

Abstract
This study evaluated the anti-cancer activity and mechanism of action of a γ-tocopherol rich tocopherol mixture, γ-TmT, in two different animal models of estrogen-induced breast cancer. The chemopreventive effect of γ-TmT at early (6 weeks), intermediate (18 weeks) and late (31 weeks) stages of mammary tumorigenesis was determined using the ACI rat model. Female rats receiving 17β-estradiol (E2) implants were administered with different doses (0, 0.05%, 0.1%, 0.3% and 0.5%) of γ-TmT diet. Treatment with 0.3% and 0.5% γ-TmT decreased tumor volume and multiplicity. At 31 weeks, serum concentrations of E2 were significantly decreased by γ-TmT. γ-TmT preferentially induced expression of the E2 metabolizing enzyme CYP1A1, over CYP1B1 in the rat mammary tissues. Nrf2-dependent antioxidant response was stimulated by γ-TmT, as evident from enhanced expression of its downstream targets, NQO1, GCLM and HMOX1. Serum concentrations of the oxidative stress marker, 8-isoprostane, were also decreased in the γ-TmT treated groups. Treatment with γ-TmT increased expression of PPARγ and its downstream genes, PTEN and p27, while the cell proliferation marker, PCNA, was significantly reduced in γ-TmT treated mammary tumors. In an orthotopic model in which human MCF-7 breast cancer cells were injected into the mammary fat pad of immunodeficient mice, γ-TmT inhibited E2-dependent tumor growth at all the doses tested. In conclusion, γ-TmT reduced mammary tumor development, in part through decreased E2 availability and reduced oxidative stress in mammary tissues; γ-TmT could thus be an effective agent for the prevention and treatment of E2-induced breast cancer.
Keywords: Breast cancer, estrogen, oxidative stress, antioxidant response, PPARγ
INTRODUCTION
Breast cancer is one of the most common malignancies in women worldwide and the second leading cause of cancer death among women in the United States (1). Prolonged exposure to estrogens has been associated with an increased risk of breast cancer (2–4). However, the precise mechanisms by which estrogens exert their carcinogenic effects are not well understood. It has been suggested that estrogens induce mammary tumor formation via two distinct mechanisms; namely, estrogen-receptor (ER)-dependent and ER-independent pathways (2–6). The ER-dependent pathway involves cell growth and proliferation triggered by the binding of estrogen to the ER (2, 4). The ER-independent mechanism depends on metabolism of endogenous estrogens by cytochrome P450 (CYP) enzymes to generate highly reactive genotoxic metabolites (3). In breast epithelium, estrogens are converted to their 2-OH and 4-OH catechol metabolites by CYP1A1 and CYP1B1, respectively (3, 5). These catechol estrogens are further oxidized to electrophilic quinones which have the potential to cause oxidative DNA damage (7). Furthermore, redox cycling of quinones results in the formation of free radicals and reactive oxygen species (ROS), which are capable of damaging cellular biomolecules like DNA, protein and lipids (8, 9). A growing body of evidence strongly supports the role of oxidative stress generated by estrogen metabolism in the initiation of mammary carcinogenesis (10–12). Estrogen-induced carcinogenesis might therefore be prevented through attenuated formation or increased detoxification of catechol estrogens, as well as trapping or clearance of quinones and ROS. Several phase II detoxification and antioxidant enzymes regulated by the transcription factor nuclear factor (erythroid derived 2)-like 2 (Nrf2) have been suggested to be protective against oxidative stress induced by estrogens (13, 14). Thus, diets rich in antioxidants could be effective for the prevention of estrogen-induced breast cancer.
Vitamin E is a family of fat soluble phenolic compounds consisting of a chromanol ring and a side chain (15). They occur in eight different forms, four tocopherols (with a saturated phytyl tail) and four tocotrienols (with an unsaturated isoprenoid side chain) designated as α, β, γ and δ, respectively (15). It has been suggested that tocopherols can reduce the risk of cancer due to their strong antioxidant properties (16, 17). Several studies show that a lower vitamin E nutritional status is associated with an increased risk of certain cancers (17, 18). In contrast, some large scale clinical trials with α-tocopherol (α-T) have failed to demonstrate its cancer preventive activity (19, 20). Such conflicting outcomes raise questions about the anti-cancer potential of α-T and emphasize the need to investigate the biological activity of other forms of tocopherols in cancer prevention.
γ-Tocopherol (γ-T) is the most abundant tocopherol in the U.S. diet, found in vegetable oils such as soybean, corn and cottonseed (21). γ-TmT, a byproduct of refined vegetable oil, is a naturally occurring tocopherol mixture rich in γ-T (22). γ-TmT has been recently shown to inhibit colon, lung and breast carcinogenesis in animal models (22–24). We have previously reported that γ-TmT suppresses N-methyl-N-nitrosourea (NMU) induced mammary tumor growth in Sprague Dawley rats (24, 25). Since estrogen-mediated animal models of breast cancer are probably more physiologically relevant to human breast cancer, August-Copenhagen Irish (ACI) rats, which exhibit 80–100% tumor incidence after chronic estrogen exposure (26) were utilized in the current study to determine the anti-cancer effects of γ-TmT and further investigate the role of tocopherols in estrogen-mediated events. In earlier studies, we demonstrated that γ-TmT inhibits oxidative/nitrosative stress and reduces cell proliferation in estrogen induced mammary hyperplasia (27, 28). However, the protective effects of γ-TmT during mammary tumorigenesis have not been evaluated.
In the present study, we determined the efficacy of dietary γ-TmT on estrogen-induced mammary tumors in two different in vivo models. In the first study, ACI rats supplemented with estrogen in silastic tubing were utilized to assess the chemopreventive effect of γ-TmT at early (6 weeks), intermediate (18 weeks) and late (31 weeks) stages of mammary tumorigenesis. The mechanism of action of γ-TmT was investigated with respect to regulation of estrogen metabolizing enzymes, antioxidant response, nuclear receptor signaling and cell proliferation. In a second study using an orthotopic model, immunodeficient mice supplemented with estrogen pellets were xenografted with MCF-7 cells and fed with γ-TmT. We demonstrate that γ-TmT inhibits estrogen-induced mammary tumor growth in both ACI rats and MCF-7 xenografts.
MATERIALS AND METHODS
Diets
Natural γ-TmT was obtained from BASF Corporation (Kankakee, Illinois; Covi-ox T-90, Batch number 0008778732). It contained 56.1% γ-T, 22.3% δ-T, 11.5% α-T and 1.2% β-T. Semi-purified diet AIN-93M obtained from Research Diets, Inc. (New Brunswick, NJ) was used as the control diet. Experimental diets were prepared by adding required percentages of γ-TmT to the AIN-93M diet. The diets were stored at 4°C in sealed containers and food cups were replenished with fresh pellets twice a week.
