

Abstract
Ubiquitination is a key protein post-translational modification that regulates many important cellular pathways and whose levels are regulated by equilibrium between the activities of ubiquitin ligases and deubiquitinases. Here we present a method to identify specific deubiquitinase substrates based on treatment of cell lysates with recombinant enzymes, immunoaffinity purification and global quantitative proteomic analysis. As a model system to identify substrates, we used a virulence-related deubiquitinase, SseL, secreted by Salmonella enterica serovar Typhimurium into host cells. Using this approach two SseL substrates were identified in RAW 264.7: murine macrophage-like cell line, S100A6 and heterogeneous nuclear ribonuclear protein K, in addition to the previously reported K63-linked ubiquitin chains. These substrates were further validated by a combination of enzymatic and binding assays. This method can be used for the systematic identification of substrates of deubiquitinases from other organisms and applied to study their functions in physiology and disease.
Keywords: ubiquitination, deubiquitinase, post-translational modification, substrate identification, mass spectrometry
INTRODUCTION
Ubiquitination is a key post-translational modification that regulates many biological pathways in eukaryotic cells.1 Protein ubiquitination is characterized by the addition of a small polypeptide, ubiquitin, to proteins by the coordinated action of 3 enzymes: E1, E2, and E3. The E3 enzymes, also known as ubiquitin protein ligases, are responsible for substrate specificity. Ubiquitination can occur in single ubiquitin (monoubiquitination – monoUb) units or as oligomeric chains (polyubiquitination – polyUb) linked through one of its lysine residues (K6, K11, K27, K29, K33, K48 or K63) or N-terminus (linear).1 Ubiquitination is also a reversible process in which ubiquitin units are removed from proteins by deubiquitinases (DUBs).2 Different cellular functions have been associated with monoUb or polyUb; furthermore, distinct polyUb linkages have also been proposed to have specific activities. While monoUb is often associated with cellular trafficking, regulation of gene expression and DNA repair, K48-linked polyUb has been shown to target proteins to degradation into the proteasome, and K63-linked polyUb regulates the activity of selected kinases.1
Recent developments in affinity purification combined to mass spectrometry have enabled the identification and quantification of thousands of ubiquitination sites from cell lysates.3–6 Enrichment of whole ubiquitinated proteins can be achieved by affinity purification with specific antibodies7 or ubiquitin-binding domains,8–10 or by expressing ubiquitin fused with an epitope tag.11 Enrichment of ubiquination sites can be performed at the peptide level using monoclonal antibodies that recognize the diglycine residues (K-ε-GG), a remnant of the C-terminus of ubiquitin following trypsin digestion.12 These recent developments in ubiquitination analysis open new opportunities for the identification of specific substrates of both E3 ligases and DUBs. Overexpression or repression of E3 ligase genes followed by ubiquitinated protein/peptide enrichment and quantitative proteomic analysis has been used to identify specific substrates of ubiquitin ligases;13–16 however, less information is available regarding the systematic identification of specific substrates of DUBs. Poulsen et al. showed the impact of knocking out each of the 20 Saccharomyces cerevisiae DUBs on protein abundances,17 but the approach failed to provide the information about specific substrates and their modification sites. Udeshi et al. used a broad spectrum DUB inhibitor, PR-619, to study global changes in protein ubiquitination under DUB inhibition.5
In an attempt to systematically identify DUB substrates and their modification sites, we developed, optimized and applied a method to identify these substrates from cell lysates. This method is based on the treatment of cell lysates with a recombinant DUB, immunoaffinity purification of K-ε-GG-containing peptides and quantitative proteomic analysis. We used the Salmonella secreted effector DUB, SseL, to identify both known and two previously unrecognized substrates in host cells. The two new substrates of SseL were further validated by a combination of enzymatic and binding assays.
EXPERIMENTAL PROCEDURES
Cloning and expression of Salmonella deubiquitinases
Salmonella enterica serovar Typhimurium (S. Typhimurium) SseL gene from LT2 strain was amplified using genomic DNA as a template and cloned into modified pET vectors, which provided His6 or His6-Streptavidin binding peptide (SBP) N-terminal fusion cleavable with Tobacco Etch Virus (TEV) protease. The C285S mutation was introduced into SseL gene sequence using the QuikChange® mutagenesis kit (Stratagene, Santa Clara, CA, USA). The N-terminal His6-tagged wild-type and mutant SseL expression constructs were transformed into E. coli BL21-CodonPlus (DE3)-RIPL (Novagen, Darmstadt, Germany). Freshly transformed cells were grown in 0.5 L of LB medium containing 100 μg/ml ampicillin. The culture was grown at 37 °C to an OD600 of 0.8, induced for 16 h at 16 °C with 1 mM isopropyl-1-thio-β-D-galactopyranoside, the cells were then harvested by centrifugation. The harvested cells were sonicated in lysis buffer consisting of 50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 5 mM imidazole supplemented with EDTA-free protease-inhibitor cocktail (Roche, Indianapolis, IN, USA) and clarified by centrifugation. The supernatant was applied onto Ni-NTA affinity resin (Qiagen, USA) pre-equilibrated with lysis buffer and washed with buffer consisting of 50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 30 mM imidazole. His-tagged protein was eluted with 250 mM imidazole, and dialyzed overnight against 3 L of 50 mM HEPES, pH 7.5, 300 mM NaCl, 5% glycerol, 1 mM tris(2-carboxyethyl)phosphine (TCEP), concentrated when needed with a Centriprep 30 concentrator, and stored at −80°C.
Enzymatic activity
Deubiquitinase activity assays were performed using K48- and K63-linked diubiquitins (Ub2) and oligoubiquitins (Ub2–7) (Enzo Lifescience, Farmingdale, NY, USA). For diubiquitin assays, 0.5 μg of each chain and 0.1 μg of SseL were added to 100 μL reaction buffer (50 mM Tris-HCl, pH 7.5, containing 1 mM DTT). Parallel reactions without enzyme were performed as negative controls. The reaction was held at 37 °C for 0–45 min and was stopped by adding gel loading buffer (Invitrogen, Grand Island, NY, USA) with incubation at 95 °C for 15 min prior to analysis by SDS-PAGE.
