

Abstract
Background
Kabuki syndrome is a rare disorder characterized by the association of mental retardation and postnatal growth deficiency with distinctive facial appearance, skeletal anomalies, cardiac and renal malformation. Two causative genes have been identified in patients with Kabuki syndrome. Mutation of KMT2D (MLL2) was identified in 55–80 % of patients, while 9–14 % of KMT2D negative patients have mutation in KDM6A gene. So far, few tumors have been reported in patients with Kabuki syndrome. We describe the first case of a patient with spinal ependymoma and Kabuki syndrome.
Case presentation
A 23 years old girl followed at our Center for KMT2D mutated Kabuki syndrome since she was 4 years old presented with acute lumbar pain and intermittent tactile hyposthenia of the feet. Spine magnetic resonance revealed a lumbar endocanalar mass. She underwent surgical resection of the lesion and histologic examination showed a tanycytic ependymoma (WHO grade II).
Conclusion
Kabuki syndrome is not considered a cancer predisposition syndrome. Nonetheless, a number of tumors have been reported in patients with Kabuki syndrome. Spinal ependymoma is a rare disease in the pediatric and young adult population. Whereas NF2 mutations are frequently associated to ependymoma such an association has never been described in Kabuki syndrome. To our knowledge this is the first case of ependymoma in a KMT2D mutated Kabuki syndrome patient. Despite KMT2D role in cancer has previously been described, no genetic data are available for previously reported Kabuki syndrome patients with tumors. Nonetheless, the association of two rare diseases raises the suspicion for a common determinant.
Keywords: Kabuki syndrome, Spinal ependymoma, KMT2D mutation, Cancer predisposition syndromes
Background
Kabuki syndrome (KS), known as Kabuki make-up syndrome or Niikawa–Kuroki syndrome, is a rare disorder (1:32000 live births) [1] firstly described by Kuroki et al. in 1981 in patients that had a distinctive facial appearance, skeletal anomalies, cardiac and renal malformations, mild to moderate mental retardation and postnatal growth deficiency [2]. KS is associated with numerous alteration in body system and apparata such as neurological abnormalities, impairment in growth, endocrinological findings, cardiac and otolaryngological malformations, and other clinical manifestations [3]. Two causative genes have been identified in KS patients. In particular, mutation in KMT2D at 12q13.12 account for 55–80 % of the patient [4], whereas 9–14 % of KMT2D negative patients have deletions or mutation in KDM6A gene at Xp11.3 [5]. The absence of genetic mutations is not an exclusion criterion for clinical diagnosis of KS.
Some cases in literature reported the association with KS and cancer even if there are no conclusive findings of the increased risk for cancer in patients with the syndrome and there are no data about the real incidence of cancer in KS [3]. Notably, only scattered case reports are found in the literature of KS associated with pre-B-ALL, hepatoblastoma, neuroblastoma, Burkitt lymphoma and synovial sarcoma [6–11].
Ependymoma is a tumor derived from the ependymal cells lining the ventricular system and is the third most common central nervous system (CNS) tumor in childhood. Most cases are located intracranially, in particular in the posterior fossa; spinal location is less frequent [12]. Ependymomas are classified as subependymomas and myxopapillary (grade I), classic (grade II), and anaplastic (grade III), according to the World Health Organization (WHO). In the pediatric population, half of the cases are diagnosed in children of less than 5 years of age [13]. Most cases of ependymoma involve adult patients, in particular the highest rates were observed in the 45–64 years age group. On the contrary, ependymomas are five times more malignant in the age group < 19 years than in adults where the incidence rate ratio (IRR) of malignant to borderline malignant tumor is about 1.5. Spinal cord/cauda equina is the primary site in 52.1 % of all cases in adults, but in children (age < 19 years) it is involved in only about 20 % of cases [14].
We describe a KS girl with the diagnosis of grade II ependymoma of the filum terminale.
Case presentation
A girl born at the 40th week of gestation with a weight of 2,380 g presented hypotonia and submucous cleft palate, surgically corrected when she was 6 years old. Physical examination at 3 years of age showed facial anomalies including arched eyebrows with lateral thinning, long palpebral fissures with lateral eversion of the lower eyelid, long eyelashes, large prominent ears with dysplastic helices, and a depressed nasal tip, suggestive for KS. She also had hypotonia, joint laxity, retarded motor developmental milestones and mild cognitive deficit. Two-dimensional color-doppler echocardiography and renal ultrasound examination were normal. Genetic testing of KMT2D gene, performed by target resequencing on the MiSeq (Illumina) platform showed the heterozygosis deletion of two bases c.16085_16086delAG; the identified variation resulted at protein level in the nonsense mutation p.Lys5362Serfs*96.
