

Abstract
Endogenous mechanisms leading to host protection and resolution of infections without immunosuppression are of wide interest1,2. Here we elucidated the structures of four new host-protective molecules produced in neutrophil-endothelial co-cultures, and present in human and mouse tissues after sterile inflammation or infection. These bioactive molecules contained conjugated triene and diene double bonds with each carrying a 13-carbon position alcohol and were derived from n-3 docosapentaenoic acid (DPA, C22:5). These compounds, termed 13-series resolvins (RvT), demonstrated potent protective actions increasing mice survival during Escherichia coli infections. RvT also regulated human and mouse phagocyte responses stimulating bacterial phagocytosis and regulating inflammasome components. Their biosynthesis during neutrophil-endothelial cell interactions was initiated by endothelial cyclooxygenase-2 (COX-2) and increased by atorvastatin via S-nitrosylation of COX-2. The actions of atorvastatin and RvT were additive in E. coli infections in mice where they accelerated resolution of inflammation and increased survival >60%. These results document novel host protective molecules in bacterial infections, namely 13-series resolvins, derived from n-3 DPA via transcellular biosynthesis and increased by atorvastatin. These novel molecules regulate key innate protective responses in the resolution of infectious-inflammation.
Infections are a leading cause of mortality worldwide, with bacterial infections, including those caused by Escherichia coli, posing an urgent health concern1,2. The prevalent approach for treating bacterial infections is administration of antibiotics, but with the rise in antibiotic-resistant bacteria, there is a pressing need for new treatment strategies1,2. In response to infection, the host mounts inflammatory responses that when self-resolving are protective3–5. We previously characterized potent endogenous anti-inflammatory and pro-resolving mediators namely resolvins, protectins and maresins known as specialized pro-resolving mediators (SPM)4. These are biosynthesized from n-3 essential fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)4. Docosapentaenoic acid (n-3, DPA), the intermediate in DHA biosynthesis from EPA, accumulates in individuals6 with single nucleotide polymorphisms in the gene encoding for fatty acid elongase-2 and is also the precursor for three new SPM families7.
During the resolution phase of a self-resolving acute inflammation, SPM are produced locally and exert protective actions on leukocytes stimulating clearance of apoptotic cells, debris and bacteria as well as promote tissue regeneration4,8. Signals produced in the early phase of infectious-inflammation can determine the amplitude and duration of the inflammatory response9. Hence, novel mediators that may be produced during early phases of self-resolving infections to fine-tune the initial response and promote resolution are of general interest.
Neutrophils are the first line of defense against invading pathogens10,11. They are rapidly recruited from the vasculature to site(s) of infection and participate in bacterial containment and clearance8. One of the first steps in recruitment is neutrophil capture onto the vascular endothelium10. To identify new molecules that may exert host protective actions produced during this key step and since resolvins and protectins enhance the host clearance of bacterial infections8, we obtained fractions from co-incubations of these cell types using C18 solid phase extraction (see methods). Given that E. coli infections are an urgent world wide health concern with at least ~270,000 new cases reported per year in the United states alone12 we next the assessed the actions of these fractions in E. coli infections in mice. When administered intravenously immediately prior to E. coli inoculation isolates from these human neutrophil-endothelial cell co-incubations significantly enhanced survival from lethal E. coli infections (Fig. 1a; p<0.05) and did not display direct antibacterial activity (Supplementary Fig. 1a).
Figure 1. Novel 13-series resolvins (RvT) from neutrophil-endothelial cell interactions are elevated in self-resolving inflammation.

(a) Fractions were extracted from human neutrophil (PMN)-endothelial cell (EC) co-incubations (see methods), administered to mice via i.v. injection 5 min prior to E. coli (2.5×107 CFU/mouse) inoculation and survival assessed. n=10 mice/group from three independent experiments. *p<0.05 vs. E. coli group. (b) Proposed structures from the novel 13-series resolvins (RvT) identified in human PMN-EC co-incubations. (c) Blood was collected from healthy volunteers pre- and post-exercise (see methods) and amounts of RvT assessed using LC-MS-MS. n=4 healthy volunteers from four independent experiments. *p<0.05 vs. pre-exercise values. (d) Plasma was obtained from the NIST repository (SRM 1950, d=3), collected from healthy volunteers (n=4) or patients diagnosed with sepsis (n=9), and RvT assessed using LC-MS-MS. Results are expressed as mean±s.e.m. from two independent experiments. *p<0.05, **p<0.01 vs. SRM 1950, # p<0.05, ## p<0.01 vs. healthy volunteers. (e) Mice were inoculated with E. coli (1×105 CFU/mouse), blood was collected at the indicated intervals and RvT amounts assessed using LC-MS-MS. (f) Mice were inoculated with 1×105 CFU/mouse E. coli (self-resolving) or 1×107 CFU/mouse E. coli (delayed-resolving), blood was collected after 4h and RvT assessed using LC-MS-MS. Results for e, f are expressed as mean± s.e.m. n=4 mice per group from two independent experiments. *p<0.05 vs 0h blood levels (e) or vs. mice receiving a self-resolving inoculum (f).
To characterize and elucidate bioactive molecule(s) within these fractions and deduce their structure(s), we initially used liquid chromatography tandem mass-spectrometry (LC-MS-MS) based lipid mediator (LM)-metabololipidomics. Along with classic eicosanoids and SPM, we identified previously unknown molecules in fractions that showed activity from neutrophil-endothelial cell co-incubations. These gave four distinct MS-MS spectra and retention times characteristic of a 22-carbon backbone with five double bonds suggesting DPA was the precursor7. Using MS-MS fragmentations we deduced the structures of these new n-3 DPA derived bioactive molecules as: 7,13,20-trihydroxy-docosapentaenoic acid, 7,12,13-trihydroxy-docosapentaenoic, 7,8,13-trihydroxy-docosapentaenoic acid and 7,13-dihydroxy-docosapentaenoic acid (Fig. 1b and Supplementary Figs. 1b and 2a–d). Their assignments were confirmed using physical characteristics including MS-MS of different chemical derivatives and reactivity (i.e.18O2 incorporation, acid methanol trapping and methyl ester vide infra and online methods) for each product. Co-incubations of neutrophil-endothelial cells with n-3 DPA, but neither DHA nor EPA precursors to SPM4,9, increased their amounts 30–50% (Supplementary Fig. 2e). Thus these new bioactive structures were denoted 13-series resolvins (RvT), RvT1, RvT2, RvT3 and RvT4 respectively, since each carried a carbon-13 position alcohol from their biosynthetic origin (Supplementary Fig. 1b).
