

SUMMARY
In contrast to the accepted pro-proliferative effect of cell-matrix adhesion, the proliferative effect of cadherin-mediated cell-cell adhesion remains unresolved. Here, we studied the effect of N-cadherin on cell proliferation in the vasculature. We show that N-cadherin is induced in smooth muscle cells (SMCs) in response to vascular injury, an in vivo model of tissue stiffening and proliferation. Complementary experiments performed with deformable substrata demonstrated that stiffness-mediated activation of a focal adhesion kinase (FAK)-p130Cas-Rac signaling pathway induces N-cadherin. Additionally, by culturing paired and unpaired SMCs on microfabricated adhesive islands of different areas, we found that N-cadherin relaxes the spreading requirement for SMC proliferation. In vivo SMC deletion of N-cadherin strongly reduced injury-induced cycling. Finally, SMC-specific deletion of FAK inhibited proliferation after vascular injury, and this was accompanied by reduced induction of N-cadherin. Thus, a stiffness-and FAK-dependent induction of N-cadherin connects cell-matrix to cell-cell adhesion and regulates the degree of cell spreading needed for cycling.
Graphical Abstract
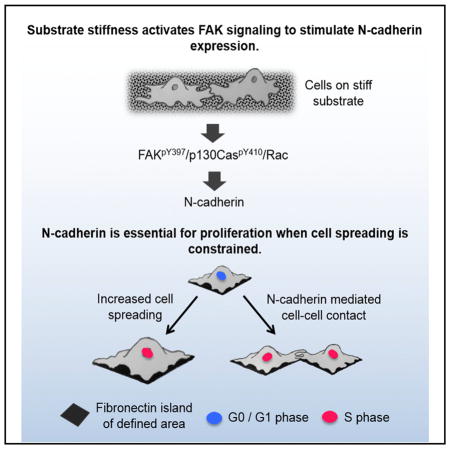
INTRODUCTION
The stimulation and arrest of cell proliferation are tightly regulated throughout embryonic and adult life. Such regulation is achieved primarily through the interaction of cells with their surrounding soluble and adhesive environment. Growth factor receptors trigger numerous mitogenic signaling pathways that are complemented by integrin-mediated adhesion to extracellular matrix (ECM), which provides necessary co-stimulatory signals for proliferation (Assoian and Klein, 2008; Chrzanowska-Wodnicka and Burridge, 1996; Geiger et al., 2001; Parsons et al., 2010; Schwartz, 2010). Additionally, early studies with drugs that disrupt actin polymerization or with micropatterned adhesive islands of distinct sizes revealed that the adhesion requirement for proliferation is directly related to the organization of the actin cytoskeleton and the degree of actin-mediated cell spreading (Assoian and Klein, 2008; Chen et al., 1997).
Compared with the mitogenic effects of growth factors and ECM, the effects of cadherins on proliferation have been more controversial. Several studies have linked cadherin engagement to inhibition of proliferation in vitro and in vivo (Baumeister et al., 2005; Chalasani and Brewster, 2011; Grazia Lampugnani et al., 2003; Haÿ et al., 2009; Kim et al., 2011; Sakane and Miyamoto, 2013; Stockinger et al., 2001; Uglow et al., 2003). However, cell-cell contact through cadherins also promotes cell cycling in vitro and in vivo (Fournier et al., 2008; Nelson and Chen, 2002; Reddy et al., 2005; Tinkle et al., 2004; Zhu and Watt, 1996). This diversity of stimulatory and inhibitory responses strongly suggests that the cellular context affects cadherin-mediated proliferation.
Here, we used complementary in vivo and in vitro approaches to examine the effect of cadherin engagement on the proliferation of vascular smooth muscle cells (SMCs). The proliferation of these cells plays critical roles in cardiovascular disease (Jones et al., 2002; Owens et al., 2004; Sabatini et al., 2008), and cell-cell adhesion in SMCs is largely mediated by a single cadherin (N-cadherin) (Resink et al., 2009). We show that N-cadherin is induced by ECM stiffness and focal adhesion kinase (FAK), and this induction is essential for FAK-mediated SMC cycling in vivo. Thus, our work reveals a new role for N-cadherin in cell-cycle control within the vasculature, a new mechanism of stiffness-dependent crosstalk between cell-substratum and cell-cell adhesion, and a basis for the stimulatory effect of cadherin on proliferation.
RESULTS
N-Cadherin Is Essential for Tamoxifen Proliferation In Vivo
We tested the physiological importance of N-cadherin expression in the in vivo response to vascular injury in the mouse. Because global deletion of N-cadherin leads to lethal cardiovascular defects during embryogenesis (Radice et al., 1997), we conditionally deleted N-cadherin in SMCs of adult mice. In healthy arteries, SMCs are in a differentiated, contractile state and exhibit a low rate of proliferation (Owens et al., 2004). Vascular injury damages the endothelial cell layer, which leads to aggregation of platelets at the sites of the injury, secretion of growth factors, and dedifferentiation, migration, and proliferation of the underlying medial SMCs. These events culminate in the formation of a neointima that is predominantly composed of vascular SMCs (Sata et al., 2000).