At the age of 23, she presented with intermittent tactile hypoesthesia of the feet and worsening lumbar pain. Magnetic resonance imaging (MRI) of the spine revealed the presence of a lumbar endocanalar mass extending from L3 to L4, isointense on T1 and T2 weighted images with peripheral contrast enhancement. The lesion had a maximum cranial-caudal diameter of 45 mm with diffuse compression and posterior displacement of spinal nerve roots. At surgery, an L3 to L5 laminotomy was performed and gross total resection of a clivable tumor arising from the filum terminale accomplished (Fig. 1). No neurological complications occurred. Histology revealed a monomorphic proliferation of elongated cells with mild nuclear atypia, surrounded by eosinophilic fibrillary stroma with a fascicular or vaguely perivascular pattern of growth. Cells showed diffuse positivity for glial fibrillary acidic protein (GFAP +++) and dot-like positivity for epithelial membrane antigen (EMA). The mitotic index assessed by immunohistochemical staining against anti-Ki67 was about 3–5 % (Fig. 2). These findings led to the diagnosis of ependymoma, likely the tanycytic type (WHO grade II). On the basis of site of the lesion, extent of resection, histology and age of the patient, no other treatment was offered after surgical resection. She remains disease free fourteen months after diagnosis.
Fig. 1.
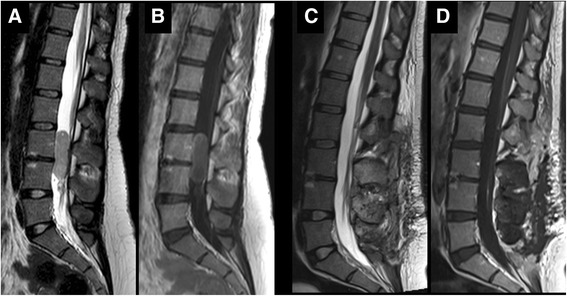
Spine MRI, sagittal pre-operative and post-operative images. Sagittal pre-operative TSE T2 WI (a) and T1 WI with Gd (b) show a lumbar endocanalar mass extending from L3 to L4, isointense on T1 and T2 WI with peripheral contrast enhancement. Sagittal post-operative TSE T2 WI (c) and T1 WI with Gd (d) show total resection of the tumor
Fig. 2.
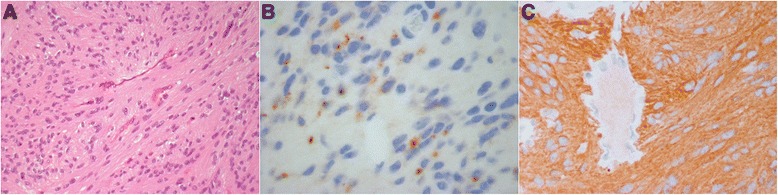
Photomicrographs of ependymoma of the filum terminale. A homogeneous proliferation of oval/elongated cells embedded in eosinophilic fibrillary stroma; a fascicular distribution and a vaguely perivascular pattern of growth are observed (a). At immunohistochemistry, a focal dot-like positivity for epithelial membrane antigen (EMA) is evident (b). Diffuse positivity of neoplastic cells for glial fibrillary acidic protein (GFAP +++) (c)
Conclusion
KS is a genetic syndrome firstly described in 1981. Patients present with mental and postnatal growth retardation associated with a characteristic facial appearance that resemble that of Kabuki Japanese theatre. Since KS is such a rare disease, no data are found in the literature about the incidence of cancer and the overall survival of patients with the syndrome. First cases described in the literature are now forty years old. These observations confirm that patients can survive to adulthood. The heterogeneity of anatomical and functional defects associated with KS can contribute in different ways to the long-term prognosis [15]. The genetic basis of KS were identified in 2010 and firstly associated to the mutation in KMT2D located in the chromosome 12 [4]. KMT2D is part of the lysine methyltransferase superfamily that are a group of evolutionarily conserved transcriptional regulators. The role of KMT2D is the methylation of histone 3 lysine 4 that acts as an activation mark [16, 17]. Guo et al. reported that the KMT2D deficiency modifies cancer cell proliferation and cell migration. Analysis of histone H3 modifications revealed that KMT2D is essential for maintaining the level of global H3K4 monomethylation and that its enzymatic SET domain is directly responsible for this function. Because they found that a majority of KMT2D binding sites were located in regions of potential enhancer elements, they supposed that these findings revealed the possible role of KMT2D in tumorigenesis [16]. Kabuki syndrome mutations C1430R and C1471Y reduce histone binding and catalytic activity of KMT2D [18]. The same reduction of function of KMT2D, due to somatic truncating mutations, has been reported in association to childhood cases of medulloblastoma and non-Hodgkin lymphoma [16].