Preparation of cell lysates
SseL substrates were identified in lysates of RAW 264.7 murine macrophage-like cells (American Type Culture Collection, ATCC, Manassas, VA, USA). RAW 264.7 cells were grown in 150-mm plates to 100% confluency in high-glucose Dulbecco’s modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum and Penicillin (100 U/mL)-Streptomycin (100 μg/mL) at 37 °C and 5% CO2 atmosphere. Cells were then stimulated for 2 h with 100 ng/mL Salmonella lipopolysaccharide (L6143, Sigma-Aldrich, Saint Louis, MO, USA), washed twice with 10 mL Dulbecco’s PBS and harvested in HEPES lysis buffer (50 mM HEPES, 5 mM EDTA, 150 mM NaCl and 1% triton X-100) supplemented with 0–0.5 mM TCEP, 1x Halt protease inhibitor cocktail (Thermo Fisher, Whaltham, MA, USA) and thiol-alkylating agents, 0–10 mM chloroacetamide (CAA), 0–10 mM iodoacetamide (IAA) or 0–10 mM N-ethylmaleimide (NEM). Proteins were then extracted by sonicating 3 times at for 3×30 s, 100% amplitude and pulse of 0.8 (UTR200, Hielscher). After sonication, extracts were centrifuged for 5 min at 4 °C and 16,000 x g, and quantified by BCA assay (Thermo Scientific). Extracted proteins were incubated for 2 h on ice to block the active sites of endogenous DUBs by alkylating the cysteine residues with NEM (concentrations as indicated in the figures). After blocking, the excess of NEM was quenched by adding a final concentration of 20 mM DTT and incubating on ice for 2 h. The efficiency of endogenous DUB inactivation was tested by incubating cell lysates at 37 °C overnight and analyzing by western blot using anti-ubiquitin antibodies.
Cell lysate treatment with DUBs
RAW 264.7 cells were harvested in HEPES lysis buffer containing 0.5 mM TCEP, 5 mM NEM and 1 mM PMSF and proteins were extracted as described above. Cell lysates were then incubated with 2 μg SseL or recombinant catalytic domain of rat USP2 (Enzo Lifesciences) per mg of protein extract for 15 min at 37 °C. The reaction was then terminated by precipitating proteins with cold acetone (−20 °C). Proteins were then separated by SDS-PAGE and analyzed by western blots using specific antibodies.
Treatment of ubiquitinated proteins with DUBs
RAW 264.7 cells were harvested in HEPES lysis buffer containing 1x protease inhibitor cocktail, 0.5 mM TCEP and 5 mM NEM and prepared according to the “Preparation of cell lysates” section. Aliquots of 1 mg of cell lysate were rotated overnight with 30 μL of agarose beads conjugated with the ubiquitin-binding domain of yeast DSK2 protein (Enzo Lifesciences). 9 Beads were washed 3 times with 1 mL HEPES lysis buffer and 2 μg of SseL or recombinant catalytic domain of rat USP2 were added. The reaction was incubated for 1h at 37 °C with shaking 800 rpm and terminated by adding SDS-PAGE loading buffer and incubating at 70 °C for 10 min. Samples were then separated by SDS-PAGE and analyzed by western blots using specific antibodies.
SseL binding to substrates
SseL-binding assay was performed by incubating with SBP-tagged C285S SseL and cell lysates prepared as described above, with the exception that Tris lysis buffer (50 mM Tris-HCl, 5 mM EDTA, 150 mM NaCl and 1% triton X-100) supplemented with 0.5 mM TCEP, 5 mM NEM, 1x Halt protease inhibitor cocktail was used. Approximately 20 mg of cell lysate was split into 2 samples and half rocked overnight at 4 °C with 25 μg SBP-tagged C285S SseL, whereas the other half was incubated without bait protein as a negative control. Then 50 μL streptavidin-conjugated agarose beads were added and rotated for 30 min at 4 °C. The beads were washed 3x by adding 1 mL of Tris lysis buffer, centrifuging for 2 min at 2,500 x g and removing the supernatant. Proteins were eluted with 50 μL SDS-PAGE loading buffer for 10 min at 70 °C. Proteins were then analyzed by SDS-PAGE and subsequent western blot with specific antibodies.
SDS-PAGE and Western blot analyses
Proteins were separated using 4–12% Nu-PAGE SDS-PAGE (Invitrogen) and stained with a silver stain kit (Thermo), or transferred onto PVDF membranes. Membranes were stained with 0.1% Ponceau S in 5% acetic acid to verify that equal amounts of proteins were loaded.18 Western blots were performed with the following antibodies: monoclonal anti-K63 (Apub3, Millipore, Darmstadt, Germany), monoclonal anti-K48, (Apub2, Millipore), and polyclonal antiubiquitin (AB1690, Millipore), anti-S100A6 (H-55, Santa Cruz Biotech, Dallas, TX, USA, and Sigma-Aldrich), anti-MHC class I (Santa Cruz Biotech) and hnRNP K (H-300, Santa Cruz Biotech and Sigma-Aldrich). Blots were developed with ECL reagent (Pierce, Rockford, IL, USA) and visualized in a Fluor-Chem Q imaging system (Alpha Innotech, Santa Clara, CA, USA).