Since in humans exercise leads to a self-resolving inflammatory state marked by increases in plasma lipid mediators13, we investigated RvT production in healthy volunteers during exercise. Significant increases in peripheral blood RvT were obtained when compared to pre-exercise levels (Fig. 1c, Supplementary Table 1a; p<0.05). Their amounts were comparable in magnitude to those from the AA, EPA or DHA bioactive-metaboIomes, namely prostaglandins, leukotriene B4, resolvins, protectins and maresins (Supplementary Table 1b). RvT were also present in human peripheral blood from sepsis patients (Fig. 1d, Supplementary Table 2) and when compared to amounts in plasma from healthy volunteers and those in a human plasma composite obtained from the National Institutes for Standards and Technologies (SRM1950). Resolvins and lipoxins were also increased in sepsis patient plasma when compared to both healthy volunteer plasma and the SRM1950 plasma composite (Supplementary Table 3).
We then assessed the temporal regulation of RvT during bacterial infections in mice. FVB mice challenged with E. coli 105 CFU E. coli/mouse had a self-resolving (i.e. self-limited) inflammatory response with PMN numbers reaching a maximum at ~12h followed by their decline8. RvT were rapidly formed during the initiation phase of inflammation with amounts in peripheral blood reaching a maximum at 4h that subsequently declined (Fig. 1e). By comparison, mediators derived from AA-, EPA- and DHA-derived mediators also increased at 4h with the majority reaching maximum at 12h. These included RvD2 and LXB4 (Supplementary Table 4). Notably, RvT were >60% lower in peripheral blood from mice challenged with a higher E. coli burden that was non-lethal but caused increased inflammation with delayed resolution (107 CFU/mouse; Fig. 1f).
We speculated based on human neutrophil-endothelial cell co-incubations that RvT were products of transcellular biosynthesis because these co-incubations gave appreciable amounts of RvT1, RvT2, RvT3 and RvT4 (Supplementary Fig. 2f). Chiral LC-MS-MS demonstrated that endothelial cells incubated with n-3 DPA gave increased 13R-hydroxy-7Z,10Z,14,16Z,19Z-docosapentaenoic acid (13R-HDPA; Supplementary Fig. 3a–d) when compared to cells kept in culture medium alone. Since IL-1β and TNF-α up-regulate endothelial COX-2 expression14, which converts DHA to 13-hydroxy-docosahexaenoic acid9, we questioned whether endothelial COX-2 also converts n-3 DPA to produce 13-HDPA. Incubations of endothelial cells with celecoxib, a COX-2 selective inhibitor, or transfection of endothelial cells with shRNA targeting COX-2, each led to significant reductions in endothelial cell derived 13-HDPA (Supplementary Fig. 3e, f, p<0.05), indicating a role for COX-2 in 13-HDPA production. Incubations of n-3 DPA with human recombinant COX-2 gave 13R-HDPA (Supplementary Fig. 3g) with KM=3.1±1.3μM (Supplementary Fig. 3h), suggesting that n-3 DPA is converted to 13R-HpDPA by endothelial COX-2. This intermediate or its reduced alcohol form may then be donated to neutrophils that convert the intermediate(s) to RvT. This proposed biosynthetic route and the RvT structures were corroborated by assessing UV chromophores, MS-MS fragmentation spectra for methyl-ester derivatives (Supplementary Fig. 4), molecular oxygen (18O2) incorporation (Supplementary Fig. 5) and acid-alcohol trapping of epoxide intermediates (Supplementary Fig. 6). These results indicate that COX-2 derived 13-HDPA is converted via lipoxygenation in human neutrophils to RvT1, RvT2, RvT3 and RvT4.
Atorvastatin is protective in bacterial infections in mice 15 and regulates COX-216. Hence we next questioned whether this statin regulates RvT biosynthesis. Incubation of neutrophil-endothelial cell co-cultures with atorvastatin, at concentrations (0.03–30μM) relevant to those reached in humans17, led to dose-dependent increases in RvT (Fig. 2a). Fractions from these co-incubations significantly increased survival in mice during E. coli infections when compared to fractions from neutrophil-endothelial cell co-cultures without atorvastatin (Fig. 2b; p<0.05) and those from either neutrophil or endothelial cells incubated separately with atorvastatin (Fig. 2b). Celecoxib significantly reduced RvT in these incubations (Fig. 2c; p<0.05) and abolished the protective actions of isolated fractions obtained from neutrophil-endothelial cell co-incubations with atorvastatin (Fig. 2c).
Figure 2. Atorvastatin increases protective actions of neutrophil-endothelia cell fractions during mouse infections and promotes RvT formation via S-nitrosylated COX-2.

(a) RvT in human neutrophil-endothelial cells co-incubations with or without atorvastatin. Mean±s.e.m.; n=4 independent cell preparations. Four independent experiments. *p<0.05 vs. EC group. (b) Fractions extracted using C18 SPE (see methods) were administered to mice (i.v.) 5 min prior to E. coli (2.5×107 CFU/mouse) inoculation and survival assessed. n= 14–17 mice/group. Three independent experiments. *p<0.05 vs. E. coli, #p<0.05 vs. E. coli plus hPMN-EC, §p<0.05 vs. E. coli plus hPMN plus atorv or E. coli plus hEC plus atorv. (c, d) Endothelial cells were incubated with IL-1β and TNF-α then vehicle, celecoxib and/or atorvastatin followed by n-3 DPA and hPMN. (c–d) Fractions were extracted (c) profiled using LM metabololipidomics. Mean±s.e.m. n=4 independent cell preparations/group. Three independent experiments. *p<0.05, **p<0.01 vs. incubations with IL-1β plus TNF-α alone; #p<0.05 vs incubations with IL-1β plus TNF-α atrovastatin. (d) administered prior to E. coli (2.5×107 CFU/mouse) and survival assessed. n=10 mice/group. Two independent experiments. *p<0.05 vs. E. coli; #p<0.05 vs. E. coli plus hPMN-EC plus atorv. (e–f) Mice were inoculated with E. coli (1×105 CFU/mouse), 1h later L-NAME (24mg/kg) or vehicle (saline) was administered followed by atorvastatin (5μg/mouse, i.v., 5h) or vehicle (saline plus 0.01% EtOH). (e) Plasma RvT were quantified using LM metabololipidomics, (f) exudate cell numbers were enumerated. Mean±s.e.m. n=4 mice/group. Two independent experiments. *p<0.05 vs. E. coli. #p< 0.05 vs. E. coli plus atorvastatin. (g) Conversion of n-3 DPA by human recombinant (hr) COX-2 and S-nitrosylated (SNO) hr-COX2. Mean±s.e.m. n=3 incubations. Three independent experiments.