Animals and experimental procedures
Female ACI rats were purchased from Harlan Laboratories (Indianapolis, IN) at 6–7 weeks of age. After two weeks of acclimatization, the rats were subcutaneously implanted with silastic tubing filled with 9 mg of 17β-estradiol (E2) (Sigma-Aldrich, St. Louis, MO) or sham implants, following a previously described method (29). Rats receiving sham implants were fed with control diet. E2 implanted rats were fed with control diet or diets containing different concentrations of γ-TmT: 0.05%, 0.1%, 0.3% and 0.5%. Diets were administered from the day of E2 implantation. Rats were sacrificed at three time points – 6 weeks, 18 weeks and 31 weeks. For 6 and 18 weeks, each treatment group consisted of 6 rats while for 31 weeks 30 rats were included per group. Body weight of the animals was measured weekly. Rats were palpated for mammary tumors weekly starting from 18 weeks after E2 implantation. Blood was collected at autopsy and serum stored at −80°C. Mammary glands, tumors and liver were frozen or fixed in 10% formalin for further analysis. For MCF-7 xenograft tumor studies, female nu/nu mice were purchased from Charles River Laboratories. After two weeks of acclimatization, the mice were implanted subcutaneously with 0.72 mg E2 pellets (Innovative Research of America, Sarasota, FL). Two days following E2 implantation, MCF-7 cells (5 × 106) were orthotopically injected into the mammary fat pad of the mice and different doses of γ-TmT diet (0.05%, 0.1%, 0.3% and 0.5%) were administered. Tumor volume was measured twice a week. Body weight was recorded weekly. The mice were sacrificed after 9 weeks of γ-TmT treatment.
Analysis of tocopherols levels in the serum and mammary glands of rats
The levels of tocopherols (α, δ, γ) and their metabolites in rat serum were analyzed by high performance liquid chromatography using previously described methods (22, 30).
Serum estradiol levels
E2 levels in the serum were analyzed using an ELISA kit from Calbiotech, Inc (Spring Valley, CA). The assay was performed according to manufacturer’s protocol. The assay was based on the principle of competitive binding between E2 in the test specimen and E2-enzyme conjugate for a constant amount of anti-estradiol polyclonal antibody. The standard curve was obtained from samples of E2 in the range of 3–300 pg/ml provided with the kit.
mRNA expression analysis using quantitative polymerase chain reaction (qPCR)
RNA was extracted from frozen mammary glands, tumors and liver. Reverse transcription and qPCR was performed as previously described (31). Labeled primers were used for cytochrome P450 1A1 (CYP1A1), cytochrome P450 1B1 (CYP1B1), nuclear factor (erythroid derived 2)-like 2 (Nrf2), NAD(P)H dehydrogenase quinone 1 (NQO1), glutamate cysteine ligase, modifier subunit (GCLM), heme oxygenase 1 (HMOX1), estrogen receptor (ER) α, ERβ, peroxisome proliferator-activated receptor γ (PPARγ), phosphatase and tensin homologue (PTEN), p27 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Western blot analysis
Mammary tumors were homogenized and the protein extracts analyzed by western blot as previously described (24). Primary antibodies against CYP1A1 (sc-25304), CYP1B1 (sc-32882), Nrf2 (sc-722), NQO1 (sc-3793), GCLM (sc-55585) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); HMOX1 (ab68477) was from Abcam (Cambridge, MA). One tumor sample from three different animals in each treatment group was pooled for analysis.
Immunohistochemical analysis
Mammary tumors were fixed in 10% formalin, embedded in paraffin and sectioned at 4 μm thickness. Sections were incubated overnight at 4°C with antibody to proliferating cell nuclear antigen (PCNA) (1:8000; M 0879, Dako, Denmark), followed by incubation with biotinylated secondary antibody and avidin/biotin peroxidase complex. The sections were then stained with 3′-diaminobenzamine substrate and counterstained with Modified Harris Haematoxylin. Tumor sections from three different animals per treatment group were stained. Representative images (40X magnification) were taken randomly and staining density was quantified using an Aperio ScanScope (Vista, CA) by counting at least 15,000 cells per slide.
Enzyme immunoassay for 8-isoprostane
EIA kit from Cayman Chemicals (Ann Arbor, MI) was used to determine 8-isoprostane levels. For the extraction of 8-isoprostane, serum samples were mixed with ethyl acetate, vortexed for 30 minutes and centrifuged at 10,000×g for 20 minutes. The organic layer was collected and dried using a speed vacuum evaporator. The dried samples were reconstituted in EIA buffer and the assay was performed following the manufacturer’s protocol.
Statistical analysis
Tumor-free survival (TFS), or time to appearance of the first tumor, was estimated by the Kaplan-Meier method. Log-rank test was used to assess the homogeneity of TFS between different treatment groups. Tumor multiplicity was analyzed using log-linear model (Poisson regression). Statistical significance was evaluated using one way analysis of variance model (ANOVA) followed by Dunnett’s multiple comparison post test, preserving the overall type-1 error at the 5% level. The data are represented as ± S.E. Differences were considered statistically significant when P < 0.05.
RESULTS
Dietary γ-TmT inhibits estrogen-induced mammary tumorigenesis in ACI rats
Female ACI rats implanted with E2 were fed with control or diets supplemented with different concentrations of γ-TmT. TFS for each treatment group was estimated (Figure 1A). The median time to the appearance of first tumor in the E2 control group was 26.5 weeks. In the 0.05%, 0.1%, 0.3% and 0.5% γ-TmT treatment groups, the median tumor free survival time was 25, 28, 27 and 29 weeks, respectively. At the end of the 31-week study, there was no overall difference in tumor free survival time between the E2 control and any of the γ-TmT treated groups (Figure 1A). Tumor volume was measured at autopsy (Figure 1B). While the treatment with two lower doses of γ-TmT (0.05% and 0.1%) had no effect on the size of E2-induced mammary tumors, 0.3% and 0.5% γ-TmT reduced tumor volume by 52% (p < 0.01) and 42% (p < 0.01), respectively. Tumor multiplicity was also reduced in the 0.3% and 0.5% γ-TmT groups (Figure 1C); in the E2 control, average tumor multiplicity was 4.4 ± 0.1 per rat while treatment with 0.3% and 0.5% γ-TmT decreased tumor multiplicity to an average of 2.7 ± 0.3 (p < 0.01) and 3.0 ± 0.2 (p < 0.05) tumors per rat, respectively. Average body weights of rats at 31 weeks were not affected by E2 or different doses of γ-TmT treatment, indicating that none of the treatments were toxic for the given duration (Figure 1D).
Figure 1.

γ-TmT inhibits estrogen-induced mammary tumorigenesis in ACI rats. ACI rats implanted with E2 were fed with control diet or diet containing different doses of γ-TmT for 31 weeks (n=30/group). (A) The tumor-free survival curve of each group is shown. (B) Average tumor volume of the different treatment groups at 31 weeks is shown. (C) Average tumor multiplicity of each group at 31 weeks is shown. (D) Average body weights of each treatment group at 31 weeks are shown. P-values are compared to E2 control. Statistical significance, *P < 0.05, **P < 0.01.
