012;72:4383–93. doi: 10.1158/0008-5472.CAN-11-3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–86. doi: 10.1038/ng.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goh XY, Rees JR, Paterson AL, Chin SF, Marioni JC, Save V, et al. Integrative analysis of array-comparative genomic hybridisation and matched gene expression profiling data reveals novel genes with prognostic significance in oesophageal adenocarcinoma. Gut. 2011;60:1317–26. doi: 10.1136/gut.2010.234179. [DOI] [PubMed] [Google Scholar]
- 16.Nancarrow DJ, Handoko HY, Smithers BM, Gotley DC, Drew PA, Watson DI, et al. Genome-wide copy number analysis in esophageal adenocarcinoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2008;68:4163–72. doi: 10.1158/0008-5472.CAN-07-6710. [DOI] [PubMed] [Google Scholar]
- 17.Frankel A, Armour N, Nancarrow D, Krause L, Hayward N, Lampe G, et al. Genome-wide analysis of esophageal adenocarcinoma yields specific copy number aberrations that correlate with prognosis. Genes Chromosomes Cancer. 2014;53:324–38. doi: 10.1002/gcc.22143. [DOI] [PubMed] [Google Scholar]
- 18.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape ofsomatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nones K, Waddell N, Wayte N, Patch AM, Bailey P, Newell F, et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nature communications. 2014;5:5224. doi: 10.1038/ncomms6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu J, Ajani JA, Hawk ET, Ye Y, Lee JH, Bhutani MS, et al. Genome-wide catalogue of chromosomal aberrations in barrett’s esophagus and esophageal adenocarcinoma: a high-density single nucleotide polymorphism array analysis. Cancer Prev Res (Phila) 2010;3:1176–86. doi: 10.1158/1940-6207.CAPR-09-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y, David S, et al. Comparative Genomic Analysis of Esophageal Adenocarcinoma and Squamous Cell Carcinoma. Cancer Discov. 2012;2:899–905. doi: 10.1158/2159-8290.CD-12-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weaver JM, Ross-Innes CS, Shannon N, Lynch AG, Forshew T, Barbera M, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet. 2014;46:837–43. doi: 10.1038/ng.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Galipeau PC, Sanchez CA, Blount PL, Maley CC, Arnaudo J, et al. Single nucleotide polymorphism-based genome-wide chromosome copy change, loss of heterozygosity, and aneuploidy in Barrett’s esophagus neoplastic progression. Cancer Prev Res (Phila Pa) 2008;1:413–23. doi: 10.1158/1940-6207.CAPR-08-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine DS, Blount PL, Rudolph RE, Reid BJ. Safety of a systematic endoscopic biopsy protocol in patients with Barrett’s esophagus. Am J Gastroenterol. 2000;95:1152–7. doi: 10.1111/j.1572-0241.2000.02002.x. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Galipeau PC, Paulson TG, Sanchez CA, Arnaudo J, Liu K, et al. Temporal and spatial evolution of somatic chromosomal alterations: a case-cohort study of Barrett’s esophagus. Cancer Prev Res (Phila) 2014;7:114–27. doi: 10.1158/1940-6207.CAPR-13-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prentice RL. A case-cohort design for epidemiologic cohort studies and disease prevention trials. Biometrika. 1986;73:1–11. [Google Scholar]
- 28.Self SG, Prentice RL. Asymptotic distribution theory and efficiency results for case-cohort studies. Annals Statistics. 1988;16:64–81. [Google Scholar]
- 29.The Cancer Genome Atlas Research Network. 2014 http://cancergenome.nih.gov.