Administration of L-NG-Nitroarginine Methyl Ester (L-NAME) prior to atorvastatin significantly reversed atorvastatin-mediated increases in plasma RvT (Fig. 2e; p<0.05) and increased neutrophil recruitment to the peritoneum during mouse infections (Fig. 2f). With human endothelial cells NOS inhibitors (L-NAME and 1400W) also significantly reduced atorvastatin-mediated RvT increases by 70–90% (Supplementary Fig. 7a; p<0.05). In addition, incubation of human recombinant COX-2 (hrCOX-2) with S-nitrosoglutathione, which S-nitrosylates COX-218 (Supplementary Fig. 7b), gave increased 13R-HDPA and hrCOX-2 catalytic activity (Fig. 2g) compared to native COX-2. These results indicated that atorvastatin increased RvT production via S-nitrosylation of endothelial COX-2.
Next we tested the bioactions of these molecules with human leukocytes. RvT were produced using recombinant enzymes and primary human neutrophils and isolated (see methods for details) using reverse phase high-pressure liquid chromatography (RP-HPLC). RvT1 and RvT4 were isolated to apparent homogeneity based on their appearance beneath a single RP-HPLC peak and characteristic UV chromophores. RvT2 and RvT3 were tested together since they eluted in the same RP-HPLC fractions. RvT1 (1pM–10nM) dose-dependently increased E. coli phagocytosis and production of intracellular reactive oxygen species (ROS) in human macrophages (Fig. 3a, b) as well as efferocytosis of apoptotic neutrophils (Fig. 3c), a key step in the resolution of inflammation4. Similar actions were obtained with human neutrophils, where RvT1 (1pM–10nM) dose-dependently increased E. coli phagocytosis (20–55%) and intracellular ROS (20–40%; n=4 donors). RvT1 (10pM–10nM) also dose-dependently blocked human macrophages activation of inflammasome components, decreasing caspase-1 and IL-1β expression and extracellular lactate dehydrogenase activity (LDH; Supplementary Fig. S8a–c). Similar results were obtained with RvT4 and RvT2 plus RvT3 (Supplementary Fig. 8a–c).
Figure 3. RvT regulate leukocyte responses and promote survival in infections.

(a) Macrophages (5×104 cells/well) were incubated with the indicated concentrations of RvT1, RvT2 plus RvT3 (1:1 ratio), RvT4 or vehicle (PBS containing 0.01% EtOH; 15min, 37°C, pH 7.45) then fluorescently labeled E. coli (2.5×106 CFU/well). (b) Macrophages (5×104 cells/well) were incubated with H2DCFDA (5μM, 30min, 37°C, pH 7.45); cells were washed, incubated with the indicated concentrations of RvT1, RvT2 plus RvT3 (1:1 ratio), RvT4 or vehicle (15 min, 37°C, pH 7.45), E. coli (2.5×106 CFU/well) were added and intracellular ROS levels determined. (c) Macrophages (5×104 cells/well) were incubated as in (a) with RvT, fluorescently-labeled apoptotic PMN (2.5×105 cells/well) were added and uptake assessed (see methods). Mean±s.e.m. n=4 donors. Three independent experiments. *p<0.05, **p<0.01 vs. macrophages plus vehicle. (d–g) Mice were given vehicle (saline containing 0.1% EtOH) or combination of RvT1, RvT2, RvT3 and RvT4 (ratio 2:1:1:8), each isolated and quantified by RP-UV-HPLC (see methods), via i.p. injection ~5min prior to E. coli (1×107 CFU/mouse) inoculation. 12h later (d) body temperatures, (e) peritoneal exudate neutrophil counts, (f) bacterial phagocytosis by peritoneal leukocytes (%E. coli+ of total CD11b+ population), (g) Macrophage efferocytosis in peritoneal exudates were assessed. Mean± s.e.m. n=5 mice/group. Two independent experiments. *p<0.05, **p<0.01 vs. E. coli mice. #p<0.05 vs. naive mice. (h) Mice were inoculated with E. coli (2.5×107 CFU/mouse); 2h later vehicle (saline+0.1% EtOH) or RvT (as in d–g; 50ng/mouse or 500ng/mouse) were administered via i.p. injection and survival assessed. n=10 mice per group. Two independent experiments *p<0.05 vs. E. coli mice.
Given RvT were each identified in human and murine infections, we assessed their combined actions in E. coli infections in mice, administering a mixture of RvT1, RvT2, RvT3 and RvT4 (RvT1-4) immediately prior to intraperitoneal E. coli inoculation. Twelve hours later these molecules afforded dose-dependent protection against hypothermia, limited further neutrophil recruitment to sites of inflammation, increased bacterial phagocytosis by peritoneal leukocytes (Fig. 3d–f), and reduced monocyte/macrophage expression of caspase-1, IL-1β levels and LDH activity (Supplementary Fig. 8d–f) without exerting direct antibacterial activities (Supplementary Fig. 9a). RvT also reduced systemic inflammation as demonstrated by reduced platelet-leukocyte aggregates7 (Supplementary Fig. 8g). LM metabololipidomics of plasma from mice given RvT1-4 (500ng/mouse) gave significant reductions in TxB2, PGD2 and PGE2 (Supplementary Fig. 9b; p<0.05) that are elevated by inflammasome activation (i.e. eicosanoid storm) during infections19. RvT also significantly increased exudate macrophage efferocytosis (Fig. 3g; p<0.05). Moreover, administration of RvT1-4 2h after E. coli inoculation dose-dependently increased mice survival (Fig. 3i).
Prolonged statin use is linked with a number of adverse effects including inflammasome activation20 and diabetes21. Therefore we next tested whether RvT could reduce the effective dose of atorvastatin needed to clear E. coli infections thus reducing statin exposure. Co-administration of RvT1-4, (~12.5ng/mouse each), and a sub-threshold dose of atorvastatin (0.5μg/mouse) immediately prior to E. coli inoculation significantly protected mice from hypothermia, reduced neutrophil recruitment (~50%; p<0.05), local and systemic bacterial loads, exudate IL-1β levels (~45%; p<0.05) and LDH activity (~40%; Supplementary Fig. 10a–e; p<0.05). This co-treatment also significantly increased bacterial phagocytosis by exudate leukocytes (~90%; p<0.05) and efferocytosis by exudate macrophages (~90%; Supplementary Fig. 10f, g; p<0.05). Thus suggesting that RvT may reduce the exposure to atorvastatin therapy potentially limiting some of the unwanted side effects of statins observed at higher statin doses20,21.