Administration of dietary γ-TmT increases the serum levels of tocopherols and their metabolites in ACI rats
To determine the serum levels of tocopherols in ACI rats fed with different doses of γ-TmT for 6, 18 and 31 weeks, serum samples collected at autopsy were analyzed for γ-, δ- and α-T and their respective short chain metabolites, carboxyethyl hydroxychromans (CEHCs) and carboxymethylbutyl hydroxychromans (CMBHCs) (Table 1). At 18 and 31 week time points, a dose-dependent increase in the serum levels of γ- and δ-T was observed in response to γ-TmT administration. The concentration of γ- and δ-T and in the 0.5% γ-TmT treated rat serum was also time-dependent, increasing progressively with 6, 18 and 31 weeks of γ-TmT supplementation. At 31 weeks, the rats fed with 0.3% and 0.5% γ-TmT exhibited a 66- and 134-fold increase in δ-T, while γ-T levels increased by 27- and 46- fold, respectively. Serum levels of α-T were increased in the 0.3% and 0.5% γ-TmT groups at 18 and 31 weeks. However, the fold increase in α-T was modest since γ-TmT has a lower content of α-T compared to δ- and γ-T. γ-TmT supplementation also increased the serum levels of γ-, δ-, α-CEHC and CMBHC metabolites in the serum of ACI rats within individual time points. At all three time points, γ- and δ-CEHC and CMBHC metabolites increased dose-dependently in response to γ-TmT administration, and the levels of these metabolites were higher at 18 and 31 weeks compared to 6 weeks. Overall, the CEHC metabolites were more abundant than the CMBHC metabolites in the serum.
Table 1.
Analysis of tocopherols and their metabolites in the serum of ACI rats.
| Time | Treatment | γ-T (μM) | δ-T (μM) | α-T (μM) | γ-CEHC (μM) | δ-CEHC (μM) | α-CEHC (μM) | γ-CMBHC (μM) | δ-CMBHC (μM) | α-CMBHC (μM) |
|---|---|---|---|---|---|---|---|---|---|---|
| 6 weeks | Negative control | 0.2 ± 0.0 | 0.1 ± 0.0 | 16.6 ± 2.1 | 0.1 ± 0.0 | 0.3 ± 0.1 | 0.8 ± 0.2 | 0.001 ± 0.0 | 0.01 ± 0.0 | 0.002 ± 0.0 |
| E2 control | 0.3 ± 0.0 | 0.1 ± 0.0 | 40.2 ± 2.7 | 0.3 ± 0.1 | 0.4 ± 0.1 | 9.2 ± 2.2 | 0.002 ± 0.0 | 0.02 ± 0.0 | 0.03 ± 0.0 | |
| E2 + 0.05% γ-TmT | 1.6 ± 0.1 *** | 0.3 ± 0.0 *** | 44.9 ± 4.2 | 11.2 ± 0.3 *** | 10.1 ± 0.0 *** | 25.5 ± 3.3 ** | 0.1 ± 0.0 *** | 0.2 ± 0.0 *** | 0.03 ± 0.0 | |
| E2 + 0.1% γ-TmT | 3.7 ± 0.5 *** | 0.9 ± 0.1 *** | 48.5 ± 2.7 | 21.9 ± 2.8 *** | 19.3 ± 1.6 *** | 28.1 ± 4.5 ** | 0.2 ± 0.0 ** | 0.3 ± 0.0 *** | 0.03 ± 0.0 | |
| E2 + 0.3% γ-TmT | 3.0 ± 0.2 *** | 0.8 ± 0.1 *** | 44.1 ± 2.6 | 48.1 ± 11.7 ** | 43.5 ± 9.9 ** | 31.0 ± 7.1 * | 0.5 ± 0.1 * | 0.9 ± 0.1 ** | 0.06 ± 0.0 | |
| E2 + 0.5% γ-TmT | 5.9 ± 0.7 *** | 1.9 ± 0.3 *** | 37.0 ± 1.6 | 61.0 ± 3.8 *** | 52.4 ± 6.3 **** | 10.5 ± 4.2 | 1.4 ± 0.2 ** | 2.6 ± 0.4 *** | 0.04 ± 0.0 | |
|
| ||||||||||
| 18 weeks | Negative control | 0.2 ± 0.0 | 0.1 ± 0.0 | 23.1 ± 0.6 | 2.9 ± 0.4 | 0.8 ± 0.1 | 4.9 ± 0.4 | 0.02 ± 0.0 | 0.03 ± 0.0 | 0.3 ± 0.1 |
| E2 control | 0.4 ± 0.1 | 0.1 ± 0.0 | 36.4 ± 1.7 | 5.3 ± 0.3 | 1.1 ± 0.1 | 8.1 ± 0.6 | 0.05 ± 0.0 | 0.02 ± 0.0 | 0.4 ± 0.0 | |
| E2 + 0.05% γ-TmT | 2.8 ± 0.5 ** | 0.8 ± 0.1 ** | 38.7 ± 1.9 | 50.3 ± 4.1 *** | 11.7 ± 1.1 *** | 11.2 ± 0.8 * | 0.2 ± 0.0 *** | 0.2 ± 0.0 *** | 1.1 ± 0.1 *** | |
| E2 + 0.1% γ-TmT | 3.7 ± 0.3 *** | 1.3 ± 0.2 *** | 60.3 ± 2.0 *** | 142.7 ± 11.5 *** | 35.1 ± 2.1 *** | 14.0 ± 0.7 *** | 0.5 ± 0.1 *** | 0.5 ± 0.0 *** | 2.3 ± 0.2 *** | |
| E2 + 0.3% γ-TmT | 8.1 ± 1.2 *** | 3.3 ± 0.5 *** | 62.2 ± 2.6 *** | 268.5 ± 5.4 *** | 74.3 ± 1.6 *** | 20.2 ± 1.0 *** | 0.8 ± 0.1 *** | 1.1 ± 0.1 *** | 5.1 ± 0.2 *** | |
| E2 + 0.5% γ-TmT | 8.2 ± 1.0 *** | 3.7 ± 0.5 *** | 50.9 ± 1.8 *** | 263.8 ± 11.9 *** | 75.9 ± 3.4 *** | 19.7 ± 1.1 *** | 0.7 ± 0.0 *** | 1.0 ± 0.1 *** | 5.0 ± 0.5 *** | |
|
| ||||||||||
| 31 weeks | Negative control | 0.2 ± 0.0 | 0.1 ± 0.0 | 34.2 ± 1.7 | 1.9 ± 0.2 | 0.7 ± 0.1 | 5.5 ± 1.0 | 0.02 ± 0.0 | 0.06 ± 0.0 | 0.3 ± 0.1 |
| E2 control | 0.3 ± 0.1 | 0.05 ± 0.0 | 38.6 ± 2.4 | 4.8 ± 1.1 | 1.1 ± 0.2 | 6.3 ± 1.1 | 0.03 ± 0.0 | 0.07 ± 0.0 | 0.4 ± 0.1 | |
| E2 + 0.05% γ-TmT | 1.6 ± 0.2 *** | 0.5 ± 0.1 *** | 43.0 ± 4.3 | 31.7 ± 3.0 *** | 8.1 ± 0.9 *** | 7.2 ± 1.1 | 0.1 ± 0.0 ** | 0.1 ± 0.0 *** | 0.8 ± 0.1 * | |