Up to now, there are no available data regarding KMT2D mutation status in patients with KS that develop cancer. One study in a large cohort of pediatric cancer patients showed that the incidence of monogenic syndromes was higher than expected in the general population and that diagnoses of these syndromes had frequently been missed. Considering that 1 patient out of 1073 had been diagnosed with KS [6], Bogershausen et al. reported that the hypothesis of a KS predisposition to malignancies remains questionable [3].
To our knowledge, this is the first case of spinal ependymoma described in KS. Spinal ependymoma is a rare tumor of adults that represents about 60 % of spinal cords tumors. Pathophysiology of this tumor is not known so far but there is a high incidence (33 %) of spinal low grade ependymoma in patients with type 2 neurofibromatosis [19].
In conclusion, despite KS is not considered to be a cancer predisposition syndrome, an increasing number of tumors is being reported in these patients. No previous genetic data are available from KS patients developing tumors. KMT2D mutation is now found in most KS cases and shows a role in cancerogenesis. The rarity and heterogeneity of tumors descripted in patients with KS doesn’t permit to indicate an active oncologic surveillance in this patients. However we think that the description of new cases and the genetic characterizations of these patients can help us to better know the role of KMT2D mutation and the natural history of KS.
Consent
Written informed consent was obtained from the patients’ parents for publication of these Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Acknowledgements
The authors would like to thank patient for collaboration.
Abbreviations
- KS
Kabuki syndrome
- CNS
Central nervous system
- WHO
World Health Organization
- IRR
Incidence rate ratio
- MRI
Magnetic resonance imaging
- EMA
Epithelial membrane antigen
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DR has given the main contribution to conception and design of the paper; PP, RC, FRL and MCD have been involved in drafting the manuscript; LFG and FDC have been involved in acquisition of neuroimaging and histological findings; RM and AC have been involved in analysis and interpretation of data. AM, FR and CEM have been involved in revising it critically; AM has given final approval of the version to be published. All authors read and approved the final manuscript.
Contributor Information
Davide Roma, Email: davidroma.865@gmail.com.
Paolo Palma, Email: paolo2.palma@opbg.net.
Rossella Capolino, Email: rossella.capolino@opbg.net.
Lorenzo Figà-Talamanca, Email: lorenzo.figatalamanca@opbg.net.
Francesca Diomedi-Camassei, Email: francesca.diomedi@opbg.net.
Francesca Romana Lepri, Email: francescaromana.lepri@opbg.net.
Maria Cristina Digilio, Email: mcristina.digilio@opbg.net.
Carlo Efisio Marras, Email: carloefisio.marras@opbg.net.
Raffaella Messina, Email: raffaella.messina@opbg.net.
Andrea Carai, Email: andrea.carai@opbg.net.
Franco Randi, Email: franco.randi@opbg.net.
Angela Mastronuzzi, Email: angela.mastronuzzi@opbg.net.