Consistent with work of Jones et al. (2002), immunohistochemical analysis of femoral artery sections from wild-type mice showed that N-cadherin was barely detectable in uninjured arteries, but was strongly induced in the media and neointima following injury (Figure 1A), with 80% of the mice having higher N-cadherin levels in injured arteries than in contralateral uninjured arteries (Figure 1B). We were able to link this increase in N-cadherin expression to in vivo cell proliferation because when we produced vascular injury in N-cadherinfl/fl;iCre mice expressing a tamoxifen-inducible SMC-specific Cre, we found that induction of N-cadherin (Figure S1A), neointimal formation (Figure S1B), and cell proliferation (Figures 1C and 1D) were all reduced. Controls showed that proliferating cells were not detected in uninjured femoral arteries (Figure S1C), apoptosis was not affected by the deletion of SMC N-cadherin (Figure S1D), and tamoxifen did not affect the number of S-phase nuclei after vascular injury in mice lacking Cre (Figure S1E).
Figure 1. N-Cadherin Is Essential for In Vivo SMC Proliferation.

(A) Uninjured and injured femoral artery sections from wild-type mice (n = 9) were immunohistochemically stained for N-cadherin. The media (M) lies between the dashed white lines, which mark the internal and external elastic laminae. The neointima (NI) seen in injured arteries is the region between internal elastic lamina and lumen. Images on the right are magnifications of the inset black boxes in the left-hand images.
(B) N-cadherin expression in the injured and uninjured femoral arteries of each mouse was scored by blinded grouping (inj, injured; uninj, uninjured).
(C) In vivo SMC proliferation was examined by in vivo BrdU labeling in injured femoral arteries from vehicle-treated (n = 8) and tamoxifen-treated (n = 7) N-cadherinfl/fl;iCre mice.
(D) Results in (C) were quantified by manual counting and plotted as mean + SD.
(E) Asynchronous SMCs were cultured on FN-coated PA hydrogels (2, 11, and 24 kPa) for 24 hr in 10% FBS and analyzed by western blot. The bar graph shows N-cadherin levels normalized to control and plots results as mean + SE; n = 3.
See also Figure S1.
N-Cadherin Gene Expression Is Regulated by the Stiffness-Sensitive FAK-Cas-Rac Pathway
Vascular injury is associated with an increase in arterial stiffness from 2–4 kPa to 10–24 kPa (Klein et al., 2009). To study the effect of ECM stiffness on the levels of N-cadherin, we cultured SMCs and mouse embryonic fibroblasts (MEFs) on fibronectin (FN)-coated polyacrylamide (PA) hydrogels spanning this pathophysiologic range of stiffness, and stimulated them with fetal bovine serum (FBS). N-cadherin expression was directly proportional to ECM stiffness in both SMCs (Figure 1E) and MEFs (Figure S1F). ECM stiffening has several effects on cells, including an increase in actin-dependent intracellular stiffness (Bae et al., 2014; Paszek et al., 2005; Solon et al., 2007), and intracellular stiffness is regulated by both Rho-GTP and Rac-GTP (Figure 2A). We inhibited Rho- and Rac-dependent stiffening, respectively, with the Rho kinase inhibitor Y-27632 (Figure 2B) or the Rac inhibitor NSC23766 (Figure 2C) to determine which small GTPase played the major role in stiffness-dependent induction of N-cadherin. MEFs treated with these inhibitors were cultured on stiff hydrogels (24 kPa). Inhibition of Rho kinase had a relatively small effect on N-cadherin expression (Figure 2B) when compared with the reduced N-cadherin levels seen upon inhibition of Rac-GTP (Figure 2C). Expression of Rac1 small interfering RNA (siRNA) or dominant-negative Rac (RacN17) in MEFs also reduced the expression of N-cadherin protein on stiff hydrogels (Figure 2D). Conversely, expression of a constitutively active Rac (RacV12) allowed for N-cadherin expression in cells cultured on a soft hydrogels (2 kPa) (Figure 2E). A similar reduction in N-cadherin expression was seen in MEFs (Figure 2F) and SMCs (Figure S2A) when Rac activity was inhibited by ectopic expression of RacN17 or β2-chimaerin (a Rac GAP) on a rigid substratum.
Figure 2. N-Cadherin Is Regulated by Rac.

(A) Serum-starved MEFs were suspended in 10% FBS, seeded on stiff FN-coated hydrogels (24 kPa), and treated with 10 μM of the Rho kinase inhibitor Y-27632 or 150 μM of the Rac inhibitor NSC23766 for 20 hr. The elastic moduli of individual cells were determined by atomic force microscopy; n = 3.
(B and C) Serum-starved MEFs treated with (B) 10 μM Y-27632 or (C) 150 μM NSC2376 on stiff FN-coated PA hydrogels (24 kPa) for 20–24 hr in 10% FBS were analyzed by western blot; n = 3–4.
(D) Serum-starved MEFs transfected with 200 nM non-specific (NS) or Rac siRNA, or infected with dominant-negative RacN17 adenovirus were cultured on stiff FN-coated PA hydrogels in 10% FBS for 9 hr. Cells were lysed and analyzed by western blot; n = 3.