Sample preparation for quantitative proteomic analysis
RAW 264.7 cells were harvested in HEPES lysis buffer containing 0.5 mM TCEP, 5 mM NEM and 1 mM PMSF and proteins were extracted as described above. Cell lysates were then incubated with 2 μg SseL per mg of protein extract for 0–4 h for western blot analysis, or 15 min for mass spectrometry analysis. After this period the reaction was stopped by adding cold acetone (−20 °C) and precipitating for 2 h at −20 °C. Precipitated proteins were either analyzed by western blot using anti-ubiquitin antibodies or submitted to digestion with trypsin. For trypsin digestion, samples were prepared in duplicates of SseL treated and mock control (without SseL) of about 8 mg of cell lysate each. Protein pellets were dissolved in 10 mL 50 mM NH4HCO3 and 1 mM CaCl2 and digested for 3 h with 40 μg sequencing-grade trypsin (Promega, Madison, WI, USA) at 37 °C and occasional vortexing. After this period, another 40 μg trypsin was added to each sample and the reaction was incubated overnight at 37 °C. The reaction was stopped by adding a final concentration of 1% trifluoroacetic acid (TFA), and peptides were clean up using C18 solid-phase extraction cartridges (Discoverer, 100 mg, 1mL, Supelco, Bellefonte, PA, USA) as previously described 19 and dried in a vacuum centrifuge. Each sample of dried peptides were dissolved in 2 mL 50 mM NH4HCO3 and 1 mM CaCl2 prepared in H2 16O or H2 18O and the trypsin catalyzed oxygens were exchanged overnight at 37 °C with 40 μg of trypsin. The reaction was then terminated by boiling the samples for 10 min. Then control and SseL-treated samples were mixed in the proportion 1:1. In the first replicate the control samples were labeled with H2 18O, whereas the label was swapped in the second replicate the the SseL-treated samples contained 18O to enable to identification of peptides that are resistant to isotope exchange. Next, peptides carrying the ubiquitin-remnant diglycine residues were captured by immunoaffinity purification using the PTMScan® Ubiquitin Remnant Motif (K-ε-GG) Kit according to the manufacturer instructions (Cell Signaling, Beverly, MA, USA). After capture, peptides were eluted with 0.15% TFA, desalted with C18 Omix tips (Agilent, Santa Clara, CA) and dried in a vacuum centrifuge before being submitted to proteomic analysis.
Quantitative proteomic analysis
Enriched peptides were loaded into capillary columns (75 μm × 100 cm, Polymicro, Phoenix, AZ, USA) packed with C18 beads (3 μm particles, Phenomenex) and connected to a nano-liquid chromatography system (nanoAcquity, Waters, Waltham, MA, USA). The elution was performed in an exponential gradient from 1–35% B mobile phase (mobile phase A: 0.1% FA; mobile phase B: 90% ACN/0.1% FA) over 300 min in a flow rate of 200 nL/min and directly analyzed by electrospray ionization-mass spectrometry (LTQ Orbitrap Velos, Thermo Scientific, San Jose, CA, USA). Full scan spectra were collected at 400–2000 m/z, and the top ten most intense ions were submitted to CID fragmentation (35% normalized collision energy), before being dynamically excluded for 60 s. Tandem mass spectra were converted into high resolution DTA files using DeconMSn20 (available at omics.pnl.gov) and searched with MS-GF+ (http://proteomics.ucsd.edu/Software/MSGFPlus.html) against the mouse Uniprot database (downloaded on August 22nd, 2011) and common contaminant sequences (human keratins and porcine trypsin) (all in forward and reversed orientations; total of 32,798 searched sequences). The database was searched using the following parameters: (i) partial tryptic digestion with 3 missed cleavage sites allowed, (ii) 20 ppm peptide mass tolerance; and (iii) cysteine NEM alkylation (C6H7NO2: +125.0477) as a fixed modification, and (iv) lysine ubiquitination (diglycine residue: +114.0429 Da) and C-terminus 18O isotope label (+4.0085) as variable modifications. Peptides were validated with mass spectrum generating function (MS-GF) probability 21 ≤ 6.0e-10, which resulted in a FDR of ≤ 0.05.
Quantitative analysis was achieved by an in-house written feature-finding tool, SIPPER C13 Detector (available at http://omics.pnl.gov/software/SIPPER.php)22 which automatically finds peaks based on peptide identifications and calculates the ratio between 16O-/18O-labeled peptides. For the quantitative analysis, we only considered peptides that 16O/18O pairs were reproducibly found in both replicates. Considering only peptides that were present in both datasets, no reverse sequences were found, resulting in a FDR much lower than 0.05.
Structural analysis
YASARA23 software was used to calculate the Surface Accessible Solvent Area (SASA) and secondary structure prediction.
RESULTS
Preparation of cell lysate for deubiquitinase activity
One of the key steps for our approach to identify specific DUB substrates is to properly prepare the cell lysate. Mammalian cells have numerous endogenous DUBs that need to be completely inactivated to prevent erroneous interpretation of results. Further, the DUB inhibitors also must be efficiently quenched before the exogenous DUB is added to the reaction. To evaluate conditions for inhibiting endogenous DUB activity, RAW 264.7 murine cells were extracted by sonication and treated with a protease inhibitor cocktail in combination with thiol-alkylating agents chloroacetamide (CAA), iodoacetamide (IAA) or N-ethylmaleimide (NEM) to minimize the activity of cysteine-dependent DUB enzymes. As a positive control RAW 264.7 cell lysates were pretreated with 20% trichloroacetic acid (TCA), which has been proposed to quench all the DUB activity. 24 However, since TCA precipitates proteins, cell lysates prepared with this method cannot be used for DUB assays. After incubation for 1 h at room temperature or overnight at 4 °C cell lysates were analyzed by western blot using anti-ubiquitin antibodies (Fig. 1). Judging the DUB inhibition by the amount the polyUb proteins preserved in the samples, the analysis suggested that the combined treatment of protease inhibitor cocktail and NEM provides strong inhibition of endogenous DUB activity (Fig. 1).
Figure 1.

Testing of different thiol-alkylating agents for inhibition of endogenous DUBs. Cells were harvested in the absence or presence of protease inhibitor cocktail (PIC) in addition to 10 mM of IAA, CAA, or NEM. A parallel control was performed by harvesting cells in 20% TCA, which was previously reported to block most of the DUB activity. After harvesting, cell lysates were incubated for 1 h at room temperature (22 °C) or for 18 h at 4 °C, and analyzed by western blot using anti-ubiquitin antibodies. (A) Ponceau S stain and (B) western blot.