In order to assess whether this treatment regime was also protective in a therapeutic setting, we treated mice with atorvastatin (0.5 μg/mouse) and/or a mixture of RvT1-4 (50 ng/mouse) 2h after E. coli inoculation. Co-administration of atorvastatin and RvT significantly reduced neutrophil recruitment to the peritoneum and reduced exudate bacterial loads (Supplementary Fig. 10h; p<0.05). To gain insight into potential mechanism(s) activated by atorvastatin and RvT, we investigated expression of host protective22,23 and pro-inflammatory factors24 regulated by atorvastatin. In mice given 0.5 μg/mouse statin alone, we measured increases in host-protective PGI2 and 15-deoxy-Δ-12,14-PGJ2, (Supplementary Fig. 10i) whereas in endothelin-1 and plasminogen activator inhibitor-1 were reduced, molecules associated with a pro-inflammatory status (Supplementary Fig. 10j). RvT administration (50ng/mouse) regulated some of these molecules, although to a lesser extent (~30–60% lower) than atorvastatin. Co-administration of atorvastatin and RvT1-4 significantly reduced these molecules when compared to vehicle- or RvT-treated mice (Supplementary Fig. 10j, k; p<0.05). Peripheral blood eicosanoids were also reduced upon RvT1-4 administration (Supplementary Fig. 10k), effects that were only in part shared with atorvastatin.
Given these protective actions we assessed whether co-administration of RvT and atorvastatin could accelerate resolution of infections. Administration of RvT1-4 (50 ng/mouse) together with atorvastatin (0.5 μg/mouse) at peak neutrophil infiltration (12h) significantly accelerated exudate neutrophil clearance, increased bacterial phagocytosis by exudate leukocytes (~50%; p<0.05), accelerated resolution of inflammation, shortening the resolution interval from ~20h to ~8h (Fig. 4a and Supplementary Fig. 10). Co-administration of RvT1-4 with atorvastatin at both high (500ng RvT plus 5μg atorvastatin) and low doses (50ng RvT plus 0.5μg atorvastatin) increased survival of mice compared to treatment with atorvastatin alone (Fig. 4b). Together these results suggest that during infections RvT lower the effective dose of atorvastatin.
Figure 4. Atorvastatin and RvT accelerate resolution of infections and promote survival in bacterial infections in mice.
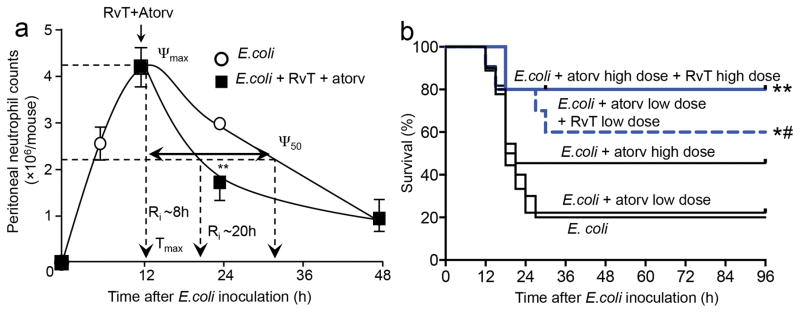
(a) Mice were inoculated with E. coli (1×105 CFU/mouse) plus RvT (combination of RvT1, RvT2, RvT3 and RvT4, ratio of 1:1:1:1, isolated and quantified by RP-UV-HPLC, total 50ng/mouse). Atorvastatin (0.5μg/mouse; i.p.) or vehicle (saline containing 0.1% EtOH) was administered and exudate neutrophil counts (CD11b+Ly6G+) assessed at the indicated time points using light microscopy and flow cytometry. Results are mean±s.e.m. n=4 mice per group from two independent experiments. **p<0.01 vs. 24h vehicle group. (b) Mice were inoculated with E. coli (2.5×107 CFU/mouse); after 3h administered vehicle (Saline+0.1% EtOH), 0.5μg/mouse (atorv low dose), 5μg/mouse (atorv high dose) atorvastatin, 0.5μg/mouse atorvastatin plus 50ng/mouse RvT (atorv low dose plus RvT low dose) or 5μg/mouse atorvastatin plus 500ng/mouse RvT (atorv high dose plus RvT high dose); each of the RvT was isolated as in panel (a) and survival assessed. n=10 mice per group from three independent experiments. *p<0.05, **p<0.01 vs. E. coli mice. #p<0.05 vs. E. coli plus atorv low dose mice.
In summary, using LC-MS-MS based lipid mediator-metabololipidomidics, we elucidated the structures of four new bioactive molecules termed RvT formed by transcellular biosynthesis 4,9,14 during human neutrophil-endothelial cell interactions (Supplementary Table.5 and Supplementary Fig. 1b). RvT production was elevated during self-resolving inflammatory challenge in humans and mice, and down-regulated in mice during delayed resolution of infections. These 13-series resolvins from n-3 DPA exerted both anti-inflammatory and potent pro-resolving activities regulating host responses during E. coli infections in mice and phagocytosis by isolated human cells, thereby fulfilling criteria as immunoresolvents4: namely, they stimulate the cardinal signs of resolution including expurgatio reliquiorum (clearance of debris), expurgatio contagionem agentis (clearance of infective agents), doloris absentia (analgesia) and muneris lucrum (gain of function)4. Given the extensive size of the vascular-endothelial system in humans and the abundance of neutrophils within the circulation, RvT formation during early stages of self-resolving acute inflammation and their regulation by atorvastatin, could provide a molecular basis for the development of new treatment strategies for infectious-inflammation. In addition, RvT may also serve as statin-markers and mediators of their actions during resolution responses.
