

Abstract
Purpose
Enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA) prolongs survival in infantile Pompe disease (IPD). However, the majority of cross reactive immunologic material (CRIM)-negative (CN) patients have immune responses with significant clinical decline despite continued ERT. We aimed to characterize immune responses in CN IPD patients receiving ERT monotherapy.
Methods
A chart review identified 20 CN IPD patients treated with ERT monotherapy for ≥6 months. Patients were stratified by anti-rhGAA antibody titers: high sustained antibody titers (HSAT) ≥51,200 at least twice; low titers (LT) <6,400 throughout treatment; or sustained intermediate titers (SIT) 6,400–25,600.
Results
Despite early initiation of treatment, the majority (85%) of CN patients developed significant antibody titers, most with HSAT associated with invasive ventilation and death. Nearly all patients with HSAT had at least one nonsense GAA mutation, while the LT group exclusively carried splice site or frameshift mutations. Only one patient in the HSAT group is currently alive after successful immune modulation in the entrenched setting.
Conclusion
Immunological responses are a significant risk in CN IPD; thus, immune tolerance induction in the naïve setting should strongly be considered. Further exploration of factors influencing immune responses is required, particularly with the advent of newborn screening for Pompe disease.
Keywords: Pompe disease, alglucosidase alfa, enzyme replacement therapy, antibodies, immune tolerance
INTRODUCTION
In Pompe disease (OMIM 232300; acid maltase deficiency, glycogen storage disease type II), a deficiency of lysosomal acid alpha-glucosidase (GAA) leads to the accumulation of glycogen in skeletal, cardiac and smooth muscle, resulting in progressive, debilitating muscle weakness. If left untreated, infants with the classic infantile form of Pompe disease typically succumb to the disease by cardiopulmonary failure within the first two years of life. However, intravenous enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA), or alglucosidase alfa (Lumizyme®), has improved clinical outcomes and prolonged survival in patients with infantile Pompe disease (IPD).
Despite improved clinical outcomes including prolonged overall and ventilator-free survival in IPD patients1–4, the efficacy of ERT is diminished for some patients who mount an immune response against rhGAA. Previous studies have demonstrated that cross reactive immunologic material (CRIM) status is highly correlated with clinical response to treatment5,6. CRIM-positive (CP) patients generally respond well to ERT due to the presence of residual GAA expression. On the other hand, patients with no residual protein expression are categorized as CRIM-negative (CN). In general, when compared to CP patients who tolerize to ERT over time, CN patients mount an immune response against rhGAA resulting in significant clinical decline and ultimately ventilator dependence or death by 27.1 months despite continuation of ERT5.
Physicians caring for patients treated with a therapeutic protein such as rhGAA in IPD are thus charged with the challenge of also managing significant immune responses. Our group has demonstrated the success of immune tolerance induction (ITI) protocols along with ERT in both the naïve setting, to prevent the development of high antibody titers7–9, and also in the entrenched setting, to suppress an immune response once high antibody titers have already developed6,10. However, ITI has proved to be most successful in the naïve setting, which requires a short 5-week regimen of immune modulation initiated at the start of ERT compared to a significantly longer, multipronged approach for treating patients with an entrenched immune response to the rhGAA6,10.
Current literature reflects how clinical outcomes of CN patients treated with ERT+ITI surpass those on ERT monotherapy9. However, three cases have been reported in which CN patients have done relatively well, clinically, in the absence of high sustained antibody titers (HSAT) without the use of ITI11,12. Yet, it is important to note that one of these patients received omalizumab, an IgG monoclonal antibody which binds IgE, for severe allergic reaction to alglucosidase alfa infusion11. The role of this drug in immunomodulation is yet to be reported.
As the use of ITI protocols is becoming more prevalent, it is important to understand the factors influencing immune responses in the CN IPD population, as such knowledge is essential for guiding further implementation of ITI protocols. However, a fundamental understanding of the long-term clinical outcomes of CN IPD patients treated with ERT alone (ERT monotherapy) is missing. In this study we aimed to retrospectively assess the immune responses in our cohort of CN IPD patients receiving rhGAA, which we believe is the largest in the world. We did this in order to determine what proportion of CN patients develop significant immune responses, which negatively affect clinical outcomes, and to consider the influence of potentially modifying factors in the development of an immune response, such as GAA genotype or age at which ERT is initiated. This cohort represents an invaluable set of data with a unique opportunity to examine responses to ERT alone, especially as the treatment paradigm for CN IPD patients continues to shift toward the implementation of ITI in the naïve setting.