After determining that NEM was an effective DUB inhibitor, we tested the ability of the quenching process to inactivate the alkylating agent. We harvested cells with various concentrations of NEM, incubated for 2 h on ice to inhibit the endogenous DUB activity and then quenched the remaining NEM by adding dithiothreitol (DTT) in a molar excess. To test if the DUB activity was efficiently inhibited, lysates were incubated overnight at 37 °C and analyzed by western blot using anti-ubiquitin antibodies (Fig. 2A). Surprisingly, we found that the addition of DTT reverted the DUB inhibition (Fig. 2B), which was an intriguing phenomenon since alkylation of DUB active cysteine residues is an irreversible process.
Figure 2.

Blocking endogenous DUB activity with NEM and TCEP. Cells were harvested in the absence or presence of TCEP, and with the addition different NEM concentrations followed by blocking the DUB activity for 2 h on ice. Then the excess of NEM was quenched with 20 mM DTT for 2 h on ice and the efficiency of DUB inactivation was tested by incubating the lysates overnight (O/N) at 37 °C. (B) Cells harvested without TCEP and (C) cells harvested with 0.5 mM TCEP.
We had an alternative hypothesis that the cells could have a pool of DUBs with an oxidized thiol at the active site.25 The cysteine residues would then be resistant to alkylation, but with the addition of reducing agent DTT become active again. To address this, we added 0.5 mM tris(2-carboxyethyl)phosphine (TCEP), a reducing agent lacking thiols, to the lysis buffer. TCEP would allow the reduction of the oxidized thiols without interfering in the alkylation process. The lysis buffer tested contained TCEP, the protease inhibitor cocktail, and NEM. Cell lysate was then incubated for 2 h on ice before being treated with DTT then analyzed by western blot as described above. The addition of TCEP did indeed cause effective and stable DUB inhibition by NEM without secondary activation by addition of DTT (Figure 2C). The results supported the hypothesis that DUBs with oxidized active sites are endogenously present in cells. These results show that the cells should be lysed in the presence of TCEP and NEM, followed by quenching of the alkylating agent with DTT.
Identification of SseL substrates by mass spectrometry
Having established conditions for cell lysate preparation, we next undertook identification of potential DUB substrates. We chose the secreted effector protein DUB SseL from pathogenic Salmonella, as an example of relatively poorly studied protein with medical relevance. In addition, SseL has one well-characterized substrate, K63-linked ubiquitin chains, but specific host cell targets remain largely unknown. On the other hand, SseL is known to not efficiently cleave K48-linked ubiquitin chains. These features provide good positive and negative controls that make SseL an ideal DUB to our analysis. To test for activity of the recombinant SseL protein preparations, we performed an enzymatic assay using either diubiquitin (Ub2) or oligoubiquitin (Ub2–7) chains as substrates. The products were then separated by SDS-PAGE and activity was assessed by a decrease in oligomeric Ub chains and the appearance of free monoUb chains. As expected, SseL cleaved K63-linked Ub chains faster than K48-linked chains (Supporting information Figure 1) in agreement with the literature26 and confirms that the recombinant protein is indeed enzymatically active.
Since SseL’s natural substrates may only be present during Salmonella infection, RAW 264.7 cells were pretreated with Salmonella lipopolysaccharide for 2 h to trigger major aspects of the cellular response that occurs during Salmonella infection.27 Since a short incubation with DUBs may not produce enough products to be measured and a prolonged treatment time could induce unspecific reactions, we attempted to find an optimal incubation time. Thus, cell lysates were treated with SseL in a time course experiment of 0–4 hours, analyzed by a western blot with anti-ubiquitin antibodies, and DUB activity was judged by the decrease in polyUb proteins. Incubation with SseL resulted in a decrease of ubiquitinated protein pools within 5 min of treatment, but surprisingly no further change was observed past this time point (Figure 3A). This observation suggested that SseL could have specific activity towards a select pool of ubiquitinated substrates rather than a broader range of substrates.
Figure 3.

Global analysis of SseL substrates in RAW 264.7 cells. (A) Kinetics of digestion of ubiquitinated conjugates by SseL. RAW 264.7 cell lysate had endogenous DUB activity blocked with NEM, followed by a quenching of the reagent excess with DTT, and then incubated for various times with SseL (see material and methods for details). A control with no SseL was run in parallel for 4 h. (B) Methodology flowchart of quantitative proteomic analysis to identify potential SseL substrates. RAW 264.7 cells were treated for 120 min with LPS and harvested in duplicates. Endogenous DUBs of cell lysates were blocked, and then the lysates were treated with SseL, digested with trypsin and submitted to isotope exchange with H2 16O and H2 18O. Isotopically labeled peptides were then mixed in the proportion 1:1 and ubiquitin signature (K-ε-GG)-containing peptides were captured with monoclonal antibodies and analyzed by nano-liquid chromatography coupled to high resolution mass spectrometry. (C) Dispersion graph show the distribution of abundance ratios (16O/18O) of peptides bearing K-ε-GG ubiquitin signature (Ub-peptides).
Based on the time course, a proteomic experiment was prepared by incubating RAW 264.7 cell lysate with SseL for 15 minutes. Proteins were next precipitated with cold acetone followed by trypsin digestion. To enable relative quantification between treated and untreated samples, tryptic peptides were submitted to exchange with H2 18O. This procedure incorporates two 18O atoms in the C-terminus of the peptides increasing their masses by 4 Da, and relative quantification is achieved by comparing the peak areas of isotopically-exchanged (18O)/non-exchanged (16O) peptides.28 Importantly, samples were prepared in biological replicate and 18O-labeling was ‘flipped’ between replicates: while in the first replicate the untreated sample had their isotopes exchanged with 18O, in the second replicate the SseL-treated sample was exchanged with 18O. This replicate labeling strategy allows for detection of contaminants and peptides with inefficient 18O-labeling (see below).29 Isotopically-exchanged and non-exchanged samples were then mixed in the proportion 1:1, and peptides bearing the ubiquitination signature K-ε-GG residue were captured using monoclonal antibodies and analyzed by high resolution LC-MS/MS (Figure 3B). After searching tandem-mass spectra against a mouse protein sequence database over 1600 unique peptides were found with a false-discovery rate ≤ 0.05 (Supporting information Tables S1 and S2). From those peptides about one half had the K-ε-GG ubiquitin signature and was selected for further analysis.