Online Methods
E. coli peritonitis
All animal experiments were conducted in accordance with the Harvard Medical Area Standing Committee on Animals (protocol no. 02570). Mice (male FvB, 6–8 weeks old, Charles River, fed lab diet containing essential fatty acids as from supplier) were anesthetized with isoflurane and microbial peritonitis initiated. Briefly, test compounds or vehicle were injected i.p. or i.v. 5 min prior to live E. coli (serotype O6:K2:H1; 1×105 or 1×107CFU/mouse). At designated time intervals mice were harvested, blood was collected via cardiac puncture in heparin, and peritoneal exudates were collected in 4ml of PBS. The cellular composition in the exudates was determined using Turks solution and light microscopy, and by flow cytometry. For flow cytometry, exudate cells were incubated with anti-mouse CD16/CD32 (eBiosciences, 20min, 4°C, in PBS containing 5% fetal calf serum-staining solution), followed by incubation with FITC-conjugated anti-mouse Ly6G antibody (clone:1A8, eBiosciences), PE-conjugated anti-mouse F4/80 antibody (clone:BM8, eBiosciences) and PerCP-Cy5.5-conjugated anti-mouse CD11b antibody (clone:M1/70, eBiosciences) for 30min (4°C, in staining solution). To assess bacterial phagocytosis in peritoneal exudate leukocytes, exudate cells were incubated with PerCP-Cy5.5-conjugated anti-mouse CD11b antibody (30 min, 4°C, in staining solution), then fixed and permeabilized using BD Perm/Wash™ Buffer (BD Biosciences) following manufacturer’s instructions and incubated with FITC-conjugated anti-E. coli antibody (GenTex, 30 min, 4°C, in BD Perm/Wash™ Buffer). Efferocytosis in infectious exudates was assessed by incubating exudate leukocytes with PE-conjugated anti-mouse F4/80 antibody (30 min, 4°C, in staining solution), then fixed and permeabilized using BD Perm/Wash™ Buffer (BD Biosciences) and incubated with FITC-conjugated anti-mouse Ly6G antibody (30 min, 4°C, in BD Perm/Wash™ Buffer). Body temperatures were assessed using an Infrared thermometer (Fluke). Bacterial counts in peripheral blood and inflammatory exudates were determined by overnight cultures (37°C) of serially diluted samples on LB agar plates. Caspase 1 levels were assessed by incubation of exudate cells with PerCP-Cy5.5-conjugated anti-mouse CD11b antibody (30 min, 4°C, in staining solution), then fixed and permeabilized using BD Perm/Wash™ Buffer (BD Biosciences), incubated with anti-caspase 1 antibody (Abcam, 30 min, 4°C, in BD Perm/Wash™ Buffer) followed by anti-rabbit Alexa 488 conjugated antibody (Life Technologies, 30 min, 4°C, in BD Perm/Wash™ Buffer). IL-1β levels were determined in cell-free supernatants using an anti-mouse IL-1β ELISA (Abcam) following manufacturer’s instructions. Lactate dehydrogenase activity was measured in cell-free supernatants assessing the formation of NADH from NAD+ using a Lactate Dehydrogenase Assay kit (Sigma) following manufacturer’s instructions.
In select experiments resolution indices were calculated as in 27. ψmax, maximal PMN numbers in exudates; Tmax, time point when PMN numbers reach maximum; R50, 50% of maximal PMN numbers; T50, time point when PMN numbers reduce to 50% of maximum; Ri (resolution interval), T50-Tmax, time period when 50% PMN are lost from the exudates. To assess the proresolving actions of the test products, these were administered 12h after E. coli (105 CFU) inoculation, and resolution indices were assessed.
For some experiments, mice were administered via i.p. injection RvT1, RvT2, RvT3 and RvT4 (that were isolated as detailed below and combined as a mixture at a ratio of 2:1:1:8 respectively) 2h after E. coli inoculation. Blood and lungs were harvested 6h after E. coli inoculation. Blood was employed to determine peripheral blood eicosanoid levels as detailed below. Lungs were placed in TriReagent (Invitrogen), RNA isolated following manufacturer’s instructions and relative mRNA expression for mouse endothelin-1 and plasminogen activator inhibitor-1 determined using Qiagen SYBR® Green ROX qPCR mastermix and QuantiTect® Primer assays following manufacturer’s instruction and using mouse Glyceraldehyde 3-phosphate dehydrogenase as house keeping gene. mRNA expression was determined using an ABI PRISM 7900HT system (Applied Biosystems). In determined experiments, peritoneal exudates were obtained from mice 12h after E. coli inoculation and bacterial loads along with leukocyte counts determined as detailed above.
For survival studies, mice were administered test compounds or biological isolates from neutrophil-endothelial cell (HUVEC) co-incubations via i.p. injection 5 min prior to inoculation with E. coli (2.5×107 CFU), and survival was assessed. In designated experiments, RvT1, RvT2, RvT3 and RvT4 were isolated as detailed below, combined as a mixture at a ratio of 2:1:1:8 respectively, and were administered via i.p. injection 2h after E. coli (2.5×107 CFU); survival was assessed for the subsequent 94h. In separate experiments, 0.5μg (lower dose) or 5μg (higher dose) of atorvastatin was administered via i.p. with or without RvT1, RvT2, RvT3 and RvT4, which were isolated and quantified as detailed below, combined as a mixture (at a ratio of 2:1:1:8) at the indicated concentrations 3h after E. coli (2.5×107 CFU) inoculation, and survival assessed for the next 93h.
In a separate set of experiments, mice were inoculated with E. coli; after 60 min they were given L-NAME (24mg/kg) and 5 min later atorvastatin (5μg) via i.v. injection. Plasma was obtained after 5h via cardiac puncture and LM levels were investigated by lipid mediator metabololipidomics. For all animal experiments mice were randomized to vehicle or treatment groups without blinding.
Biosynthesis and lipid mediator metabololipidomics
Isolated n-3 DPA (Cayman Chemicals) was incubated with human recombinant COX-2 (Cayman Chemicals; in 0.1M Tris-HCl, pH 8.0, 20μM porcine hematin, 0.67mM phenol) for 30min at RT. Incubations were stopped with 1 volume of methanol and products extracted using diethyl ether as in 28. 13-HDPA was isolated using RP-HPLC (1100 Series; Agilent Technologies) and an Agilent C18 Poroshell column (2.7μm × 4.6 mm × 150 mm) with a mobile phase consisting of methanol/water (60:40, vol:vol) at 0.5 ml/min that was ramped up to 98:2 (vol:vol) for 20 min. 13-HDPA (10μM) was incubated with potato 5-LOX (Cayman Chemicals) for 30 min (4°C, 0.1M phosphate buffer, pH 6.3, 0.03% Tween 20). Incubations were stopped with 1 volume of ice-cold methanol, products were reduced using NaBH4 (Sigma-Aldrich) and extracted as in 28.