(E) Serum-starved MEFs were infected with adenoviral vectors expressing LacZ or RacV12, and cultured on soft FN-coated PA hydrogels (2 kPa) with 10% FBS for 9 hr and analyzed by western blot; n = 4.
(F) MEFs in 10% FBS were infected with adenovirus expressing GFP, RacN17, β2-chimaerin (β2-ch), FRNK, or FAKY397F (Y397F) for 24 hr, lysed, and examined by western blot; n = 3.
See also Figure S2.
FAK and p130Cas are upstream of stiffness-sensitive Rac activity (Bae et al., 2014). We inhibited FAK activity by ectopic expression of dominant-negative constructs (FRNK or FAKY397F) or depleted FAK with siRNA, and found that these two treatments reduced N-cadherin levels in both cell types on a rigid substratum (Figures 2F, S2B, 3A, and S3A), as did depletion of FAK in cells on stiff hydrogels (Figure S3B). Furthermore, exposing MEFs to the pharmacologic FAK inhibitor PF573228 also reduced N-cadherin expression (Figure 3B), and the effect was proportional to the inhibition of FAK activity as judged by phosphorylation at Y397 (Figure 3B). In contrast, Cre-mediated excision of the floxed N-cadherin allele in N-cadherinfl/fl MEFs yielded no changes in either FAK autophosphorylation or the overall levels of FAK (Figure 3C). Thus, a unidirectional crosstalk from FAK to N-cadherin connects cell-substratum adhesion to cell-cell adhesion.
Figure 3. N-Cadherin Is Induced by FAK-Src-Cas Signaling.

(A) MEFs were transfected with 200 nM of two distinct FAK siRNAs or a nonspecific (NS) siRNA. The cells were then serum starved for 48 hr, incubated in 10% FBS for 20 hr, and analyzed by western blot; n = 3.
(B) Serum-starved MEFs were treated with different concentrations of the small-molecule FAK inhibitor PF573228 for 20 hr in 10% FBS and analyzed by western blot; n = 4.
(C) N-cadherinfl/fl MEFs in 10% FBS were infected overnight with 1,000 moi of adenovirus expressing GFP or Cre and analyzed by western blot; n = 3.
(D) Serum-starved MEFs were either transfected with 200 nM of two distinct siRNAs against p130Cas or paxillin, or a non-specific (NS) siRNA, or infected with 1,000 moi adenoviral FRNK. Cells were then incubated with 10% FBS for 24 hr, lysed, and examined by western blot; n = 3.
(E) Serum-starved SYF null MEFs cultured on stiff FN-coated PA hydrogels (24 kPa) were incubated with 10% FBS for 9 hr, lysed, and examined by western blot; n = 3. The bar graphs under (A)–(E) show N-cadherin levels normalized to control and are plotted as mean + SE.
(F) MEFs in 10% FBS were treated with 20 μM PF573228 for 20 hr or infected with FAKY397F adenovirus for 24 hr. The relative expression of N-cadherin mRNA was examined by quantitative RT-PCR. Results are shown as mean + SE; n = 3.
See also Figure S3.
FAK activity leads to the phosphorylation of paxillin (Turner, 2000) and p130Cas (Cary et al., 1998; Fonseca et al., 2004). Knockdown of p130Cas, but not paxillin, with siRNA decreased N-cadherin levels in MEFs cultured on a rigid substratum (Figure 3D), and the p130Cas effect on N-cadherin was confirmed in SMCs (Figure S3C). Moreover, we were able to link this effect of p130Cas to FAK because inhibition of FAK with FRNK reduced the levels of both activated p130CaspY410 and N-cadherin (Figure 3D, FRNK). Src has been implicated in the phosphorylation of FAK and Cas (Ruest et al., 2001; Schlaepfer and Hunter, 1996). Even though Src expression and activity are not strongly sensitive to stiffness (Bae et al., 2014), FAK phosphorylation on Src sites (Y576 and Y861) and N-cadherin expression were low in SYF null cells (which are deficient in the Src family kinase proteins Src, Yes, and Fyn) when cultured on stiff hydrogels and restored by reconstitution of the cells with c-Src (Figure 3E). Because Src is recruited to FAK autophosphorylated at Y397 (Mitra and Schlaepfer, 2006; Schaller et al., 1994), our interpretation of these results is that the stiffness sensitivity of FAK autophosphorylation renders Src-dependent FAK phosphorylation sensitive to stiffness despite the constitutively expressed Src. Collectively, these results indicate that the stiffness-sensitive FAK/Src-Cas-Rac signaling pathway regulates stiffness-dependent N-cadherin gene expression.
We used Ingenuity Pathway Analysis to identify other possible signaling pathways downstream of p130Cas (here called by its gene name, BCAR1) and upstream of N-cadherin (CDH2) gene expression. In addition to Rac, this global approach identified transforming growth factor (TGF)-β/Smad signaling and c-Jun N-terminal kinase (JNK) as potential upstream regulators of N-cadherin (Figure S3D). However, N-cadherin gene expression was minimally affected by treatment with TGF-β (Figure S3E) or by JNK inhibition with SP600125 (Figure S3F). Thus, stiffness-dependent activation of the FAK-Cas-Rac pathway plays the major role in inducing N-cadherin in response to increased substratum stiffness.