For the quantitative analysis the 16O/18O isotope ratios were extracted, and only peptides with 16O/18O pairs consistently found in both replicates were considered for further analyses. Since we plotted the ratios as 16O/18O pairs and the labeling was flipped between both biological replicates, potential SseL substrates have opposite ratios when transformed by log2. On the other hand, peptides with inefficient 18O exchange have more natural (16O) isotopes, thus will have high (> 0) log2 ratios in both replicates. When these data are plotted into a dispersion graph, the unchanged peptides are at the center of the graph, the SseL-specific substrates are present at the upper-left quadrant, and the inefficiently 18O exchanged peptides, at the upper-right quadrant (Figure 3C and 3D). Figure 3C shows that the 18O exchange efficiency was indeed very high, leading to ratios close to 1 (log2 ratios = 0), although a few peptides were resistant to exchange (Figure 3C). As expected, K63-linked ubiquitin chains (UBB-K63) were found among the SseL specific substrates (Figure 3D). Only 3 other ubiquitin signature-containing peptides were identified as potential by SseL substrates: lysine 47 of S100A6 (S100A6-K47), lysine 405 of heterogeneous nuclear ribonucleoprotein K (Hnrnpk-K405) and lysine 340 of major histocompatibility complex (MHC) class I (H-2D-K340) (Figure 3D). The fact that most of the ubiquitination sites remained unchanged with SseL treatment suggests that this enzyme is highly selective to only a few substrates.
Validation of SseL substrates
In line with our in vitro assays (Supporting information Figure S1), the proteomic analysis showed that the amount of K63-linked Ub chains were reduced 3-fold after SseL treatment, while other polyUb chains remained unchanged (Table 1). To confirm these results all spectra derived from potential SseL substrates were individually inspected (Supporting information Figure S2). The first column of Supporting information Figure S2 shows an example of an ubiquitination site that is unaffected by SseL treatment (ubiquitination at lysine 6 of ubiquitin – UBB-K6), whereas all the SseL substrate candidates had their abundance substantially reduced. To further validate SseL activity we next compared the effect of SseL on ubiquitinated proteins in macrophage cell lysate to that of USP2 catalytic domain, a well-characterized DUB from rat with broad substrate specificity.3 The increase of lower and/or decrease in higher molecular weight ubiquitinated protein after SseL or USP2 treatment were evaluated by western blotting using anti-ubiquitin antibodies. In line with our previous results SseL demonstrated lower activity against K48-linked polyUb compared to USP2 DUB (Figure 4). Conversely, SseL activity against K63-linked polyUb was comparable to that of USP2 (Figure 4). Our experiment also confirmed S100A6 and hnRNP K, but not MHC class I (H-2D), as SseL substrates, judging by the bands lower molecular mass that increase in intensity after enzymatic treatment (Figure 4, Supporting information Figure S3). Although some of these bands are more intense when cell lysates were treated with USP2, they are clearly present in SseL treatment (Figure 4, Supporting information Figure S3). It is also worth to note that some of the bands that appear upon DUB treatment may not represent a completely unmodified protein since the molecular is a little higher than expected. We speculate that those bands correspond to other post-translational modifications, as these proteins are known to be further modified (Uniprot accession numbers: P14069 and P61979). More importantly, these bands are also present in the positive control with USP2, which leads to the conclusion that they represent the cleavage products of (poly)ubiquitinated proteins.
Table 1.
Quantification of ubiquitination sites on ubiquitin upon SseL treatment by proteomic analysis
| Site | Peptide | Average fold reduction with SseL treatment |
|---|---|---|
| K6 | MQIFVK#TLTGK | 1.13 |
| K11 | TLTGK#TITLEVEPSDTIENVK | 0.85* |
| K27 | TITLEVEPSDTIENVK#AK | 1.01 |
| K48 | LIFAGK#QLEDGR | 0.76 |
| K63 | TLSDYNIQK#ESTLHLVLR | 2.77 |
K# - ubiquitinated lysine residue
Inefficient 18O-labeling
Figure 4.

Validation of SseL substrates in RAW 264.7 cells. RAW 264.7 cell lysates were incubated with no enzyme (lane 1) or SseL (lane 2) or USP2 (lane 3) and analyzed by western blots with specific antibodies. The asterisks show the mass of the unmodified protein, whereas the arrows show the protein bands that increase with the enzymatic treatment, which validates S100A6 and hnRNP K, but not MHC classI (H-2D) as SseL substrates. # Deubiquitinated product of a truncated form of hnRNP K. For overexposed versions of anti-S100A6 and anti-Hnrnpk western blots see Supporting information figure S4.
To further support these results we performed another experiment by capturing ubiquitinated proteins from RAW 264.7 cell lysate by affinity purification using an ubiquitin-binding domain, treating with the enzyme and visualizing by western blots. When probing with anti-hnRNP K antibody, bands corresponding to the unmodified mass of the protein clearly appear when treated with SseL and USP2 deubiquitinases (Supporting information Figure S4). Unfortunately, ubiquitinated forms of hnRNP K are polydisperse and could not be detected. Thus, we reprobed the western blot using a combination of anti hnRNP K and anti-ubiquitin antibodies that clearly showed that ubiquitinated proteins were enriched in the affinity purification (Supporting information Figure S4). These results support that deubiquitinase products can be detected by mass shift on western blot experiments.
Since substrate recognition is an important step for enzymatic activity, we further validated the identified substrates by evaluating their binding to SseL. SseL interactions with S100A6 and hnRNP K were tested by incubating a SBP-tagged catalytically inactive C285S mutant with RAW 264.7 cell lysates and then precipitated with streptavidin-conjugated agarose beads. The captured proteins were analyzed by western blot using S100A6 and hnRNP K specific antibodies. As a positive control, the first western blot was probed against K63-linked polyUb chains (Figure 5). The pulldown experiment showed that SseL is capable of interacting with the newly identified substrates S100A6 and hnRNP K. Interestingly, SseL seems to bind to a small population of modified proteins, for which the increase in mass is consistent with the addition of one ubiquitin unit (Figure 5). These results suggest that SseL binds to ubiquitinated S100A6 and hnRNP K, but not their unmodified isoforms. Taken together the enzymatic and binding assays support the findings of the proteomic analysis and validated S100A6 and hnRNP K as new SseL substrates.