PATIENTS AND METHODS
This was a retrospective chart review of our large cohort of 183 total IPD patients from our previously published cohort5 and enrolled in Duke Institutional Review Board (IRB)-approved protocol 00001562 [LDN6709 Site 206; Clinicaltrials.gov Identifier: NCT01665326] for CRIM determination and longitudinal follow-up. Of those, we identified 58 patients who were determined to be CN, 20 of whom were treated with alglucosidase alfa ERT monotherapy for at least 6 months at cumulative doses of 20 to 40 milligrams per kilogram every two weeks. We included patients who had been on ITI regimens that were unsuccessful in preventing the development of HSAT. We also included one patient (Patient 10; HSAT group) who was successfully treated with ITI in the entrenched setting; data were used until ITI was initiated at week 87. Patients were excluded from data analysis if there was administration of ITI in the naïve setting.
Laboratory Methods
CN status was first determined based on reactivity of a pool of monoclonal and/or polyclonal anti-rhGAA antibodies capable of recognizing both native and recombinant GAA protein bands, using western blot analysis on skin fibroblast cell extracts. If none of the GAA protein forms (unprocessed precursor band at 110 kDa or processed forms bands at 95, 76 and 70 kDa) were detected, the patient is categorized as CN5. In one case, CN status was determined using western blot analysis of amniocytes following the prior diagnosis of the proband sibling12. For all patients, final CN status was assigned once the results of western blot analysis were correlated with GAA mutation genotype13.
Anti-rhGAA IgG Antibodies
Anti-rhGAA IgG antibodies were ascertained using enzyme-linked immunosorbent assays and confirmed using radioimmunoprecipitation as described previously2 and assessed by Genzyme Corporation (Framingham, MA) at baseline and at regular intervals throughout treatment. CN IPD patients were further divided into three groups based on anti-rhGAA antibody titer levels and the need for further clinical intervention6,14:
HSAT group: Anti-rhGAA IgG antibodies measured ≥51,200 on two or more occasions after six months on ERT;
Low titer (LT) group: IgG antibodies remained <6,400 throughout the course of ERT; and
Sustained intermediate titer (SIT) group: IgG antibodies ranged 6,400–25,600.
Clinical Parameters
Baseline and follow-up data were obtained to assess treatment outcomes in these three groups including age at diagnosis, age at initiation of ERT, invasive ventilator-free survival, overall survival and left ventricular mass index (LVMI).
Statistical Analysis
Survival data were analyzed using the Kaplan–Meier method with two-tailed P values generated using the log-rank test15. Analyses were performed with STATA version 13.1 (StataCorp LP, College Station, Texas). Due to small sample size, descriptive data are presented as medians.
RESULTS
Of our total IPD cohort, established between 1999 through February 1, 2014 (n=183), 58 were identified as CN, which to our knowledge represents the largest CN IPD cohort in current literature. From this group, 23 CN IPD were excluded from analysis because ITI treatment was administered in the naïve setting; for 15 other CN IPD patients, death occurred before six months on ERT or there was insufficient follow-up data; thus, the resulting data analysis included 20 CN IPD patients (Figure 1).
Figure 1. Cohort of infantile Pompe disease patients.