| E2 + 0.1% γ-TmT | 3.1 ± 0.6 *** | 0.9 ± 0.1 *** | 35.7 ± 4.7 | 60.3 ± 4.6 *** | 16.8 ± 1.1 *** | 9.2 ± 0.9 | 0.2 ± 0.0 *** | 0.2 ± 0.0 *** | 0.9 ± 0.1 ** | |
| E2 + 0.3% γ-TmT | 8.1 ± 1.5 *** | 3.3 ± 0.7 ** | 49.3 ± 2.3 * | 167.4 ± 19.9 *** | 50.7 ± 6.7 *** | 13.1 ± 1.7 ** | 0.4 ± 0.1 *** | 0.7 ± 0.1 ** | 2.4 ± 0.4 *** | |
| E2 + 0.5% γ-TmT | 13.9 ± 2.6 *** | 6.7 ± 1.4 ** | 51.4 ± 3.9 * | 261.9 ± 13.9 *** | 87.7 ± 4.7 *** | 14.9 ± 1.6 ** | 0.6 ± 0.1 *** | 1.3 ± 0.2 *** | 3.6 ± 0.6 *** | |
The effect of γ-TmT supplementation on the levels of γ-, δ-, α-T and their short chain CEHC and CMBHC metabolites in the serum (μMol/l) of ACI rats were analyzed at 6, 18 and 31 weeks. P-values are compared to the E2 control of the respective time points.
Statistical significance
P < 0.05,
P < 0.01,
P < 0.001.
γ-TmT decreases serum estradiol levels and induces the estrogen metabolizing enzyme CYP1A1 in ACI rat tissues
Our data showed that 0.3% and 0.5% were the doses of γ-TmT capable of inhibiting estrogen-induced mammary tumorigenesis in ACI rats. Hence, all subsequent molecular analyses were performed on these two treatment groups and controls. Circulating levels of E2 in the negative control (no E2 supplementation), E2 control and γ-TmT treated ACI rats were measured at 6, 18 and 31 weeks (Figure 2A). At 6 weeks, the average serum E2 level of the negative control group was 39.3 ± 2.5 pg/ml, which was increased to 54.7 ± 4.4 pg/ml with E2 treatment. At 18 weeks, serum E2 level of the negative control group was 32.2 ± 1.9 pg/ml while that of the E2 control was 48.4 ± 2.4 pg/ml (p < 0.01). At the 31 week time point, serum E2 increased from 33 ± 1.9 pg/ml in the negative control to 82.4 ± 5 pg/ml in the E2 control group (p < 0.001). This corresponds to a 1.3-, 1.5- and 2.4-fold increase of serum E2 levels in the E2 control group at 6, 18 and 31 weeks, respectively, compared to the negative control. At 6 and 18 weeks, γ-TmT treatment had no effect on the serum E2 levels. However, at 31 weeks, 0.3% and 0.5% γ-TmT lowered the average levels of serum E2 to 56 ± 5.7 pg/ml (32%, p < 0.01) and 48.4 ± 5.1 pg/ml (41%, p < 0.001), respectively.
Figure 2.

γ-TmT reduces serum E2 levels and induces estrogen metabolizing enzyme CYP1A1 in E2-treated ACI rats. (A) Analysis of E2 levels in ACI rat serum at 6, 18 and 31 weeks (n=6/group for 6, 18 weeks; n=9/group for 31 weeks). (B) qPCR analysis of CYP1A1 and CYP1B1 mRNA expression in the mammary glands (6, 18 weeks), tumors (31 weeks) and liver (31 weeks) of ACI rats (n=6–8/group). Statistical significance, *P < 0.05, **P < 0.01, ***P < 0.001. (C) Western blot analysis of CYP1A1 and CYP1B1 in the mammary tumors of ACI rats at 31 weeks (n=3/group).
To elucidate how γ-TmT decreases serum E2 levels in ACI rats, expression of the genes for two major estrogen metabolizing enzymes, CYP1A1 and CYP1B1, in rat tissues was analyzed by qPCR (Figure 2B). For 6 and 18 weeks, analysis was done on rat mammary glands while for the 31 week time point, mammary tumors and liver tissues were analyzed. At all three time points, γ-TmT induced expression of CYP1A1 mRNA compared to the E2 control. Treatment with 0.3% and 0.5% γ-TmT induced CYP1A1 mRNA by 3.3 ± 0.7 (p < 0.05) and 9.9 ± 0.9 (p < 0.001) fold, respectively, at 6 weeks and 1.5 ± 0.1 (p < 0.05) and 2.0 ± 0.1 (p < 0.01) fold, respectively, at 18 weeks. Induction of CYP1A1 mRNA expression was highest in mammary tumors at 31 weeks, showing an increase of 112.5 ± 46.7 (p < 0.05) fold with 0.3% γ-TmT and 41.9 ± 23.6 fold with 0.5% γ-TmT. γ-TmT did not alter the levels of CYP1B1 mRNA in mammary tissues at 6, 18 or 31 weeks. As the liver is a major site of estrogen metabolism, CYP1A1 and CYP1B1 mRNA levels were analyzed in the rat livers for the 31 week time point. Treatment with γ-TmT significantly induced expression of CYP1A1 mRNA but had no effect on the levels of CYP1B1 mRNA in rat livers. γ-TmT also increased protein expression of CYP1A1 in the mammary tumors while CYP1B1 levels remained fairly constant (Figure 2C).