Figure 5.
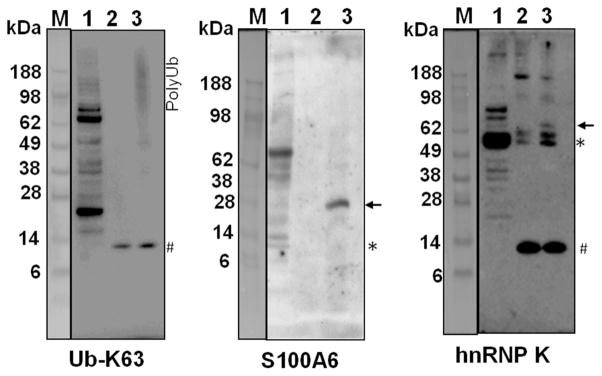
SseL binding to substrates in RAW 264.7 cells. SseL-interacting proteins were captured from RAW 264.7 cell lysate using the catalytically inactive SBP-C285S SseL and analyzed by western blots with specific antibodies. M – molecular weight marker, 1 – lysate prior to affinity purification capture, 2 – control affinity purification without SBP-C285S SseL, 3 – affinity purification in the presence of SBP-C285S SseL. The asterisks show the expected mass of the unmodified protein, the arrows show bands of proteins that specifically bind to SseL, and the pound sign represents unspecific reactions.
Potential substrate motif recognized by SseL
Common features of the amino acid residues surrounding the ubiquitination sites targeted by SseL were investigated to identify possible motifs that drive the enzyme specificity. A sequence alignment showed that the ubiquitination sites targeted by SseL do not have an obvious sequence motif (Figure 6A). We also performed a motif enrichment analysis with all the mapped ubiquitination sites in our dataset using the motif-X tool.30 However, none of the SseL substrates were among the enriched ubiquitination motifs (Supporting information Figure S5), supporting the idea that this enzyme is specific to a few targets inside the host cells.
Figure 6.

Structural analysis of SseL substrates and the Lys48-linked diubiquitin. (A) Sequence alignment of amino acid residues surrounding SseL deubiquitination sites and the lysine 48 from the ubiquitin (PDBid 3M3J). (B) Structure of K48-linked diubiquitin (PDB ID: 3M3J). (C) Structure of K63-linked diubiquitin (PDB ID: 2JF5). (D) Structure of S100A6 (PDB ID: 1K96). (E) Structure of heterogeneous ribonucleoprotein K (hnRNP K) (PDB ID: 1J5K). (F) Surface accessible solvent area of the lysine residues from the Bos Taurus and Homo sapiens diubiquitins, and S100A6 and hnRNP K proteins. The ubiquitinated lysine residues are highlighted in bold.
We also investigated if there might be common features in the secondary and tertiary structures of the SseL substrates. Thus, we evaluated the structure of K63-linked diubiquitin (PDB ID: 2JF5), S100A6 (PDB ID: 1K96) and hnRNP K (PDB ID: 1J5K). Interestingly, all the ubiquitination sites targeted by SseL are located in unstructured loops that were predicted as coils with high accuracy (68.4–95.1%) (Figure 6C–E). As a comparison, the structure of modified residue K48-linked diubiquitin (Figure 6B), a poor substrate of SseL, is located at a sheet (estimated prediction accuracy of 81.90%). We calculated the solvent accessible surface area (SASA) of the lysine residues in each protein. Taking into consideration that the maximum SASA of a lysine side chain is 171.9 Å2, the ubiquitinated residues targeted by SseL are indeed highly exposed (Figure 6F). In contrast, the K48 residue of ubiquitin is only partially exposed, which may explain in part the poor activity of SseL at this site. Together, these analyses suggest that SseL recognizes ubiquitin chains that were attached to lysine residues that are highly accessible to solvents and located on unstructured loops. This feature does not necessary confers specificity to the SseL interaction, but could be a fundamental condition for cleaving the ubiquitin.
DISCUSSION
There are several strategies for identifying substrates of enzymes that target specific post-translational modifications (PTMs). One of the most popular approaches includes the overexpression or repression of the enzyme expression followed by PTM enrichment and quantitative proteomic analysis. This approach has been successfully used for the analysis of many PTM-regulating enzymes, such as kinases, 31 phosphatases32 and deacetylases.33 This approach can be problematic for ubiquitinated proteins since ubiquitination is well known to regulate gene expression34 and protein degradation,35 two processes that effectively change the abundances of proteins. Since most of the proteomic approaches that analyze PTMs use relative quantification between samples, it would be hard to distinguish if changes in ubiquitinated peptides would reflect alterations in protein abundances rather than variations in modification levels induced by the studied enzyme. Thus, to identify specific substrates of DUBs we used cell lysates, since protein expression and degradation can be better controlled.
One of the challenges of identifying DUB substrates from cell lysates is the need to block the activity of the endogenous enzymes in a fashion that does not affect the activity of the exogenously added DUB. Since all known DUBs are either metallo- or cysteine-proteases2 we added EDTA and thiol-alkylating agents to inhibit those enzymes. Since SseL is a cysteine-protease, it was necessary to quench the alkylating agent before adding the enzyme to the cell lysate. During this process we found that NEM had the best performance at inhibiting endogenous DUB activity. Furthermore, a small pool of DUBs seemed to be oxidized at the active enzymatic cysteine residue and can be efficiently blocked by NEM when TCEP is added to the lysis buffer. This phenomenon has major implications for proteomic analysis to characterize ubiquitinated proteins,24 since these oxidized DUBs can become active when proteins are reduced to break disulfide bonds.