FAK and Rac inhibition reduced the levels of N-cadherin mRNA (Figures 3F and S3G). Twist is mechanically regulated (Desprat et al., 2008) and is one of the transcription factors that stimulate N-cadherin gene transcription (Oda et al., 1998; Yang et al., 2004). Inhibition of FAK or Rac suppressed the expression of Twist1 mRNA (Figure S3H). Moreover, siRNA-mediated knockdown of Twist1 mRNA decreased N-cadherin mRNA levels in MEFs (Figure S3I) and SMCs (Figure S3J). Thus, a FAK-Rac-Twist pathway likely contributes to the regulation of N-cadherin.
N-Cadherin Attenuates the Spreading Requirement for Proliferation
Our data suggest that ECM stiffening at sites of vascular injury leads to the induction of N-cadherin, and the induced N-cadherin stimulates SMC proliferation in vivo. We tried to model this effect in vitro by performing proliferation assays in N-cadherinfl/fl MEFs or SMCs cultured on plastic or hydrogels and infected with adenoviral vectors expressing GFP (Ad-GFP) or Cre recombinase (Ad-Cre). However, the random distribution and migration of cells led to inconsistent cell-cell contacts and N-cadherin signaling. Therefore, we fabricated unpaired and paired micro-patterned islands on a glass substrate, which allowed us to systematically control cell-cell contact (Figure S4A) while preserving the stiff surface that is permissive for FAK activation and N-cadherin expression. We then compared the S-phase entry of primary N-cadherinfl/fl SMCs that were infected with Ad-GFP or Ad-Cre and seeded on FN-coated micropatterned islands with an area of 4,900 μm2 per cell. In cells expressing Ad-GFP, physical contact between paired cells increased S-phase entry as compared with unpaired cells (Figure 4A). This effect was greatly reduced in cells infected with Ad-Cre (Figure 4A), demonstrating that the stimulatory effect of cell-cell adhesion in this system is largely mediated by N-cadherin.
Figure 4. N-Cadherin Determines the Spreading Requirement for Proliferation.

(A) Serum-starved N-cadherinfl/fl SMCs infected with adenoviral GFP or Cre were cultured on unpaired or paired micropatterned adhesive islands (4,900 μm2), stimulated with 10% FBS for 48 hr, and analyzed for S-phase entry by EdU incorporation.
(B) Representative images of EdU-positive SMCs on 10,000 μm2 unpaired or paired micropatterned adhesive islands. Cells were immunofluorescently co-stained for DAPI and EdU.
(C) SMCs were cultured on different-sized micropatterned adhesive islands (2,500 and 10,000 μm2) with 10% FBS for 48 hr and analyzed for S-phase entry by EdU incorporation.
(D) Serum-starved SMCs were infected with adenoviral GFP, FRNK or FAKY397F (Y397F) without or with adenoviral N-cadherin, seeded on micropatterned adhesive islands (2,500 μm2) with 10% FBS for 48 hr, and analyzed for S-phase entry. All S-phase results are plotted as mean + SE; n = 3.
See also Figure S4.
ECM stiffening leads to cell spreading (Pelham and Wang, 1997), and cell spreading on ECM drives proliferation (Chen et al., 1997). Although one can manipulate cell spreading by culturing cells on ECM-coated hydrogels of varying stiffness, the stiffness-dependent induction of N-cadherin precluded us from using hydrogels to study the effect of endogenous N-cadherin on cell cycling. Therefore, we asked how the engagement of N-cadherin affected this spreading requirement for proliferation using unpaired and paired adhesive islands of distinct areas (both smaller [2,500 μm2] and larger [10,000 μm2] than those used in Figure 4A). FAK activation and N-cadherin-containing adherens junctions were readily detected in micropatterned islands of both sizes (Figure S4B). Curiously, FAK activity (as judged by the intensity of FAKpY397) was somewhat greater in SMCs on smaller micropatterned islands. The N-cadherin signal at adherens junctions was also much stronger in the smaller micropatterned islands.
Serum-starved SMCs in small and large micropatterns were incubated with 10% FBS, and S-phase entry was monitored by 5-ethynyl-2′-deoxyuridine (EdU) incorporation (Figure 4B). S-phase entry was directly related to the degree of cell spreading in unpaired cells (Figure 4C), and FAK was required for this effect (Figure S4C), consistent with earlier work (Bae et al., 2014; Chen et al., 1997; Klein et al., 2009). However, when cells were cultured in the paired islands, where N-cadherin-mediated cell-cell adhesion is functionally significant (refer to Figure 4A), cell cycling became much less dependent on cell spreading (Figure 4C). Thus, this micropatterning system successfully modeled the pro-proliferative effect of N-cadherin on SMCs that we observed after vascular injury (Figures 1C and 1D). Moreover, the results suggest that the increased presence of N-cadherin after vascular injury may contribute to SMC proliferation by relaxing the spreading requirement for cycling of SMCs within the arterial wall.