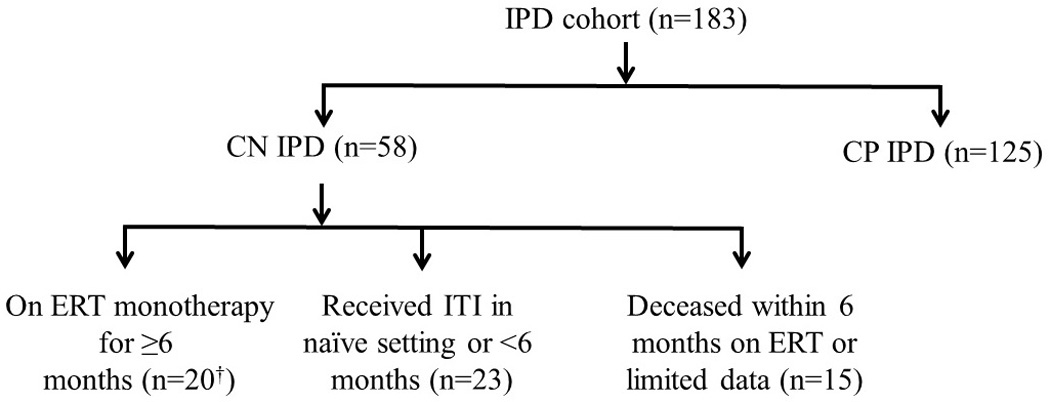
† n=1 rescued with an ITI protocol in the entrenched setting after six months on ERT; therefore, data for this patient was included up until the initiation of ITI10; n=2 included as ITI regimen was unsuccessful with persistence of antibody titers16,17
IPD, infantile Pompe disease; CN, CRIM, cross reactive immunologic material (CRIM); CN, CRIM-negative; CP, CRIM-positive; ERT, enzyme replacement therapy; ITI, immune tolerance induction
Anti-rhGAA antibody titers
The majority (85%) of CN patients (17/20) developed clinically significant IgG antibody titers to rhGAA. Thirteen mounted HSAT with median peak titers of 204,800 by 24 weeks on ERT (Table 1). Four patients had SIT with peak titers of 25,600 and 51,200. Three patients had LT peaking at 200 (n=2) and 800 (n=1).
Table 1.
Antibody response in HSAT, SIT and LT groups at peak titer and last titer measurements
| Group | Patient | Peak titers | Weeks on ERT at peak titers |
Last titers | Weeks on ERT at last titers |
|---|---|---|---|---|---|
| HSAT | 1 | 204 800 | 78 | 102 400 | 152 |
| 2 | 204 800 | 60 | 102 400 | 181 | |
| 3 | 102 400 | 24 | 51 200 | 36 | |
| 4 | 204 800 | 64 | 204 800 | 64 | |
| 5 | 409 600 | 64 | 409 600 | 82 | |
| 6 | 1 638 400 | 52 | 1 638 400 | 64 | |
| 7 | 204 800 | 38 | 102 400 | 52 | |
| 8 | 51 200 | 24 | 25 600 | 38 | |
| 9 | 409 600 | 64 | 409 600 | 64 | |
| 10 | 819 200 | 86 | 819 200* | 86 | |
| 11 | 51 200 | 28 | 51 200 | 63 | |
| 12 | 819 200 | 130 | 409 600 | 138 | |
| 13 | 819 200 | 29 | 819 200 | 29 | |
| Median | 204 800 | 60 | 204 800 | 64 | |
| Range | 51 200 – 1 638 400 | 24 – 130 | 25 600 – 1 638 400 | 29 – 181 | |
| SIT | 14 | 51 200 | 15 | 6 400 | 202 |
| 15 | 25 600 | 16 | 6 400 | 90 | |
| 16 | 25 600 | 117 | 6 400 | 194 | |
| 17 | 25 600 | 24 | 12 800 | 132 | |
| Median | 25 600 | 20 | 6 400 | 145 | |
| Range | 25 600 – 51 200 | 15 – 117 | 6 400 – 25 600 | 90 – 202 | |
| LT | 18 | 800 | 26 | 100 | 112 |
| 19 | 200 | 100 | 100 | 110 | |
| 20 | 200 | 48 | 0 | 84 | |
| Median | 200 | 48 | 100 | 110 | |
| Range | 200 – 800 | 26 – 100 | 0 – 100 | 84 – 112 |
Indicates the last titer value before the initiation of ITI