After treating the cell lysate with SseL, the ubiquitination sites targeted by the enzyme were identified by a combination of immunoaffinity purification and quantitative proteomic analysis using 18O labeling to quantify the K-ε-GG-containing peptides. It is worth noting that isotope labeled reagent that derivatizes the primary amines would also modify the K-ε-GG epitope that would no longer be recognized by the monoclonal antibody, unless the derivation step is done post enrichment.
Using our approach, two novel SseL substrates were identified and validated, S100A6 and hnRNP K, in addition to the previously characterized K63-linked Ub chains. The fact that only a few SseL substrates were identified suggests that this enzyme has very narrow specificity. DUBs differ greatly in their substrate specificities; whereas some DUBs are highly specific for one or a few substrates, others have a broad range of specificity and are able to cleave a wide range of ubiquitinated proteins. For instance, the human DUB Otulin has been proposed to only hydrolyze linear Ub chains,36 whereas rat USP2 is able to cleave thousands of ubiquitinated proteins.3 Another example is USP5, also known as isopeptidase T, which can cleave different Ub chains, but it is restricted to unanchored chains since it bears a ZnF ubiquitin-binding domain that recognizes the C-terminus of ubiquitin that is normally covalently attached to proteins.37 The examination of the substrate structures suggests that SseL cleaves ubiquitination sites that are highly accessible to solvents. However, the fine mechanism of SseL specificity to its substrates is still a topic of further investigation.
To conclude, in this paper we describe a method for identifying DUB substrates in cell lysates. The application of this method led to the identification and validation of two novel substrates of the Salmonella DUB SseL in host cells. This method can also be used to determine the landscape of DUB substrates of different organisms.
Supplementary Material
Acknowledgments
Funding Sources
This research has been funded in part by a grant from the National Institutes of Health GM094585 and GM094623, and by P41 GM103493-10. Work was partially performed in the Environmental Molecular Sciences Laboratory (EMSL), a DOE-BER national scientific user facility at Pacific Northwest National Laboratory (PNNL). PNNL is a multi-program national laboratory operated by Battelle Memorial Institute for the DOE under contract DE-AC05-76RLO 1830.
The authors thank Drs. John Cort, Matt Monroe, and Vamsi Kodali from Pacific Northwest National Laboratory, and George Niemann and Fred Heffron from Oregon Health & Science University for their constructive comments, inputs and suggestions.
ABBREVIATIONS
- ACN
acetonitrile
- ATCC
American Type Culture Collection
- CAA
chloroacetamide
- DTT
dithiothreitol
- DUB
deubiquitinase
- IAA
iodoacetamide
- MonoUb
monoubiquitination
- NEM
N-ethylmaleimide
- PolyUb
polyubiquitination
- PTM
protein post-translational modification
- TCEP
tris(2-carboxyethyl)phosphine
- TFA
trifluoroacetic acid
- Ub2
diubiquitin
Footnotes
The authors declare no competing financial interests.
Data availability. The raw data files of LC-MS/MS runs are available at Peptide Atlas (Dataset identifier: PASS00509) and the PNNL omics distribution webpage at http://omics.pnl.gov/node/440.
Supporting information, this material is available free of charge via the Internet at http://pubs.acs.org. Supporting Information Figure S1 – SseL activity on diubiquitin (Ub2) and oligoubiquitin (Ub2–7) chains. Supporting Information Figure S2 – Mass spectra for all SseL substrate candidates. Supporting Information Figure S3 – Validation of SseL substrates in RAW 264.7 cells with over exposed versions of anti-S100A6 and Hnrnpk Western blots. Supporting Information Figure S4 – Digestion of ubiquitinated hnRNP K protein by SseL. Supporting Information Figure S5 – Overrepresented motifs in the complete ubiquitinome dataset. Supporting information table S1–S2 – identified peptides with their identification scores and abundance ratios.
References
- 1.Behrends C, Harper JW. Constructing and decoding unconventional ubiquitin chains. Nat Struct Mol Biol. 2011;18:520–8. doi: 10.1038/nsmb.2066. [DOI] [PubMed] [Google Scholar]
- 2.Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–97. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–40. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10:M111 013284. doi: 10.1074/mcp.M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Udeshi ND, Mani DR, Eisenhaure T, Mertins P, Jaffe JD, Clauser KR, Hacohen N, Carr SA. Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol Cell Proteomics. 2012;11:148–59. doi: 10.1074/mcp.M111.016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, Qiao JW, Carr SA. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics. 2013;12:825–31. doi: 10.1074/mcp.O112.027094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsumoto M, Hatakeyama S, Oyamada K, Oda Y, Nishimura T, Nakayama KI. Large-scale analysis of the human ubiquitin-related proteome. Proteomics. 2005;5:4145–51. doi: 10.1002/pmic.200401280. [DOI] [PubMed] [Google Scholar]
- 8.Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, Rodriguez MS. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009;10:1250–8. doi: 10.1038/embor.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakayasu ES, Ansong C, Brown JN, Yang F, Lopez-Ferrer D, Qian WJ, Smith RD, Adkins JN. Evaluation of selected binding domains for the analysis of ubiquitinated proteomes. J Am Soc Mass Spectrom. 2013;24:1214–23. doi: 10.1007/s13361-013-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Y, Chan DW, Jung SY, Malovannaya A, Wang Y, Qin J. A data set of human endogenous protein ubiquitination sites. Mol Cell Proteomics. 2011;10:M110 002089. doi: 10.1074/mcp.M110.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–6. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 12.Xu G, Paige JS, Jaffrey SR. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat Biotechnol. 2010;28:868–73. doi: 10.1038/nbt.