γ-TmT induces Nrf2-dependent antioxidant response and decreases the oxidative stress marker 8-isoprostane in ACI rats
Oxidative metabolism of estrogen generates genotoxic metabolites, free radicals and ROS (3). Nrf2-dependent phase II and antioxidant enzymes detoxify estrogen metabolites and act as scavengers for free radicals and ROS (13, 14). In the present study, γ-TmT was found to induce the estrogen metabolizing enzyme, CYP1A1. To investigate if γ-TmT detoxifies E2 metabolites and reduces oxidative stress through induction of Nrf2-dependent antioxidant response, gene expression of Nrf2 and some of its downstream enzymes in rat mammary tissues were analyzed by qPCR (Figure 3A). No significant change in the mRNA levels of Nrf2 was observed with γ-TmT treatment at 6, 18 and 31 weeks. However, at 6 and 31 weeks, γ-TmT induced mRNA expression of the phase II detoxification enzyme NQO1, and the antioxidant enzymes, GCLM and HMOX1. Western blot analysis of mammary tumors showed increased protein levels of Nrf2, NQO1, GCLM and HMOX1 with γ-TmT supplementation (Figure 3B). The oxidative stress marker, 8-isoprostane, was quantified in rat serum by EIA (Figure 3C). Treatment with E2 significantly increased the concentrations of 8-isoprostane in the serum of ACI rats at each experimental time point. γ-TmT decreased 8-isoprostane levels from as early as 6 weeks. Treatment with 0.3% and 0.5% γ-TmT reduced serum 8-isoprostane levels by 59% (p < 0.001) and 63% (p < 0.001) at 6 weeks, 78% (p < 0.001) and 78% (p < 0.001) at 18 weeks, 77% (p < 0.001) and 76% (p < 0.001) at 31 weeks, respectively.
Figure 3.

γ-TmT induces Nrf2-mediated antioxidant response and reduces serum 8-isoprostane in E2-treated ACI rats. (A) qPCR analysis of mRNA expression of Nrf2 and its downstream targets, NQO1, GCLM and HMOX1, in rat mammary glands (6 and 18 weeks) and mammary tumors (31 weeks) (n=6–8/group). (B) Western blot analysis of Nrf2, NQO1, GCLM and HMOX1 in the mammary tumors of ACI rats at 31 weeks (n=3/group). (C) Analysis of the 8-isoprostane levels in the serum of ACI rats at 6, 18 and 31 weeks (n=6/group). Statistical significance, *P < 0.05, ***P < 0.001.
γ-TmT induces ERβ and PPARγ nuclear receptor signaling and reduces cell proliferation in mammary tumors of ACI rats
To further investigate how γ-TmT inhibits mammary tumorigenesis in ACI rats, expression of the genes for nuclear receptors, ERβ and PPARγ were analyzed by qPCR in rat mammary tissues (Figure 4A). ERβ is known to inhibit proliferation and invasion of breast cancer cells (32). While γ-TmT did not affect ERβ expression in the mammary glands at 6 and 18 weeks, mRNA expression of ERβ was significantly induced in mammary tumors by γ-TmT at 31 weeks. No change in ERα was observed at any of the three time points (data not shown). PPARγ is a ligand-activated transcription factor, belonging to the family of nuclear receptors and activation of PPARγ has been reported to inhibit mammary carcinogenesis (33, 34). γ-TmT induced PPARγ mRNA in the tumors of ACI rats (Figure 4A). mRNA expression of the tumor suppressor, PTEN, which is a downstream target of PPARγ and the cell cycle regulator, p27, were also upregulated in the tumors by γ-TmT. Immunohistochemical analysis also showed that γ-TmT reduced the cell proliferation marker PCNA in the mammary tumors of ACI rats (Figure 4B). Administration of 0.3% and 0.5% γ-TmT reduced PCNA levels by 63% (p < 0.05) and 55% (p < 0.05), respectively.
Figure 4.

γ-TmT regulates nuclear receptor signaling and cell proliferation in E2-treated ACI rats. (A) qPCR analysis of mRNA expression of ERβ, PPARγ, PTEN, and p27 in the mammary glands (6 and 18 weeks) and mammary tumors (31 weeks) of ACI rats (n=6–8/group). (B) Representative images of PCNA staining in the mammary tumors of ACI rats (40X magnification). The scale bar represents 50 μm. Quantification of nuclear PCNA staining in the mammary tumors (n=3/group) is shown. Statistical significance, *P < 0.05, **P < 0.01.
Dietary γ-TmT inhibits estrogen mediated mammary tumor growth in MCF-7 xenografts
The ACI rat study demonstrated that γ-TmT could be useful for the prevention of E2-mediated mammary cancer. To investigate if γ-TmT could also be a potential agent for the inhibition of E2-dependent mammary cancer, the effect of dietary γ-TmT was studied in a second animal model, namely, an orthotopic mouse model using MCF-7 cells. The ER-positive breast cancer cell line, MCF-7, was injected into mammary fat pads of nu/nu mice implanted with estrogen pellets. Mice were fed different dietary doses of γ-TmT and the effects of γ-TmT on tumor growth were assessed. Administration of dietary γ-TmT for 9 weeks at doses of 0.05%, 0.1%, 0.3% and 0.5% reduced mammary tumor volume by 74% (p < 0.05), 54% (p < 0.05), 82% (p < 0.01) and 71% (p < 0.05), respectively (Figure 5A). Tumor weights were also decreased by 79% (p < 0.05), 64% (p < 0.05), 87% (p < 0.01) and 78% (p < 0.01) with 0.05%, 0.1%, 0.3% and 0.5% γ-TmT, respectively (Figure 5B). The 0.05–0.5% dietary doses of γ-TmT were all effective as inhibitors of mammary tumor growth in these xenografted mice, showing a greater level of responsiveness than we observed than in the ACI rat model.
Figure 5.
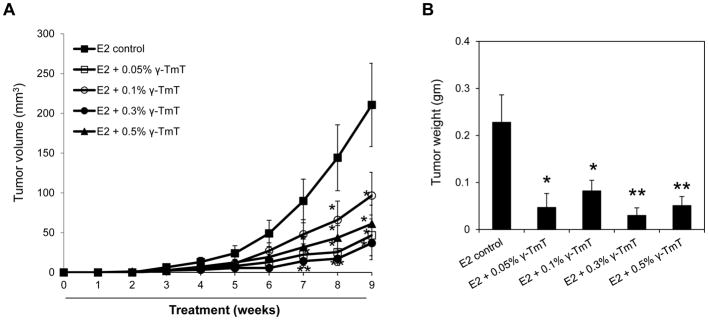
γ-TmT inhibits E2-induced mammary tumor growth in MCF-7 xenografted mice. Nu/nu mice were implanted with E2 pellets (0.72 mg). After 2 days, MCF-7 cells were injected into the mammary fat pad of the mice. From the day of MCF-7 cell injection, mice were fed with control diet or different doses of γ-TmT diet for 9 weeks (n=10/group). (A) Average tumor volume at weekly time points for the different treatment groups is shown. (B) Average tumor weight at 9 weeks is shown. Statistical significance, *P < 0.05, **P < 0.01.
DISCUSSION
The association between estrogen exposure and increased breast cancer risk has been known for decades (11, 35). ER positive breast cancer accounts for two-third of all initially diagnosed breast cancers (36). In this study, the effects of four doses of γ-TmT (0.05%, 0.1%, 0.3% and 0.5%) on estrogen-induced mammary cancer formation and growth were assessed using two different animal models of breast cancer. The cancer preventive effects of γ-TmT at early, intermediate and late stages of mammary tumorigenesis were studied using ACI rats which are an established model of E2-induced rodent mammary carcinogenesis. Previously, 6 and 18 weeks have been reported as important time points to study early and intermediate events of mammary tumorigenesis in ACI rats (37). Prolonged exposure of ACI rats to high doses of E2 (27 mg) induces pituitary tumors which can interfere with breast cancer prevention studies (38). To avoid this problem, a low dose of E2 (9 mg) in silastic tubing was used in the present study to induce mammary tumors.