Consistent with the reduced expression of N-cadherin, inhibition of endogenous FAK activity with FRNK or FAKY397F reduced the number of S-phase cells in the small, paired micropatterned islands where the stimulatory effect of N-cadherin on cell cycling is most evident (Figure 4D). Similarly, siRNA-mediated knockdown of FAK also reduced S-phase entry in cells on these small, paired micropatterned islands, although the effect was somewhat less pronounced than that seen with FRNK or FAKY397F (Figure S4D). FRNK or FAKY397F also inhibited S-phase entry on the small islands when N-cadherin was ectopically expressed (Figure 4D). Our interpretation of these results is that N-cadherin is necessary, but not sufficient, for FAK-mediated proliferation when cell spreading is constrained.
N-Cadherin Is an Essential Effector of FAK-Mediated Cell Cycling In Vivo
Since N-cadherin induction is FAK dependent, deletion of FAK should also inhibit in vivo SMC proliferation. Initial experiments showed that FAK phosphorylation at Y397 was increased after vascular injury in wild-type mice (Figures S5A and S5B). We then determined the effect of SMC-specific FAK deletion during the response to vascular injury by treating FAKfl/fl;iCre mice with vehicle or tamoxifen (Figure S5C). S-phase nuclei were not detected in uninjured vehicle- or tamoxifen-treated FAKfl/fl;iCre mice (Figure S5D). However, SMC-specific deletion of FAK with tamoxifen strongly reduced the proliferative response to injury (Figures 5A and 5B) and neointimal formation (Figure S5E). The reduction in S-phase nuclei was similar to that seen after SMC-specific deletion of N-cadherin (refer to Figures 1C and 1D). Moreover, N-cadherin upregulation after injury was apparent in vehicle-treated FAKfl/fl;iCre mice (Figure 5C), but was blunted when the mice were given tamoxifen (Figure 5D). Blinded quantification of these data indicated that the N-cadherin signal intensity after injury greatly exceeded that in the uninjured contralateral controls in 60% of vehicle-treated mice, but in only 12.5% of tamoxifen-treated mice (Figure 5E). In striking contrast, deletion of N-cadherin reduced SMC proliferation after injury without inhibiting the phosphorylation of FAK (Figures 5F and 5G) or p130Cas (Figures 5H and 5I). This in vivo epistatic relationship is remarkably consistent with our in vitro epistasis data, and indicates that unidirectional crosstalk from FAK to N-cadherin is essential for FAK-dependent proliferation during the in vivo proliferative response to vascular injury.
Figure 5. N-Cadherin Is an Essential Effector of FAK-Regulated SMC Cycling In Vivo.

(A) Injured femoral arteries from vehicle-treated (n = 7) and tamoxifen-treated (n = 8) FAKfl/fl;iCre mice were stained for incorporated BrdU. Images on the right are magnifications of the inset black boxes in the left-hand images.
(B) The number of S-phase nuclei in the neointima (NI) and media (M) of vehicle-treated (n = 7) and tamoxifen-treated (n = 8) mice was determined by manual counting and plotted as mean + SD.
(C and D) Femoral artery sections from (C) vehicle- and (D) tamoxifen-treated FAKfl/fl;iCre mice were stained for N-cadherin.
(E) Blinded grouping of the vehicle (n = 5) and tamoxifen (n = 8) data in (C) and (D). Statistical significance was determined by chi-square test; p < 000.1.
(F and H) Injured femoral arteries from vehicle- and tamoxifen-treated N-cadherinfl/fl;iCre mice were stained for (F) FAKpY397 and (H) p130CaspY410. The media (M) lies between the dashed white lines, which mark the internal and external elastic laminae. The neointima (NI) is the region between the internal elastic lamina and lumen.
(G) Quantification of results from (F); vehicle n = 4, tamoxifen n = 5.
(I) Quantification of results from (H); vehicle n = 5, tamoxifen n = 5. Results in (G) and (I) are plotted as mean + SE.
See also Figure S5.
DISCUSSION
Our work establishes the proliferative effect of N-cadherin in vascular SMCs, reveals the regulation of N-cadherin by a FAK-p130Cas-Rac pathway, and indicates that this crosstalk between cell-substratum and cell-cell adhesion is essential for FAK-dependent SMC proliferation in vivo. Our in vitro experiments show that a unidirectional crosstalk from FAK to N-cadherin regulates cell cycling in a spreading-dependent manner. In line with recent evidence demonstrating that cadherins are mechanosensors (Chopra et al., 2011; Conway et al., 2013; le Duc et al., 2010; Tzima et al., 2005), we show that N-cadherin expression changes with substrate stiffness. This crosstalk between ECM stiffness and N-cadherin may also occur in vivo as increased N-cadherin levels are linked to increased tissue stiffness during the response to vascular injury.
The small GTPases Rho and Rac are well-known regulators of cadherin adhesiveness and signaling. Coordinated signaling among different Rho family GTPases regulates cell-cell adhesions across different cell types (Braga et al., 2000; Takaishi et al., 1997; Watanabe et al., 2009; Wildenberg et al., 2006). In mesenchymal stem cells, N-cadherin-mediated cell-cell adhesion and expression are modulated by Rac signaling (Gao et al., 2010; Woods et al., 2007). We also found that Rac, rather than Rho, plays the major role in regulating stiffness-induced N-cadherin expression. As we recently demonstrated that Rac is a target of FAK and p130Cas in mesenchymal cells (Bae et al., 2014), it is fitting that Rac regulates N-cadherin expression.