Methanol (2 volumes) was added to cell incubations, plasma (mouse and human) and infectious exudates, and samples stored at −20°C until extraction. All samples for LC-MS-MS-based metabololipidomics were extracted with solid-phase extraction columns as previously reported 29. Prior to sample extraction, deuterated internal standards (d4-PGE2, d5-LXA4, d4-RvD2, d4-LTB4 and d8-5S-HETE) representing regions of interest in the chromatographic analysis (500 pg each) were added to facilitate quantification. Extracted samples were analyzed by a LC-MS-MS system, Qtrap 6500 (AB Sciex) equipped with a Shimadzu SIL-20AC autoinjector and LC-20AD binary pump (Shimadzu Corp.). An Agilent Eclipse Plus C18 column (100×4.6mm×1.8μm) was used with a gradient of methanol/water/acetic acid of 55:45:0.01 (vol:vol:vol) that was ramped to 85:15:0.01 (vol:vol:vol) over 10min and then to 98:2:0.01 (vol:vol:vol) for the next 8min. This was subsequently maintained at 98:2:0.01 (vol:vol:vol) for 2 min. The flow rate was maintained at 0.4ml/min. To monitor and quantify the levels of lipid mediators, a multiple reaction monitoring (MRM) method was developed with signature ion fragments (m/z) for each molecule monitoring the parent ion (Q1) and a characteristic daughter ion (Q3). Identification was conducted using published criteria where a minimum of 6 diagnostic ions were employed 29. Calibration curves were determined using a mixture of lipid mediators obtained via total organic synthesis, where these lipid mediators were not available (RvT1, RvT2, RvT3, RvT4 and 13-HDPA); known mediators with similar chromatographic properties (RvD2 for RvT1, RvD1 for RvT2 and RvT3; RvD5 for RvT4; 13-HDHA for 13-HDPA) were used. Linear calibration curve for each compound was obtained with r2 values ranging from 0.98 to 0.99. Detection limit was ~0.1 pg. Quantification was carried out as in 29. To measure 6-keto-PGF1α levels, samples were extracted as outlined above, suspended in water, and 6-keto-PGF1α levels measured using a 6-keto-PGF1α ELISA (Neogen).
For chiral lipidomic analysis, a Chiralpak AD-RH column (150 mm × 2.1 mm × 5 μm) was used with isocratic methanol/water/acetic acid 95:5:0.01 (v/v/v) at 0.15 ml/min. To monitor isobaric monohydroxy docosapentaenoic acid levels, a multiple reaction monitoring (MRM) method was developed using signature ion fragments for each molecule as in 30.
In select experiments, RvT1, RvT2, RvT3 and RvT4 were incubated with diazomethane in ether, prepared as in 28 for 30 min at 37°C and products assessed by LC-MS-MS, operating the mass spectrometer in positive ion mode.
In order to extract products from neutrophil-endothelial cell co-incubations for biological evaluation, 2 volumes of ice-cold methanol were added to stop the incubations and the samples placed at −20°C for at least 30 min to allow for protein precipitation. Products were then extracted using C18 SPE as detailed above and eluted using methyl formate. The solvent was then evaporated under nitrogen and products suspended in ethanol for biological evaluation.
13R-HDPA and 13S-HDPA were obtained by incubating 13-HDHA with Dess Martin Periodinane (0.03M in methylene chloride, 30min, RT); the resulting 13-oxo-DPA was then incubated with either (R)-2-methyl-CBS-oxazaborolidine or (S)-2-methyl-CBS-oxazaborolidine as in 31.
In some experiments, human peripheral neutrophils (5×107 cells/ml) were incubated with 13R-HDPA (1μM, 37°C, PBS pH 7.45) and E. coli (1×109 CFU/ml, 2 min, 37°C). Incubations were stopped with 2 volumes of acidified methanol (apparent pH 3), products extracted and profiled using LM metabololipidomics.
Human neutrophils (4×107 cells/ml) were incubated with 13R-HDPA (300nM) in PBS (pH 7.45, 37°C) in an 18O2 enriched environment; E. coli (2×109 CFU/ml) were then added. The incubations were quenched after 30 min using 2 volumes of ice-cold methanol, then incubated with sodium borohydride (1μg/ml, 15min, 4°C), products extracted and profiled using LM metabololipidomics. The following MRM transitions were employed to monitor each of the new products: RvT1 - 381>145, RvT2 - 379>145, RvT3 - 227 and RvT4 - 363 -145.
Whole blood from healthy volunteers (HV), who declared to exercise for at least 45 mins 2–3 times per week and that they had not taken medications including nonsteroidal anti-inflammatory drugs, ASA-containing products or statins 10 –14 days before venipuncture, was obtained after obtaining written consent in accordance with the Declaration of Helsinki and Partners Human Research Committee Protocol 1999P001279 for discarded materials (CNS). Participants rested for 5–10 min in a seated position prior to peripheral blood sampling pre-exercise. Immediately after, the exercise protocol commenced involving 10 min warming up and stretching exercises followed by 30–45 min vigorous intensity continuous cycling or cross-training terminating the exercise when subjects reached ~90% of their theoretical maximum. This was calculated using the formula 220 – age in years as in 32. Blood was collected from these subjects within 15 min of exercise termination. All bloods were collected in heparin, then placed in 4 volumes of ice-cold methanol containing internal standards, and mediator levels were assessed by LC-MS-MS as detailed above.
Plasma from patients diagnosed with sepsis was obtained from Dx Biosamples (see Table S3 for demographics). Plasma from healthy volunteers (HV) was collected as outlined above. Human reference plasma denoted SRM 1950 was purchased from the National Institute of Standards and Technology (NIST). Plasma from these patients and volunteers was placed in 2 volumes of ice-cold methanol containing internal standards, and mediator levels assessed by LC-MS-MS as detailed above.
Whole blood was collected in heparin at determined time intervals during E. coli infections in mice. This was then placed in 4 volumes of ice-cold methanol containing internal standards, and mediator levels assessed by LC-MS-MS as detailed above.
Neutrophil-endothelial co-incubations
Human umbilical endothelial cells (HUVEC; 8.5×105 cells/9.6cm2) were incubated with IL-1β (10ng/ml) and TNF-α (10ng/ml; 16h, 37°C, 5% CO2). Cells were then incubated with atorvastatin (30nM–30μM), celecoxib (25μM), L-NAME (25μM), 1400 W (10μM) or vehicle (PBS containing 0.01% DMSO) followed by addition of n-3 DPA (1μM, 1h, 37°C, 5% CO2). In select experiments, 30 min after n-3 DPA addition, human neutrophils (1×107 cells/ml) were added and cells incubated for 60 min (37°C, 5% CO2). Incubations were stopped with 2 volumes of ice-cold methanol and products extracted as detailed above. In determined experiments after the addition of human PMN, E. coli (5×108 CFU/ml) were also added and cells incubated 60 min (37°C, 5% CO2). Incubations were then stopped with 2 volumes ice-cold methanol and LM profiles assessed using LM-metabololipidomics.