HSAT, high sustained antibody titers; LT, low titers; SIT, sustained intermediate titers; ITI, immune tolerance induction
Spectrum of GAA Mutations
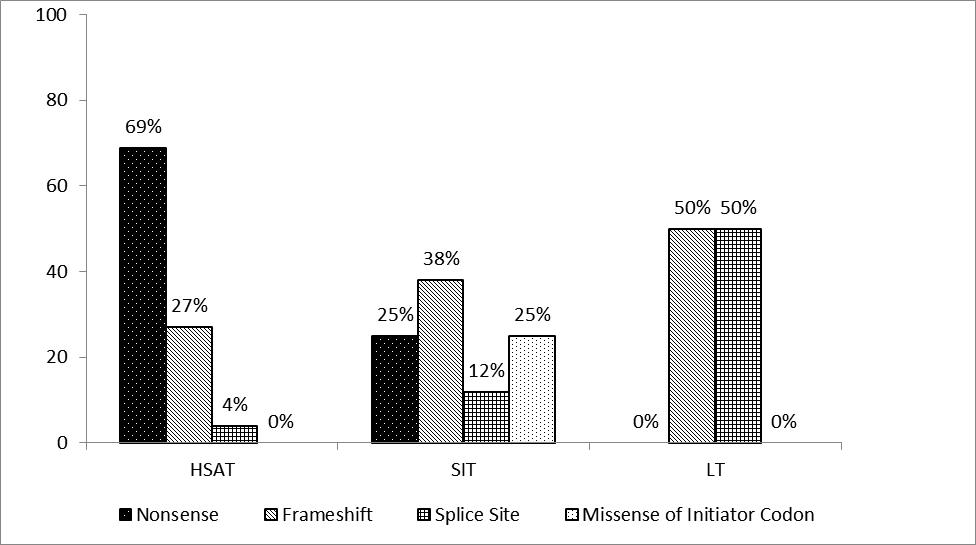
We identified GAA mutations in our cohort (Table 2) and assessed the types of mutations seen in each titer group (Figure S1). Ten of 13 patients in the HSAT group carried at least one nonsense mutation. Total nonsense mutations frequency observed in HSAT group was 18 /26 total alleles (69%), frameshift mutation allele frequency was 7/26 alleles (27%) and splice site mutation allele frequency was 1/26 alleles (4%). Three of eight alleles (38%) in the SIT group were frameshift mutations, two (25%) were missense initiator codon mutations, two (25%) were nonsense mutations and one (13%) was a splice site mutation. Three out of six alleles (50%) observed in the LT group were splice site mutations and three (50%) were frameshift mutations. All patients were unrelated with the exception of one sib-pair (Patient 14 of the SIT group and Patient 18 of the LT group) who were compound heterozygotes for the c.546+2_+5delTGGG splice site mutation and c.1650dupG frameshift mutation, as first reported by Al Khallaf et al12. Unlike the SIT and HSAT groups, no patient in the LT group carried a nonsense mutation.
Table 2.
GAA mutations identified in CRIM-negative infantile Pompe disease cohort
| Group | Patient | Allele 1 cDNA change | Allele 2 cDNA change | Allele 1 amino acid change |
Allele 2 amino acid change |
Allele 1 mutation type |
Allele 2 mutation type |
Allele 1 mutation effect† |
Allele 2 mutation effect† |
|---|---|---|---|---|---|---|---|---|---|
| HSAT | 1 | c.722_723delTT | c.1687C>T | p.Phe241CysfsX88 | p.Gln563X | Frameshift | Nonsense | very severe | very severe |
| 2 | c.1687C>T | c.1687C>T | p.Gln563X | p.Gln563X | Nonsense | Nonsense | very severe | very severe | |
| 3 | c.148_859-11del | c.685_686insCGGC | p.Glu50HisfsX37 | p.Arg229ProfsX102 | Frameshift | Frameshift | very severe | very severe | |
| 4 | c.2560C>T | c.2560C>T | p.Arg854X | p.Arg854X | Nonsense | Nonsense | very severe | very severe | |
| 5 | c.1075G>T | c.1075G>T | p.Gly359X | p.Gly359X | Nonsense | Nonsense | unknown | unknown | |
| 6 | c.2560C>T | c.2560C>T | p.Arg854X | p.Arg854X | Nonsense | Nonsense | very severe | very severe | |