ECM stiffness and cell spreading are reported to stimulate YAP and TAZ translocation into the nucleus, where they regulate gene transcription (Dupont et al., 2011; Tang et al., 2013). Cadherins and catenins are also important molecular players that regulate the localization of YAP/TAZ to modulate their transcriptional activity (Kim et al., 2011; Robinson and Moberg, 2011; Silvis et al., 2011). It is interesting to posit that crosstalk between cadherin and integrin signaling may culminate in the transcription of cell-cycle-regulatory genes by YAP/TAZ. We are currently investigating the role of YAP as well as other mechanoresponsive transcriptional regulators, such MRTF-A and NFκB (Janmey et al., 2013), in N-cadherin stimulated cell cycling.
While cadherins have been widely reported to arrest cell growth by regulating contact inhibition (Kim et al., 2011; McClatchey and Yap, 2012), pro-proliferative effects have also been observed (Fournier et al., 2008; Nelson et al., 2004; Reddy et al., 2005; Zhu and Watt, 1996). Our data suggest that cadherins are intrinsically neither pro- nor anti-proliferative. Rather, we propose that the proliferative outcome of cadherin engagement is modulated by contextual cues from the microenvironment. Our studies show that one of these contextual cues is the degree of cell spreading. Cell spreading is affected by the ECM composition and the differential expression or engagement of specific integrins. These processes themselves can be dynamic and cell-type specific, and likely contribute to the different proliferative effects of cadherin-mediated cell-cell adhesion.
EXPERIMENTAL PROCEDURES
In Vivo Vascular Injury
Femoral artery injuries were produced in wild-type mice and mice in which N-cadherin or FAK had been selectively deleted from SMCs. The number of bromodeoxyuridine (BrdU)- or EdU-positive nuclei per peak section was determined by using a method similar to that described by Kothapalli et al. (2007). Mouse procedures and protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Cell Proliferation on Micropatterned Adhesive Islands
Micropatterned adhesive islands were prepared as previously described (Nelson et al., 2007) and coated with 50 μg/ml FN. Serum-starved SMCs (2 × 104) were seeded on micropatterns in DMEM/F12 containing 10% FBS with EdU for 48 hr. Cells were then fixed in 3.7% formaldehyde in PBS and analyzed for EdU incorporation using the Click-iT EdU Imaging kit (Invitrogen) according to the manufacturer’s instructions. Only single or paired cells were analyzed in the unpaired and paired micropatterns, respectively. The percentage of EdU-incorporated cells was determined relative to DAPI-stained nuclei. At least 100 cells were counted per condition.
Analysis of Confocal Microscopy Images
Confocal images from fluorescently stained SMCs cultured on unpaired and paired micropatterned adhesive islands of different areas (2,500 μm2 and 10,000 μm2) were acquired using a Leica TCS SP8 laser scanning confocal with a 20×, 0.75 NA air immersion objective. FAKpY397 and N-cadherin fluorescence signal intensities for each unpaired cell or pair of cells were summed from three consecutive sections using FIJI software. The total signal intensity for FAKpY397 was then normalized to the spread area of each cell or pair of cells. Total N-cadherin signal intensity was measured from traced regions of cell-cell contact for paired cells (n = 5–9).
Supplementary Material
Highlights.
N-cadherin expression is regulated by stiffness-sensitive FAK signaling
N-cadherin is essential for in vivo vascular smooth muscle cell proliferation
N-cadherin overrides the spreading requirement for cell cycling
Acknowledgments
We thank Stefan Offermanns (Max Planck Institute for Heart and Lung Research) for generously providing the tamoxifen-inducible, smooth muscle-specific Cre mouse line; John Tobias for performing the bioinformatics analysis; and Paola Castagnino for the FAK western blot analysis of FAKfl/fl;iCre mouse aortas. This work was supported by NIH grants HL115553 and HL094491. K.L.M. was supported by NIH training grant R25CA101871. Y.H.B. was supported by a post-doctoral fellowship from the American Heart Association.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.02.023.
AUTHOR CONTRIBUTIONS
K.L.M., Y.H.B., L.G., C.S.C., and R.K.A. designed research. K.L.M., Y.H.B., L.G., S.-L.L., and T.X. performed experiments. K.L.M., Y.H.B., L.G., and R.K.A. performed data analyses and statistical tests. K.L.M., G.L.R., C.S.C., and R.K.A. wrote and edited the manuscript.