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee KA, Hammerle LP, Andrews PS, Stokes MP, Mustelin T, Silva JC, Black RA, Doedens JR. Ubiquitin ligase substrate identification through quantitative proteomics at both the protein and peptide levels. J Biol Chem. 2011;286:41530–8. doi: 10.1074/jbc.M111.248856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song M, Hakala K, Weintraub ST, Shiio Y. Quantitative proteomic identification of the BRCA1 ubiquitination substrates. J Proteome Res. 2011;10:5191–8. doi: 10.1021/pr200662b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubel CE, Schisler JC, Hamlett ED, DeKroon RM, Gautel M, Alzate O, Patterson C. Diggin’ on u(biquitin): a novel method for the identification of physiological E3 ubiquitin ligase substrates. Cell Biochem Biophys. 2013;67:127–38. doi: 10.1007/s12013-013-9624-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–6. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poulsen JW, Madsen CT, Young C, Kelstrup CD, Grell HC, Henriksen P, Juhl-Jensen L, Nielsen ML. Comprehensive profiling of proteome changes upon sequential deletion of deubiquitylating enzymes. J Proteomics. 2012;75:3886–97. doi: 10.1016/j.jprot.2012.04.055. [DOI] [PubMed] [Google Scholar]
- 18.Romero-Calvo I, Ocon B, Martinez-Moya P, Suarez MD, Zarzuelo A, Martinez-Augustin O, de Medina FS. Reversible Ponceau staining as a loading control alternative to actin in Western blots. Anal Biochem. 2010;401:318–20. doi: 10.1016/j.ab.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 19.Ansong C, Yoon H, Porwollik S, Mottaz-Brewer H, Petritis BO, Jaitly N, Adkins JN, McClelland M, Heffron F, Smith RD. Global systems-level analysis of Hfq and SmpB deletion mutants in Salmonella: implications for virulence and global protein translation. PLoS One. 2009;4:e4809. doi: 10.1371/journal.pone.0004809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayampurath AM, Jaitly N, Purvine SO, Monroe ME, Auberry KJ, Adkins JN, Smith RD. DeconMSn: a software tool for accurate parent ion monoisotopic mass determination for tandem mass spectra. Bioinformatics. 2008;24:1021–3. doi: 10.1093/bioinformatics/btn063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim S, Gupta N, Pevzner PA. Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J Proteome Res. 2008;7:3354–63. doi: 10.1021/pr8001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slysz GW, Steinke L, Ward DM, Klatt CG, Clauss TR, Purvine SO, Payne SH, Anderson GA, Smith RD, Lipton MS. Automated data extraction from in situ protein-stable isotope probing studies. J Proteome Res. 2014;13:1200–10. doi: 10.1021/pr400633j. [DOI] [PubMed] [Google Scholar]
- 23.Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 24.Ziv I, Matiuhin Y, Kirkpatrick DS, Erpapazoglou Z, Leon S, Pantazopoulou M, Kim W, Gygi SP, Haguenauer-Tsapis R, Reis N, Glickman MH, Kleifeld O. A perturbed ubiquitin landscape distinguishes between ubiquitin in trafficking and in proteolysis. Mol Cell Proteomics. 2011;10:M111 009753. doi: 10.1074/mcp.M111.009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cotto-Rios XM, Bekes M, Chapman J, Ueberheide B, Huang TT. Deubiquitinases as a signaling target of oxidative stress. Cell Rep. 2012;2:1475–84. doi: 10.1016/j.celrep.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rytkonen A, Poh J, Garmendia J, Boyle C, Thompson A, Liu M, Freemont P, Hinton JC, Holden DW. SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc Natl Acad Sci U S A. 2007;104:3502–7. doi: 10.1073/pnas.0610095104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakayasu ES, Brown RN, Ansong C, Sydor MA, Imtiaz S, Mihai C, Sontag R, Hixson KK, Monroe ME, Sobreira TJ, Orr G, Petyuk VA, Yang F, Smith RD, Adkins JN. Multi-omic data integration links deleted in breast cancer 1 (DBC1) degradation to chromatin remodeling in inflammatory response. Mol Cell Proteomics. 2013;12:2136–47. doi: 10.1074/mcp.M112.026138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyagi M, Rao KC. Proteolytic 18O-labeling strategies for quantitative proteomics. Mass Spectrom Rev. 2007;26:121–36. doi: 10.1002/mas.20116. [DOI] [PubMed] [Google Scholar]
- 29.Scheibe M, Arnoult N, Kappei D, Buchholz F, Decottignies A, Butter F, Mann M. Quantitative interaction screen of telomeric repeat-containing RNA reveals novel TERRA regulators. Genome Res. 2013 doi: 10.1101/gr.151878.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartz D, Gygi SP. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol. 2005;23:1391–8. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 31.Wang P, Xue L, Batelli G, Lee S, Hou YJ, Van Oosten MJ, Zhang H, Tao WA, Zhu JK. Quantitative phosphoproteomics identifies SnRK2 protein kinase substrates and reveals the effectors of abscisic acid action. Proc Natl Acad Sci U S A. 2013;110:11205–10. doi: 10.1073/pnas.1308974110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mertins P, Eberl HC, Renkawitz J, Olsen JV, Tremblay ML, Mann M, Ullrich A, Daub H. Investigation of protein-tyrosine phosphatase 1B function by quantitative proteomics. Mol Cell Proteomics. 2008;7:1763–77. doi: 10.1074/mcp.M800196-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y, Zhao W, Yang JS, Cheng Z, Luo H, Lu Z, Tan M, Gu W, Zhao Y. Quantitative acetylome analysis reveals the roles of SIRT1 in regulating diverse substrates and cellular pathways. Mol Cell Proteomics. 2012;11:1048–62. doi: 10.1074/mcp.M112.019547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weake VM, Workman JL. Histone ubiquitination: triggering gene activity. Mol Cell. 2008;29:653–63. doi: 10.1016/j.molcel.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 35.Kravtsova-Ivantsiv Y, Ciechanover A. Non-canonical ubiquitin-based signals for proteasomal degradation. J Cell Sci. 2012;125:539–48. doi: 10.1242/jcs.093567. [DOI] [PubMed] [Google Scholar]
- 36.Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, Wauer T, Hospenthal MK, Gyrd-Hansen M, Krappmann D, Hofmann K, Komander D. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell. 2013;153:1312–26. doi: 10.1016/j.cell.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reyes-Turcu FE, Horton JR, Mullally JE, Heroux A, Cheng X, Wilkinson KD. The ubiquitin binding domain ZnF UBP recognizes the C-terminal diglycine motif of unanchored ubiquitin. Cell. 2006;124:1197–208. doi: 10.1016/j.cell.2006.02.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.