| 7 | c.2560C>T | c.2560C>T | p.Arg854X | p.Arg854X | Nonsense | Nonsense | very severe | very severe | |
| 8 | c.722_723delTT | c.1754 +1G>A | p.Phe241CysfsX88 | Splice Site | Frameshift | Splice Site | very severe | unknown | |
| 9 | c.1496G>A | c.1496G>A | p.Trp499X | p.Trp499X | Nonsense | Nonsense | very severe | very severe | |
| 10 | c.1654delC | c.2560C>T | p.Leu552SerfsX26 | p.Arg854X | Frameshift | Nonsense | unknown | very severe | |
| 11 | c.2560C>T | c.2560C>T | p.Arg854X | p.Arg854X | Nonsense | Nonsense | very severe | very severe | |
| 12 | c.1826dupA | c.2238G>A | p.Tyr609X | p.Trp746X | Nonsense | Nonsense | very severe | very severe | |
| 13 | c.2222_*549+214delins13 | c.2222_*549+214delins13 | p.Asp741AlafsX28 | p.Asp741AlafsX28 | Frameshift | Frameshift | unknown | unknown | |
| SIT | 14 | c.546+2_546+5delTGGG | c.1650dupG | Splice Site | p.Thr551AspfsX85 | Splice Site | Frameshift | very severe | unknown |
| 15 | c.1209delC | c.1209delC | p.Asn403LysfsX37 | p.Asn403LysfsX37 | Frameshift | Frameshift | unknown | unknown | |
| 16 | c.2560C>T | c.2560C>T | p.Arg854X | p.Arg854X | Nonsense | Nonsense | very severe | very severe | |
| 17 | c.1A>G | c.1A>G | p.Met1? | p.Met1? | Initiator codon | Initiator codon | unknown | unknown | |
| LT | 18 | c.546+2_546+5delTGGG | c.1650dupG | Splice Site | p.Thr551AspfsX85 | Splice Site | Frameshift | very severe | unknown |
| 19 | c.2495_2496delCA | c.2495_2496delCA | p.Thr832AsnfsX51 | p.Thr832AsnfsX51 | Frameshift | Frameshift | very severe | very severe | |
| 20 | c.1637-2A>G | c.1637-2A>G | Splice Site | Splice Site | Splice Site | Splice Site | very severe | very severe |
Source: Erasmus MC Rotterdam; http://www.pompecenter.nl
HSAT, high sustained antibody titers; LT, low titers; SIT, sustained intermediate titers
Clinical Parameters
Table 3 provides demographic and clinical parameters for this international cohort of CN IPD on ERT monotherapy. Four patients started ERT before one month of age with variable clinical outcomes: while two have not developed an immune response against rhGAA (Patient 18 and 20), two others responded poorly with death at 18 and 45 months of age (Patient 9 and 16). Thus, it appears that early initiation of ERT does not necessarily prevent development of an antibody response to ERT.