References
- Assoian RK, Klein EA. Growth control by intracellular tension and extracellular stiffness. Trends Cell Biol. 2008;7:24–27. doi: 10.1016/j.tcb.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae YH, Mui KL, Hsu BY, Liu S-L, Cretu A, Razinia Z, Xu T, Puré E, Assoian RK. A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci Signal. 2014;7:ra57. doi: 10.1126/scisignal.2004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister U, Funke R, Ebnet K, Vorschmitt H, Koch S, Vestweber D. Association of Csk to VE-cadherin and inhibition of cell proliferation. EMBO J. 2005;24:1686–1695. doi: 10.1038/sj.emboj.7600647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga VM, Betson M, Li X, Lamarche-Vane N. Activation of the small GTPase Rac is sufficient to disrupt cadherin-dependent cell-cell adhesion in normal human keratinocytes. Mol Biol Cell. 2000;11:3703–3721. doi: 10.1091/mbc.11.11.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary LA, Han DC, Polte TR, Hanks SK, Guan JL. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J Cell Biol. 1998;140:211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalasani K, Brewster RM. N-cadherin-mediated cell adhesion restricts cell proliferation in the dorsal neural tube. Mol Biol Cell. 2011;22:1505–1515. doi: 10.1091/mbc.E10-08-0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- Chopra A, Tabdanov E, Patel H, Janmey PA, Kresh JY. Cardiac myocyte remodeling mediated by N-cadherin-dependent mechanosensing. Am J Physiol Heart Circ Physiol. 2011;300:H1252–H1266. doi: 10.1152/ajpheart.00515.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol. 2013;23:1024–1030. doi: 10.1016/j.cub.2013.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desprat N, Supatto W, Pouille PA, Beaurepaire E, Farge E. Tissue deformation modulates twist expression to determine anterior midgut differentiation in Drosophila embryos. Dev Cell. 2008;15:470–477. doi: 10.1016/j.devcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Fonseca PM, Shin NY, Brábek J, Ryzhova L, Wu J, Hanks SK. Regulation and localization of CAS substrate domain tyrosine phosphorylation. Cell Signal. 2004;16:621–629. doi: 10.1016/j.cellsig.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Fournier AK, Campbell LE, Castagnino P, Liu WF, Chung BM, Weaver VM, Chen CS, Assoian RK. Rac-dependent cyclin D1 gene expression regulated by cadherin- and integrin-mediated adhesion. J Cell Sci. 2008;121:226–233. doi: 10.1242/jcs.017012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, McBeath R, Chen CS. Stem cell shape regulates a chondrogenic versus myogenic fate through Rac1 and N-cadherin. Stem Cells. 2010;28:564–572. doi: 10.1002/stem.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix—cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- Grazia Lampugnani M, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, Orsenigo F, Cattelino A, Kemler R, Daniel TO, Dejana E. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J Cell Biol. 2003;161:793–804. doi: 10.1083/jcb.200209019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haÿ E, Nouraud A, Marie PJ. N-cadherin negatively regulates osteoblast proliferation and survival by antagonizing Wnt, ERK and PI3K/Akt signalling. PLoS ONE. 2009;4:e8284. doi: 10.1371/journal.pone.0008284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janmey PA, Wells RG, Assoian RK, McCulloch CA. From tissue mechanics to transcription factors. Differentiation. 2013;86:112–120. doi: 10.1016/j.diff.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M, Sabatini PJ, Lee FS, Bendeck MP, Langille BL. N-cadherin upregulation and function in response of smooth muscle cells to arterial injury. Arterioscler Thromb Vasc Biol. 2002;22:1972–1977. doi: 10.1161/01.atv.0000036416.14084.5a. [DOI] [PubMed] [Google Scholar]
- Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci USA. 2011;108:11930–11935. doi: 10.1073/pnas.1103345108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA, Yin L, Kothapalli D, Castagnino P, Byfield FJ, Xu T, Levental I, Hawthorne E, Janmey PA, Assoian RK. Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr Biol. 2009;19:1511–1518. doi: 10.1016/j.cub.2009.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothapalli D, Zhao L, Hawthorne EA, Cheng Y, Lee E, Puré E, Assoian RK. Hyaluronan and CD44 antagonize mitogen-dependent cyclin D1 expression in mesenchymal cells. J Cell Biol. 2007;176:535–544. doi: 10.1083/jcb.200611058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Duc Q, Shi Q, Blonk I, Sonnenberg A, Wang N, Leckband D, de Rooij J. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol. 2010;189:1107–1115. doi: 10.1083/jcb.201001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey AI, Yap AS. Contact inhibition (of proliferation) redux. Curr Opin Cell Biol. 2012;24:685–694. doi: 10.1016/j.ceb.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Nelson CM, Chen CS. Cell-cell signaling by direct contact increases cell proliferation via a PI3K-dependent signal. FEBS Lett. 2002;514:238–242. doi: 10.1016/s0014-5793(02)02370-0. [DOI] [PubMed] [Google Scholar]
- Nelson CM, Pirone DM, Tan JL, Chen CS. Vascular endothelial-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol Biol Cell. 2004;15:2943–2953. doi: 10.1091/mbc.E03-10-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Liu WF, Chen CS. Manipulation of cell-cell adhesion using bowtie-shaped microwells. Methods Mol Biol. 2007;370:1–10. doi: 10.1007/978-1-59745-353-0_1. [DOI] [PubMed] [Google Scholar]