Endothelial cells were transfected with control scrambled sh-RNA or human COX-2 sh-RNA (Origene) using jetPEI®-HUVEC (PolyPlus) following manufacturer’s instructions, and then incubated with IL-1β (10ng/ml) and TNF-α (10ng/ml; 16h, 37°C, 5% CO2). Cells were then incubated with n-3 DPA (1μM, 37°C, 5% CO2, in 0.1% EGM). Incubations were stopped with 2 volumes of ice-cold methanol and lipid mediators assessed by lipid mediator metabololipidomics.
Human neutrophil and macrophage phagocytosis and ROS
Macrophages were prepared from peripheral blood mononuclear cells (PBMC) purchased from Children’s Hospital Blood Bank, Boston, and phagocytosis was assessed as in 31. Briefly, macrophages (5×104 cells/well) were incubated with RvT1 (1pM–10nM), 1:1 mixture RvT2 and RvT3 (1pM–10nM), RvT4 (1pM–10nM) or vehicle (0.1% EtOH in DPBS) for 15 min at 37°C, then fluorescent labeled apoptotic cells were added and cells incubated 45 min at 37°C. Extracellular fluorescence was quenched using Trypan blue (1:15 dilution) and phagocytosis assessed using an M3 SpectraMax plate reader (Molecular Devices). In select experiments, macrophages (prepared as detailed above, 5×104 cells/well) or neutrophils (1×105 cells/well), obtained from human healthy volunteers and isolated as in 31 in accordance with Partners Human Research Committee Protocol (number 1999P001297), were incubated with H2DCFDA (5μM, 30min, 37°C), excess dye was washed off and cells incubated with RvT1 (1pM–10nM), 1:1 mixture RvT2 and RvT3 (1pM–10nM), RvT4 (1pM–10nM) or vehicle (0.1% EtOH in DPBS, 15 min, 37°C), then incubated with E. coli (1:50 leukocytes to E. coli, 45 min, 37°C). Intracellular reactive oxygen species were determined by measuring fluorescence using an M3 SpectraMax plate reader. To assess bacterial phagocytosis, macrophages (5×104 cells/well) or neutrophils (1×105 cells/well) were incubated with RvT1 (1pM–10nM), 1:1 mixture RvT2 and RvT3 (1pM–10nM), RvT4 (1pM–10nM) or vehicle (0.1% EtOH in DPBS, 15 min, 37°C), then incubated with BacLight Green (Molecular Probes) labeled E. coli (1:50 leukocytes to E. coli, 45 min, 37°C). Extracellular fluorescence was then quenched using Trypan blue (1:15 dilution), and phagocytosis was assessed using an M3 SpectraMax plate reader.
Human macrophage caspase 1, IL-1β and lactate dehydrogenase
Human macrophages (1.5×104 cells/well) were incubated with RvT1 (10pM–10nM), 1:1 mixture RvT2 and RvT3 (10pM–10nM), RvT4 (10pM–10nM) or vehicle (0.1% EtOH in DPBS, 15 min, 37°C), then E. coli (7.5×105 cells/well, in DPBS, 16h, 37°C). Caspase 1 levels were assessed in macrophages by flow cytometry after cell fixing and permeabilization, staining with an anti-caspase 1 (Abcam; 4°C, 30min) and anti-rabbit-Alexa 488 conjugated antibody (Molecular Probes; 4°C, 30min). IL-1β levels were determined in cell-free supernatants using an anti-human IL-1β ELISA (eBiosciences) following manufacturer’s instructions. Lactate dehydrogenase activity was measured in cell-free supernatants assessing the formation of NADH from NAD+ using a Lactate Dehydrogenase Assay kit (Sigma) following manufacturer’s instructions.
Recombinant enzyme incubations
Human recombinant COX-2 (10 units; Cayman Chemicals; in 0.1M Tris-HCl, pH 8.0, 20μM porcine hematin, 0.67mM phenol as in 33) was incubated with the indicated concentrations of AA or n-3 DPA (1h, RT) and absorbance at 235nM was investigated at 1 min intervals using a Cary UV-Vis Spectrophotometer (Agilent).
In designated experiments, n-3 DPA was incubated with hrCOX2, hrCOX-2 that was previously incubated with S-nitrosoglutathione (30 min RT), prepared by incubating 100mM glutathione with 100 mM sodium nitrite in 200mM hydrochloric acid at RT as in 34 or S-nitrosoglutathione. After 20 min the incubations were quenched with methanol and 13-HDPA levels were assessed using LC-MS-MS-based LM metabololipidomics.
COX-2 S-nitrosylation was assessed using a Pierce Western Blot S-nitrosylation Kit (Thermo Fisher) following manufacturer’s instructions, and total COX-2 levels were determined using a rabbit anti-COX-2 (Cell Signaling) antibody and a goat anti-rabbit-HRP conjugated antibody (eBioscience).
Anti-bacterial actions
Neutrophil-endothelial cell isolates, RvT1 (10μM), RvT2 plus RvT3 (10μM), RvT4 (10μM) or ampicillin (1 or 10mM) were placed on GF-C filters (Waters) that were then transferred onto LB agar plates containing E. coli (1×107 CFU). The zone of clearance was assessed after overnight incubation at 37°C.
Statistics
All results are expressed as means ± s.e.m.. We assumed normality and equal distribution of variance between the different groups analyzed. Differences between groups were compared using Student’s t test (2 groups) or 1-way ANOVA (multiple groups) followed by post hoc Bonferroni test. Survival in mouse experiments was demonstrated with Kaplan-Meier curves and analyzed using log-rank (Mantel-Cox). Investigators were not blinded to group allocation or outcome assessment. Sample sizes for each experiment were determined on the variability observed in preliminary experiments and prior experience with the experimental systems. The criterion for statistical significance was p<0.05.
Supplementary Material
Acknowledgments
The authors thank Dr. Romain Colas and Iliyan Vlasakov for assistance with material preparation and lipid mediator metabololipidomics. This work was supported by the National Institutes of Health (grant P01GM095467).