References
- 1.Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H, et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet. 1988;31(3):565–89. doi: 10.1002/ajmg.1320310312. [DOI] [PubMed] [Google Scholar]
- 2.Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99:570–3. doi: 10.1016/S0022-3476(81)80256-9. [DOI] [PubMed] [Google Scholar]
- 3.Bögershausen N, Wollnik B. Unmasking Kabuki syndrome. Clin Genet. 2013;83(3):201–11. doi: 10.1111/cge.12051. [DOI] [PubMed] [Google Scholar]
- 4.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42(9):790–3. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dentici ML, Di Pede A, Lepri FR, Gnazzo M, Lombardi MH, Auriti C, et al. Kabuki syndrome: clinical and molecular diagnosis in the first year of life. Arch Dis Child. 2015;100(2):158–64. doi: 10.1136/archdischild-2013-305858. [DOI] [PubMed] [Google Scholar]
- 6.Merks JH, Caron HN, Hennekam RC. High incidence of malformation syndromes in a series of 1073 children with cancer. Am J Med Genet A. 2005;134A:132–43. doi: 10.1002/ajmg.a.30603. [DOI] [PubMed] [Google Scholar]
- 7.Tumino M, Licciardello M, Sorge G, Cutrupi MC, Di Benedetto F, Amoroso L, et al. Kabuki syndrome and cancer in two patients. Am J Med Genet A. 2010;152A:1536–9. doi: 10.1002/ajmg.a.33405. [DOI] [PubMed] [Google Scholar]
- 8.Scherer S, Theile U, Beyer V, Ferrari R, Kreck C, Rister M. Patient with Kabuki syndrome and acute leukemia. Am J Med Genet A. 2003;122A:76–9. doi: 10.1002/ajmg.a.20261. [DOI] [PubMed] [Google Scholar]
- 9.Ijichi O, Kawakami K, Matsuda Y, Ikarimoto N, Miyata K, Takamatsu H, et al. A case of Kabuki make-up syndrome with EBV + Burkitt’s lymphoma. Acta Paediatr Jpn. 1996;38:66–8. doi: 10.1111/j.1442-200X.1996.tb03439.x. [DOI] [PubMed] [Google Scholar]
- 10.Shahdadpuri R, O’Meara A, O’Sullivan M, Reardon W. Low-grade fibromyxoid sarcoma: yet another malignancy associated with Kabuki syndrome. Clin Dysmorphol. 2008;17:199–202. doi: 10.1097/MCD.0b013e3282f5f4e3. [DOI] [PubMed] [Google Scholar]
- 11.Casanova M, Selicorni A, Ferrari A. Cancer predisposition in children with Kabuki syndrome. Am J Med Genet A. 2011;155A:1504. doi: 10.1002/ajmg.a.33711. [DOI] [PubMed] [Google Scholar]
- 12.Kuttesh JF, Zieber Rush S, Ater JL. Brain tumors in childhood. In: Kliegman RM, Behrman RE, Stanton BF, St. Geme JW III, Schor NF, Behrman RE, editors. Nelson textbook of pediatrics. 19. Philadelphia, PA: Saunders Elsevier; 2011. pp. 1746–53. [Google Scholar]
- 13.Ailon T, Dunham C, Carret AS, Tabori U, Mcneely PD, Zelcer S, et al. The role of resection alone in select children with intracranial ependymoma: the Canadian Pediatric Brain Tumour Consortium experience. Childs Nerv Syst. 2015;31(1):57–65. doi: 10.1007/s00381-014-2575-4. [DOI] [PubMed] [Google Scholar]
- 14.Villano JL, Parker CK, Dolecek TA. Descriptive epidemiology of ependymal tumours in the United States. Br J Cancer. 2013;108(11):2367–71. doi: 10.1038/bjc.2013.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasdon BD, Fox JE. Kabuki syndrome: diagnostic and treatment considerations. Ment Health Fam Med. 2012;9(3):171–9. [PMC free article] [PubMed] [Google Scholar]
- 16.Guo C, Chen LH, Huang Y, Chang CC, Wang P, Pirozzi CJ, et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget. 2013;4(11):2144–53. doi: 10.18632/oncotarget.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ali M, Hom RA, Blakeslee W, Ikenouye L, Kutateladze TG. Diverse functions of PHD fingers of the MLL/KMT2 subfamily. Biochim Biophys Acta. 2014;1843(2):366–71. doi: 10.1016/j.bbamcr.2013.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X, et al. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26:2749–62. doi: 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zemmoura I, Vourc’h P, Paubel A, Ikenouye L, Kutateladze TG. A deletion causing NF2 exon 9 skipping is associated with familial autosomal dominant intramedullary ependymoma. Neuro Oncol. 2014;16(2):250–5. doi: 10.1093/neuonc/not165. [DOI] [PMC free article] [PubMed] [Google Scholar]