- Oda H, Tsukita S, Takeichi M. Dynamic behavior of the cadherin-based cell-cell adhesion system during Drosophila gastrulation. Dev Biol. 1998;203:435–450. doi: 10.1006/dbio.1998.9047. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pelham RJ, Jr, Wang Yl. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci USA. 1997;94:13661–13665. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M, Hynes RO. Developmental defects in mouse embryos lacking N-cadherin. Dev Biol. 1997;181:64–78. doi: 10.1006/dbio.1996.8443. [DOI] [PubMed] [Google Scholar]
- Reddy P, Liu L, Ren C, Lindgren P, Boman K, Shen Y, Lundin E, Ottander U, Rytinki M, Liu K. Formation of E-cadherin-mediated cell-cell adhesion activates AKT and mitogen activated protein kinase via phosphatidylinositol 3 kinase and ligand-independent activation of epidermal growth factor receptor in ovarian cancer cells. Mol Endocrinol. 2005;19:2564–2578. doi: 10.1210/me.2004-0342. [DOI] [PubMed] [Google Scholar]
- Resink TJ, Philippova M, Joshi MB, Kyriakakis E, Erne P. Cadherins and cardiovascular disease. Swiss Med Wkly. 2009;139:122–134. doi: 10.4414/smw.2009.12429. [DOI] [PubMed] [Google Scholar]
- Robinson BS, Moberg KH. Cell-cell junctions: α-catenin and E-cadherin help fence in Yap1. Curr Biol. 2011;21:R890–R892. doi: 10.1016/j.cub.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruest PJ, Shin NY, Polte TR, Zhang X, Hanks SK. Mechanisms of CAS substrate domain tyrosine phosphorylation by FAK and Src. Mol Cell Biol. 2001;21:7641–7652. doi: 10.1128/MCB.21.22.7641-7652.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini PJB, Zhang M, Silverman-Gavrila R, Bendeck MP, Langille BL. Homotypic and endothelial cell adhesions via N-cadherin determine polarity and regulate migration of vascular smooth muscle cells. Circ Res. 2008;103:405–412. doi: 10.1161/CIRCRESAHA.108.175307. [DOI] [PubMed] [Google Scholar]
- Sakane F, Miyamoto Y. N-cadherin regulates the proliferation and differentiation of ventral midbrain dopaminergic progenitors. Dev Neurobiol. 2013;73:518–529. doi: 10.1002/dneu.22077. [DOI] [PubMed] [Google Scholar]
- Sata M, Maejima Y, Adachi F, Fukino K, Saiura A, Sugiura S, Aoyagi T, Imai Y, Kurihara H, Kimura K, et al. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol. 2000;32:2097–2104. doi: 10.1006/jmcc.2000.1238. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol. 2010;2:a005066. doi: 10.1101/cshperspect.a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvis MR, Kreger BT, Lien W-H, Klezovitch O, Rudakova GM, Camargo FD, Lantz DM, Seykora JT, Vasioukhin V. α-Catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal. 2011;4:ra33. doi: 10.1126/scisignal.2001823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solon J, Levental I, Sengupta K, Georges PC, Janmey PA. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys J. 2007;93:4453–4461. doi: 10.1529/biophysj.106.101386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger A, Eger A, Wolf J, Beug H, Foisner R. E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J Cell Biol. 2001;154:1185–1196. doi: 10.1083/jcb.200104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaishi K, Sasaki T, Kotani H, Nishioka H, Takai Y. Regulation of cell-cell adhesion by rac and rho small G proteins in MDCK cells. J Cell Biol. 1997;139:1047–1059. doi: 10.1083/jcb.139.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Rowe RG, Botvinick EL, Kurup A, Putnam AJ, Seiki M, Weaver VM, Keller ET, Goldstein S, Dai J, et al. MT1-MMP-dependent control of skeletal stem cell commitment via a β1-integrin/YAP/TAZ signaling axis. Dev Cell. 2013;25:402–416. doi: 10.1016/j.devcel.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinkle CL, Lechler T, Pasolli HA, Fuchs E. Conditional targeting of E-cadherin in skin: insights into hyperproliferative and degenerative responses. Proc Natl Acad Sci USA. 2004;101:552–557. doi: 10.1073/pnas.0307437100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CE. Paxillin and focal adhesion signalling. Nat Cell Biol. 2000;2:E231–E236. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- Uglow EB, Slater S, Sala-Newby GB, Aguilera-Garcia CM, Angelini GD, Newby AC, George SJ. Dismantling of cadherin-mediated cell-cell contacts modulates smooth muscle cell proliferation. Circ Res. 2003;92:1314–1321. doi: 10.1161/01.RES.0000079027.44309.53. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Sato K, Kaibuchi K. Cadherin-mediated intercellular adhesion and signaling cascades involving small GTPases. Cold Spring Harb Perspect Biol. 2009;1:a003020. doi: 10.1101/cshperspect.a003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildenberg GA, Dohn MR, Carnahan RH, Davis MA, Lobdell NA, Settleman J, Reynolds AB. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127:1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- Woods A, Wang G, Dupuis H, Shao Z, Beier F. Rac1 signaling stimulates N-cadherin expression, mesenchymal condensation, and chondrogenesis. J Biol Chem. 2007;282:23500–23508. doi: 10.1074/jbc.M700680200. [DOI] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Zhu AJ, Watt FM. Expression of a dominant negative cadherin mutant inhibits proliferation and stimulates terminal differentiation of human epidermal keratinocytes. J Cell Sci. 1996;109:3013–3023. doi: 10.1242/jcs.109.13.3013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.