We investigated the chemopreventive activity of the naturally occurring tocopherol mixture, γ-TmT which contains 56% γ-T. In a published study with type 2 diabetic human subjects (39), 500 mg mixed tocopherols/day (mixed tocopherols containing 60% γ-T) resulted in serum γ-T levels (approximately 9 μM) similar to that observed with 0.3% γ-TmT administration in rats (8.2 μM) in our study (Table 1). While treatment with lower doses of γ-TmT (0.05% and 0.1%) had no inhibitory effect on mammary tumor growth, 0.3% and 0.5% γ-TmT reduced tumor burden and multiplicity in the ACI rats. Treatment with 0.3% γ-TmT was more effective than 0.5% γ-TmT in inhibiting E2-induced mammary tumors, suggesting that γ-TmT may have a biphasic effect at higher concentrations.
Elevated serum E2 concentrations are associated with a higher risk of breast cancer in women (40). At 31 weeks, γ-TmT decreased the circulating levels of E2 in ACI rats which could contribute to its inhibitory effect on mammary tumorigenesis. To determine the mechanism of E2 decrease, we examined expression of CYP1A1 and CYP1B1, the major phase I enzymes expressed in breast and liver tissues (41). CYP1A1 and CYP1B1 metabolize E2 to the catechol E2 2-hydroxyestradiol (2-OHE2) and 4-hydroxyestradiol (4-OHE2), respectively (3). 2-OHE2 lacks tumorigenic activity and is considered to be the ‘good metabolite’ of estrogen (41). It is rapidly converted to the non-toxic 2-methoxyestradiol by catechol-O-methyltransferase (COMT) (41). On the other hand, 4-OHE2 is highly genotoxic and possesses carcinogenic potential due to its ability to form depurinating DNA adducts (4). γ-TmT induced gene expression of CYP1A1 without altering the expression of CYP1B1 at all three stages of mammary tumorigenesis (Figure 2). At 31 weeks, the dramatic induction of CYP1A1 by γ-TmT in mammary tumors could lead to increased E2 metabolism, contributing to reduction in levels of serum E2. Induction of CYP1A1 and CYP1B1 is typically observed after exposure to aryl hydrocarbon receptor (AhR) ligands and the differential induction of CYP1A1 compared to CYP1B1, as observed in this study, has been previously reported with AhR active pharmaceuticals (42). There are no reports on the role of γ-TmT as an AhR ligand suggesting that CYP1A1 induction by γ-TmT could be indirectly AhR-dependent or AhR-independent and is currently being investigated. CYP1A1 is known to be highly expressed in breast epithelial cells while CYP1B1 is found in mesenchymal cells (43), suggesting that factors other than AhR could also regulate the expression of these CYP genes. Whether γ-TmT specifically targets epithelial cells to induce CYP1A1 is being studied. Preferential induction of CYP1A1 over CYP1B1 by γ-TmT may shift E2 metabolism to a more protective pathway through increased generation of 2-OHE2, thereby inhibiting mammary tumorigenesis.
Oxidation of catechol E2 produces genotoxic quinones, which undergo redox cycling to form free radicals and ROS capable of damaging cellular macromolecules (7–9). 8-Isoprostane is a marker of oxidative stress produced in vivo by free radical catalyzed peroxidation of arachidonic acid (44). Reduced concentrations of serum 8-isoprostane in γ-TmT treated ACI rats is suggestive of the protective role of γ-TmT against E2-induced lipid peroxidation. The Nrf2-mediated antioxidant pathway is an important cellular defense mechanism against oxidative damage (45). Upon activation, the transcription factor Nrf2 binds to antioxidant responsive elements, inducing the expression of phase II detoxification and antioxidant enzymes (45). We have previously reported that γ-TmT induces Nrf2-dependent antioxidant response in mammary hyperplasia of ACI rats (27, 28). In the present study, γ-TmT induced expression of downstream targets of Nrf2, such as NQO1, GCLM and HMOX1. Antioxidant response was stimulated by γ-TmT in the mammary glands at 6 weeks, while no significant effect was observed at 18 weeks. γ-TmT-induced antioxidant response was highest in the mammary tumors at 31 weeks. This differential response could be explained by the changes in serum E2 levels of ACI rats and resultant oxidative stress at the different stages of mammary carcinogenesis. The levels of serum E2 at the 6 week time point (54.7 pg/ml) was higher than that at 18 weeks (48.4 pg/ml), leading to increased antioxidant response at 6 weeks compared to 18 weeks. At 31 weeks when serum E2 levels were highest (82.4 pg/ml), a dramatic induction of Nrf2-dependent genes was observed in response to γ-TmT treatment. NQO1 is a key phase II detoxification enzyme involved in the clearance of genotoxic quinones and reduction of oxidative stress. It catalyzes the reduction of quinones back to catechol E2, making them unavailable for reaction with DNA (46). Upregulation of the antioxidant enzymes, GCLM and HMOX1, by γ-TmT could help in maintaining redox homeostasis in cells.
In addition to its effect on E2 availability and antioxidant response, γ-TmT probably inhibits mammary tumorigenesis in ACI rats by modulating additional cellular pathways. PPARγ is a ligand activated nuclear receptor with diverse cellular functions (47). Activation of PPARγ reduces cell proliferation through upregulation of the tumor suppressor PTEN and subsequent inhibition of PI3K/Akt pathway (48). Further, PPARγ induces the cyclin dependent kinase inhibitors (p21 and p27), apoptosis and cell differentiation markers (48). γ-T has been reported to activate PPARγ signaling in cancer cell lines (49, 50). We have previously shown that γ-TmT induced PPARγ during mammary hyperplasia in ACI rats (28). In the current study using silastic tubing, E2 release is gradual and induction of PPARγ was minimal at 6 and 18 weeks but significant at 31 weeks of treatment. Here, we report that γ-TmT induced mRNA expression of PPARγ, PTEN and p27 in the mammary tumors of ACI rats. In addition, γ-TmT inhibited cell proliferation in E2-induced mammary tumors, as evident from reduced percentage of PCNA positive cells. Further, γ-TmT reduced growth of MCF-7 xenografted mammary tumors in immunodeficient mice at all the doses tested.
In conclusion, γ-TmT inhibits E2-induced mammary tumorigenesis by lowering the levels of circulating E2, increasing E2 metabolism via CYP1A1 and facilitating the clearance of toxic metabolites and ROS by stimulation of antioxidant response. Activation of PPARγ signaling and inhibition of cell proliferation could also contribute to the chemopreventive effects of γ-TmT. Thus, γ-TmT could be a safe and potential agent for the prevention of E2 mediated breast cancer.