- 30.Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med. 1997;16:385–95. doi: 10.1002/(sici)1097-0258(19970228)16:4<385::aid-sim380>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 31.Rabinovitch PS, Longton G, Blount PL, Levine DS, Reid BJ. Predictors of progression in Barrett’s esophagus III: baseline flow cytometric variables. American Journal of Gastroenterology. 2001;96:3071–83. doi: 10.1111/j.1572-0241.2001.05261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reid BJ, Levine DS, Longton G, Blount PL, Rabinovitch PS. Predictors of progression to cancer in Barrett’s esophagus: baseline histology and flow cytometry identify low- and high-risk patient subsets. American Journal of Gastroenterology. 2000;95:1669–76. doi: 10.1111/j.1572-0241.2000.02196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galipeau PC, Li X, Blount PL, Maley CC, Sanchez CA, Odze RD, et al. NSAIDs modulate CDKN2A, TP53, and DNA content risk for future esophageal adenocarcinoma. PLoS Med. 2007;4:e67. doi: 10.1371/journal.pmed.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–6. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foster JM, Oumie A, Togneri FS, Vasques FR, Hau D, Taylor M, et al. Cross-laboratory validation of the OncoScan(R) FFPE Assay, a multiplex tool for whole genome tumour profiling. BMC Med Genomics. 2015;8:5. doi: 10.1186/s12920-015-0079-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hagenkord JM, Monzon FA, Kash SF, Lilleberg S, Xie Q, Kant JA. Array-based karyotyping for prognostic assessment in chronic lymphocytic leukemia: performance comparison of Affymetrix 10K2.0, 250K Nsp, and SNP6.0 arrays. J Mol Diagn. 2010;12:184–96. doi: 10.2353/jmoldx.2010.090118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med. 1996;125:605–13. doi: 10.7326/0003-4819-125-7-199610010-00011. [DOI] [PubMed] [Google Scholar]
- 41.Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8:431–40. doi: 10.1002/sim.4780080407. [DOI] [PubMed] [Google Scholar]
- 42.Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–73. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 43.Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014;4:175–85. doi: 10.1158/2159-8290.CD-13-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–40. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thrift AP, Kendall BJ, Pandeya N, Whiteman DC. A model to determine absolute risk for esophageal adenocarcinoma. Clin Gastroenterol Hepatol. 2013;11:138–44 e2. doi: 10.1016/j.cgh.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 46.Gatenby RA, Cunningham JJ, Brown JS. Evolutionary triage governs fitness in driver and passenger mutations and suggests targeting never mutations. Nature communications. 2014;5:5499. doi: 10.1038/ncomms6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelloff GJ, Lippman SM, Dannenberg AJ, Sigman CC, Pearce HL, Reid BJ, et al. Progress in chemoprevention drug development: the promise of molecular biomarkers for prevention of intraepithelial neoplasia and cancer-a plan to move forward. Clin Cancer Res. 2006;12:3661–97. doi: 10.1158/1078-0432.CCR-06-1104. [DOI] [PubMed] [Google Scholar]
- 48.Ek WE, Levine DM, D’Amato M, Pedersen NL, Magnusson PK, Bresso F, et al. Germline genetic contributions to risk for esophageal adenocarcinoma, Barrett’s esophagus, and gastroesophageal reflux. J Natl Cancer Inst. 2013;105:1711–8. doi: 10.1093/jnci/djt303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levine DM, Ek WE, Zhang R, Liu X, Onstad L, Sather C, et al. A genome-wide association study identifies new susceptibility loci for esophageal adenocarcinoma and Barrett’s esophagus. Nat Genet. 2013;45:1487–93. doi: 10.1038/ng.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kadri SR, Lao-Sirieix P, O’Donovan M, Debiram I, Das M, Blazeby JM, et al. Acceptability and accuracy of a non-endoscopic screening test for Barrett’s oesophagus in primary care: cohort study. BMJ. 2010;341:c4372. doi: 10.1136/bmj.c4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X, Blount PL, Reid BJ, Vaughan TL. Quantification of population benefit in evaluation of biomarkers: practical implications for disease detection and prevention. BMC medical informatics and decision making. 2014;14:15. doi: 10.1186/1472-6947-14-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.