Footnotes
Author Contributions
All authors contributed to manuscript and figure preparations. J.D. and N.C. designed and carried out experiments and analyzed data; J.D. and C.N.S. conceived the overall research plan and experimental design.
Competing financial interests
The authors do not have any competing financial interests to disclose.
References
- 1.Ward PA. New approaches to the study of sepsis. EMBO molecular medicine. 2012;4:1234–1243. doi: 10.1002/emmm.201201375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magill SS, et al. Multistate point-prevalence survey of health care-associated infections. The New England journal of medicine. 2014;370:1198–1208. doi: 10.1056/NEJMoa1306801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fullerton JN, O’Brien AJ, Gilroy DW. Lipid mediators in immune dysfunction after severe inflammation. Trends in immunology. 2014;35:12–21. doi: 10.1016/j.it.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemaitre RN, et al. Genetic loci associated with plasma phospholipid n-3 fatty acids: a meta-analysis of genome-wide association studies from the CHARGE Consortium. PLoS genetics. 2011;7:e1002193. doi: 10.1371/journal.pgen.1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalli J, Colas RA, Serhan CN. Novel n-3 immunoresolvents: structures and actions. Sci Rep. 2013;3:1940. doi: 10.1038/srep01940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiang N, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serhan CN, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. The Journal of experimental medicine. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends in immunology. 2011;32:452–460. doi: 10.1016/j.it.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Mead PS, et al. Food-related illness and death in the United States. Emerg Infect Dis. 1999;5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markworth JF, et al. Human inflammatory and resolving lipid mediator responses to resistance exercise and ibuprofen treatment. American journal of physiology Regulatory, integrative and comparative physiology. 2013;305:R1281–1296. doi: 10.1152/ajpregu.00128.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kandasamy K, et al. Atorvastatin prevents vascular hyporeactivity to norepinephrine in sepsis: role of nitric oxide and alpha(1)-adrenoceptor mRNA expression. Shock. 2011;36:76–82. doi: 10.1097/SHK.0b013e31821a4002. [DOI] [PubMed] [Google Scholar]
- 16.Atar S, et al. Atorvastatin-induced cardioprotection is mediated by increasing inducible nitric oxide synthase and consequent S-nitrosylation of cyclooxygenase-2. American journal of physiology Heart and circulatory physiology. 2006;290:H1960–1968. doi: 10.1152/ajpheart.01137.2005. [DOI] [PubMed] [Google Scholar]
- 17.Lins RL, et al. Pharmacokinetics of atorvastatin and its metabolites after single and multiple dosing in hypercholesterolaemic haemodialysis patients. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2003;18:967–976. doi: 10.1093/ndt/gfg048. [DOI] [PubMed] [Google Scholar]
- 18.Samuelsson B. Role of basic science in the development of new medicines: examples from the eicosanoid field. The Journal of biological chemistry. 2012;287:10070–10080. doi: 10.1074/jbc.X112.351437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji Y, Akerboom TP, Sies H, Thomas JA. S-nitrosylation and S-glutathiolation of protein sulfhydryls by S-nitroso glutathione. Archives of biochemistry and biophysics. 1999;362:67–78. doi: 10.1006/abbi.1998.1013. [DOI] [PubMed] [Google Scholar]
- 20.von Moltke J, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henriksbo BD, et al. Fluvastatin causes NLRP3 inflammasome-mediated adipose insulin resistance. Diabetes. 2014 doi: 10.2337/db13-1398. [DOI] [PubMed] [Google Scholar]
- 22.Cederberg H, et al. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: a 6 year follow-up study of the METSIM cohort. Diabetologia. 2015;58:1109–1117. doi: 10.1007/s00125-015-3528-5. [DOI] [PubMed] [Google Scholar]
- 23.Gryglewski RJ, Mackiewicz Z. Vane’s blood-bathed organ technique adapted to examine the endothelial effects of cardiovascular drugs in vivo. Pharmacological reports: PR. 2010;62:462–467. doi: 10.1016/s1734-1140(10)70301-7. [DOI] [PubMed] [Google Scholar]
- 24.Ye Y, et al. Activation of peroxisome proliferator-activated receptor-gamma (PPAR-gamma) by atorvastatin is mediated by 15-deoxy-delta-12,14-PGJ2. Prostaglandins & other lipid mediators. 2007;84:43–53. doi: 10.1016/j.prostaglandins.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Morikawa S, et al. The effect of statins on mRNA levels of genes related to inflammation, coagulation, and vascular constriction in HUVEC. Human umbilical vein endothelial cells. Journal of atherosclerosis and thrombosis. 2002;9:178–183. doi: 10.5551/jat.9.178. [DOI] [PubMed] [Google Scholar]
- 26.Ye Y, et al. Phosphorylation of 5-lipoxygenase at ser523 by protein kinase A determines whether pioglitazone and atorvastatin induce proinflammatory leukotriene B4 or anti-inflammatory 15-epi-lipoxin a4 production. Journal of immunology. 2008;181:3515–3523. doi: 10.4049/jimmunol.181.5.3515. [DOI] [PubMed] [Google Scholar]
- 27.Chiang N, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serhan CN, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. The Journal of experimental medicine. 2002;196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. American journal of physiology. Cell physiology. 2014 doi: 10.1152/ajpcell.00024.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh SF, Pillai PS, Recchiuti A, Yang R, Serhan CN. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. The Journal of clinical investigation. 2011;121:569–581. doi: 10.1172/JCI42545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corej EJ, Bakshi RK, Shibata S. Highly Enantioselective Borane Reduction of Ketones Catalyzed by Chiral Oxazaborolidines. Mechanism and Synthetic Implications. J Am Chem Soc. 1987;109:5551–5553. [Google Scholar]
- 32.Gangemi S, et al. Physical exercise increases urinary excretion of lipoxin A4 and related compounds. Journal of applied physiology. 2003;94:2237–2240. doi: 10.1152/japplphysiol.01004.2002. [DOI] [PubMed] [Google Scholar]
- 33.Hemler ME, Lands WE. Protection of cyclooxygenase activity during heme-induced destabilization. Archives of biochemistry and biophysics. 1980;201:586–593. doi: 10.1016/0003-9861(80)90548-2. [DOI] [PubMed] [Google Scholar]
- 34.Ji Y, Akerboom TP, Sies H, Thomas JA. S-nitrosylation and S-glutathiolation of protein sulfhydryls by S-nitroso glutathione. Archives of biochemistry and biophysics. 1999;362:67–78. doi: 10.1006/abbi.1998.1013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.