Acknowledgments
Financial Support: This work was supported by the National Institutes of Health Grant R01 AT007036, the National Institute of Environmental Health Sciences Grant ES005022, P30 ES023512, Charles and Johanna Busch Memorial Fund at Rutgers University, the Trustees Research Fellowship Program at Rutgers and the New Jersey Commission on Cancer Research Postdoctoral Fellowship to Soumyasri Das Gupta.
We sincerely thank Dr. Philip Furmanski for his critical comments on the manuscript. We thank the Laboratory of Animal Service at the Department of Chemical Biology, Rutgers University, for taking care of the animals.
Footnotes
Disclosure of potential conflicts of interest: No potential conflicts of interest were disclosed.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Clemons M, Goss P. Estrogen and the risk of breast cancer. N Engl J Med. 2001;344:276–85. doi: 10.1056/NEJM200101253440407. [DOI] [PubMed] [Google Scholar]
- 3.Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R, et al. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta. 2006;1766:63–78. doi: 10.1016/j.bbcan.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–82. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 5.Cavalieri E, Rogan E. Catechol quinones of estrogens in the initiation of breast, prostate, and other human cancers: keynote lecture. Ann N Y Acad Sci. 2006;1089:286–301. doi: 10.1196/annals.1386.042. [DOI] [PubMed] [Google Scholar]
- 6.Yue W, Yager JD, Wang JP, Jupe ER, Santen RJ. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids. 2013;78:161–70. doi: 10.1016/j.steroids.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Bolton JL, Thatcher GR. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol. 2008;21:93–101. doi: 10.1021/tx700191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fussell KC, Udasin RG, Smith PJ, Gallo MA, Laskin JD. Catechol metabolites of endogenous estrogens induce redox cycling and generate reactive oxygen species in breast epithelial cells. Carcinogenesis. 2011;32:1285–93. doi: 10.1093/carcin/bgr109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Chandrasena ER, Yuan Y, Peng KW, van Breemen RB, Thatcher GR, et al. Redox cycling of catechol estrogens generating apurinic/apyrimidinic sites and 8-oxo-deoxyguanosine via reactive oxygen species differentiates equine and human estrogens. Chem Res Toxicol. 2010;23:1365–73. doi: 10.1021/tx1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhat HK, Calaf G, Hei TK, Loya T, Vadgama JV. Critical role of oxidative stress in estrogen-induced carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:3913–8. doi: 10.1073/pnas.0437929100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mense SM, Remotti F, Bhan A, Singh B, El-Tamer M, Hei TK, et al. Estrogen-induced breast cancer: alterations in breast morphology and oxidative stress as a function of estrogen exposure. Toxicol Appl Pharmacol. 2008;232:78–85. doi: 10.1016/j.taap.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaikwad NW, Yang L, Muti P, Meza JL, Pruthi S, Ingle JN, et al. The molecular etiology of breast cancer: evidence from biomarkers of risk. Int J Cancer. 2008;122:1949–57. doi: 10.1002/ijc.23329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh B, Bhat NK, Bhat HK. Induction of NAD (P) H-quinone oxidoreductase 1 by antioxidants in female ACI rats is associated with decrease in oxidative DNA damage and inhibition of estrogen-induced breast cancer. Carcinogenesis. 2012;33:156–63. doi: 10.1093/carcin/bgr237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh B, Bhat HK. Superoxide dismutase 3 is induced by antioxidants, inhibits oxidative DNA damage and is associated with inhibition of estrogen-induced breast cancer. Carcinogenesis. 2012;33:2601–10. doi: 10.1093/carcin/bgs300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Constantinou C, Papas A, Constantinou AI. Vitamin E and cancer: An insight into the anticancer activities of vitamin E isomers and analogs. Int J Cancer. 2008;123:739–52. doi: 10.1002/ijc.23689. [DOI] [PubMed] [Google Scholar]
- 16.Ju J, Picinich SC, Yang Z, Zhao Y, Suh N, Kong AN, et al. Cancer-preventive activities of tocopherols and tocotrienols. Carcinogenesis. 2010;31:533–42. doi: 10.1093/carcin/bgp205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang CS, Suh N, Kong A-NT. Does vitamin E prevent or promote cancer? Cancer prevention research. 2012;5:701–5. doi: 10.1158/1940-6207.CAPR-12-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinstein SJ, Peters U, Ahn J, Friesen MD, Riboli E, Hayes RB, et al. Serum α-tocopherol and γ-tocopherol concentrations and prostate cancer risk in the PLCO Screening Trial: a nested case-control study. PloS one. 2012;7:e40204. doi: 10.1371/journal.pone.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee IM, Cook NR, Gaziano JM, Gordon D, Ridker PM, Manson JE, et al. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women’s Health Study: a randomized controlled trial. JAMA. 2005;294:56–65. doi: 10.1001/jama.294.1.56. [DOI] [PubMed] [Google Scholar]
- 20.Gaziano JM, Glynn RJ, Christen WG, Kurth T, Belanger C, MacFadyen J, et al. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2009;301:52–62. doi: 10.1001/jama.2008.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang Q, Christen S, Shigenaga MK, Ames BN. gamma-tocopherol, the major form of vitamin E in the US diet, deserves more attention. Am J Clin Nutr. 2001;74:714–22. doi: 10.1093/ajcn/74.6.714. [DOI] [PubMed] [Google Scholar]
- 22.Ju J, Hao X, Lee MJ, Lambert JD, Lu G, Xiao H, et al. A gamma-tocopherol-rich mixture of tocopherols inhibits colon inflammation and carcinogenesis in azoxymethane and dextran sulfate sodium-treated mice. Cancer Prev Res (Phila) 2009;2:143–52. doi: 10.1158/1940-6207.CAPR-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu G, Xiao H, Li G-X, Picinich SC, Chen Y-K, Liu A, et al. A γ-tocopherol-rich mixture of tocopherols inhibits chemically induced lung tumorigenesis in A/J mice and xenograft tumor growth. Carcinogenesis. 2010;31:687–94. doi: 10.1093/carcin/bgp332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee HJ, Ju J, Paul S, So JY, DeCastro A, Smolarek A, et al. Mixed tocopherols prevent mammary tumorigenesis by inhibiting estrogen action and activating PPAR-gamma. Clin Cancer Res. 2009;15:4242–9. doi: 10.1158/1078-0432.CCR-08-3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smolarek AK, So JY, Burgess B, Kong AN, Reuhl K, Lin Y, et al. Dietary administration of delta- and gamma-tocopherol inhibits tumorigenesis in the animal model of estrogen receptor-positive, but not HER-2 breast cancer. Cancer Prev Res (Phila) 2012;5:1310–20. doi: 10.1158/1940-6207.CAPR-12-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shull JD, Spady TJ, Snyder MC, Johansson SL, Pennington KL. Ovary-intact, but not ovariectomized female ACI rats treated with 17beta-estradiol rapidly develop mammary carcinoma. Carcinogenesis. 1997;18:1595–601. doi: 10.1093/carcin/18.8.1595. [DOI] [PubMed] [Google Scholar]
- 27.Das Gupta S, So JY, Wall B, Wahler J, Smolarek AK, Sae-Tan S, et al. Tocopherols inhibit oxidative and nitrosative stress in estrogen-induced early mammary hyperplasia in ACI rats. Mol Carcinog. 2014 doi: 10.1002/mc.22164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smolarek AK, So JY, Thomas PE, Lee HJ, Paul S, Dombrowski A, et al. Dietary tocopherols inhibit cell proliferation, regulate expression of ERalpha, PPARgamma, and Nrf2, and decrease serum inflammatory markers during the development of mammary hyperplasia. Mol Carcinog. 2013;52:514–25. doi: 10.1002/mc.21886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turan VK, Sanchez RI, Li JJ, Li SA, Reuhl KR, Thomas PE, et al. The effects of steroidal estrogens in ACI rat mammary carcinogenesis: 17beta-estradiol, 2-hydroxyestradiol, 4-hydroxyestradiol, 16alpha-hydroxyestradiol, and 4-hydroxyestrone. J Endocrinol. 2004;183:91–9. doi: 10.1677/joe.1.05802. [DOI] [PubMed] [Google Scholar]
- 30.Zhao Y, Lee MJ, Cheung C, Ju JH, Chen YK, Liu B, et al. Analysis of multiple metabolites of tocopherols and tocotrienols in mice and humans. J Agric Food Chem. 2010;58:4844–52. doi: 10.1021/jf904464u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee HJ, Liu H, Goodman C, Ji Y, Maehr H, Uskokovic M, et al. Gene expression profiling changes induced by a novel Gemini Vitamin D derivative during the progression of breast cancer. Biochem Pharmacol. 2006;72:332–43. doi: 10.1016/j.bcp.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 32.Lazennec G, Bresson D, Lucas A, Chauveau C, Vignon F. ER beta inhibits proliferation and invasion of breast cancer cells. Endocrinology. 2001;142:4120–30. doi: 10.1210/endo.142.9.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elstner E, Muller C, Koshizuka K, Williamson EA, Park D, Asou H, et al. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci U S A. 1998;95:8806–11. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suh N, Wang Y, Williams CR, Risingsong R, Gilmer T, Willson TM, et al. A new ligand for the peroxisome proliferator-activated receptor-gamma (PPAR-gamma), GW7845, inhibits rat mammary carcinogenesis. Cancer Res. 1999;59:5671–3. [PubMed] [Google Scholar]
- 35.Cavalieri EL, Stack DE, Devanesan PD, Todorovic R, Dwivedy I, Higginbotham S, et al. Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc Natl Acad Sci U S A. 1997;94:10937–42. doi: 10.1073/pnas.94.20.10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim E, Metzger-Filho O, Winer EP. The natural history of hormone receptor-positive breast cancer. Oncology (Williston Park) 2012;26:688–94. 96. [PubMed] [Google Scholar]
- 37.Aiyer HS, Gupta RC. Berries and ellagic acid prevent estrogen-induced mammary tumorigenesis by modulating enzymes of estrogen metabolism. Cancer Prev Res (Phila) 2010;3:727–37. doi: 10.1158/1940-6207.CAPR-09-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurz SG, Hansen KK, McLaughlin MT, Shivaswamy V, Schaffer BS, Gould KA, et al. Tissue-specific actions of the Ept1, Ept2, Ept6, and Ept9 genetic determinants of responsiveness to estrogens in the female rat. Endocrinology. 2008;149:3850–9. doi: 10.1210/en.2008-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clarke MW, Ward NC, Wu JH, Hodgson JM, Puddey IB, Croft KD. Supplementation with mixed tocopherols increases serum and blood cell γ-tocopherol but does not alter biomarkers of platelet activation in subjects with type 2 diabetes. The American journal of clinical nutrition. 2006;83:95–102. doi: 10.1093/ajcn/83.1.95. [DOI] [PubMed] [Google Scholar]
- 40.Cauley JA, Lucas FL, Kuller LH, Stone K, Browner W, Cummings SR. Elevated serum estradiol and testosterone concentrations are associated with a high risk for breast cancer. Study of Osteoporotic Fractures Research Group. Ann Intern Med. 1999;130:270–7. doi: 10.7326/0003-4819-130-4_part_1-199902160-00004. [DOI] [PubMed] [Google Scholar]
- 41.Samavat H, Kurzer MS. Estrogen metabolism and breast cancer. Cancer Lett. 2015;356:231–43. doi: 10.1016/j.canlet.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin U-H, Lee S-o, Safe S. Aryl hydrocarbon receptor (AHR)-active pharmaceuticals are selective AHR modulators in MDA-MB-468 and BT474 breast cancer cells. Journal of Pharmacology and Experimental Therapeutics. 2012;343:333–41. doi: 10.1124/jpet.112.195339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spink DC, Spink BC, Cao JQ, DePasquale JA, Pentecost BT, Fasco MJ, et al. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis. 1998;19:291–8. doi: 10.1093/carcin/19.2.291. [DOI] [PubMed] [Google Scholar]
- 44.Montuschi P, Barnes PJ, Roberts LJ., 2nd Isoprostanes: markers and mediators of oxidative stress. FASEB J. 2004;18:1791–800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- 45.Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–71. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaikwad NW, Rogan EG, Cavalieri EL. Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen ortho-quinones. Free Radic Biol Med. 2007;43:1289–98. doi: 10.1016/j.freeradbiomed.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res. 2003;9:1–9. [PubMed] [Google Scholar]
- 48.Mansure JJ, Nassim R, Kassouf W. Peroxisome proliferator-activated receptor gamma in bladder cancer: a promising therapeutic target. Cancer Biol Ther. 2009;8:6–15. doi: 10.4161/cbt.8.7.7853. [DOI] [PubMed] [Google Scholar]
- 49.Campbell SE, Musich PR, Whaley SG, Stimmel JB, Leesnitzer LM, Dessus-Babus S, et al. Gamma tocopherol upregulates the expression of 15-S-HETE and induces growth arrest through a PPAR gamma-dependent mechanism in PC-3 human prostate cancer cells. Nutr Cancer. 2009;61:649–62. doi: 10.1080/01635580902825654. [DOI] [PubMed] [Google Scholar]
- 50.Campbell SE, Stone WL, Whaley SG, Qui M, Krishnan K. Gamma (gamma) tocopherol upregulates peroxisome proliferator activated receptor (PPAR) gamma (gamma) expression in SW 480 human colon cancer cell lines. BMC Cancer. 2003;3:25. doi: 10.1186/1471-2407-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]