Table 3.
Demographic and Clinical Parameters of CN IPD Cohort
| Group | Patient | Gender | Race | Age at ERT† (months) |
Baseline invasive ventilation (Y/N) |
Baseline LVMI (g/m2) |
Age at death/current age (months) |
Deceased (Y/N) |
|---|---|---|---|---|---|---|---|---|
| HSAT | 1 | M | W | 3.5 | N | 253.0 | 50.2 | Y |
| 2 | M | B | 2.8 | N | 160.0 | 44.8 | Y | |
| 3 | M | A | 3.9 | N | 157.2 | 14.7 | Y | |
| 4 | F | A | 2.7 | N | 246.7 | 23.3 | Y | |
| 5 | F | W | 6.1 | N | N/A | 25.5 | Y | |
| 6 | F | B | 1.8 | N | 153.0 | 32.0 | Y | |
| 7 | M | B | 5.6 | N | 202.1 | 45.0 | Y | |
| 8 | F | W | 1.8 | N | 233.4 | 44.6 | Y | |
| 9 | M | W | −0.3 | N | 89.0 | 18.0 | Y | |
| 10 | M | W | 3.4 | N | 200.9 | 92.0 | N | |
| 11 | F | B | 8.9 | N | 175.0 | 33.2 | Y | |
| 12 | M | W | 1.6 | N | 141.0 | 45.8 | Y | |
| 13 | M | A | 4.0 | N | N/A | 19.0 | Y | |
| SIT | 14 | M | B | 7.5 | N | N/A | 84.7 | N |
| 15 | F | A | 3.7 | N | 203.1 | 27.1 | Y | |
| 16 | F | B | 0.3 | N | 141.5 | 45.0 | Y | |
| 17 | F | A | 16.4 | N | N/A | 65.0 | N | |
| LT | 18 | F | B | −0.3 | N | 191.1 | 55.4 | N |
| 19 | F | W | 3.3 | N | N/A | 77.8 | N | |
| 20 | M | W | −0.8 | N | N/A | 89.2 | N |
Age at ERT corrected by gestational age
HSAT, high sustained antibody titers; LT, low titers; SIT, sustained intermediate titers; ERT, enzyme replacement therapy; LVMI, left ventricular mass index; M, male; F, female; W, White; B, Black; A, Asian; N/A, not available; Y, yes; N, no
Invasive Ventilator-Free and Overall Survival
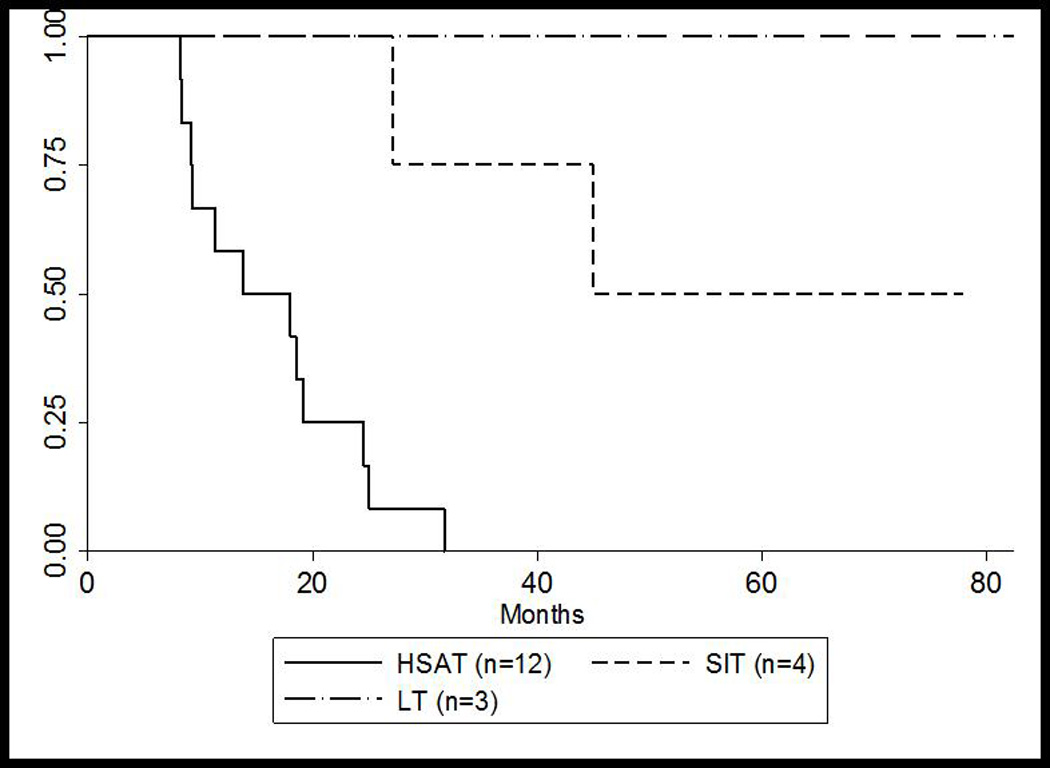
No patients required invasive ventilator support at baseline; however, 55% required invasive ventilation by a median age of 13.8 months (range: 8.2–33.6 months, n=11) (Figure 2A). Four patients (20%) died (median age of death: 23.1 months; range: 18.0–45.0 months) prior to requiring invasive ventilation and the remaining five patients (25%) are alive and invasive ventilator free. Among the patients who died during the study period, the median age of death was 32.5 months (range: 14.7–50.2 months, n=14) (Figure 2B). For the HSAT group, the median age of death was also 32.5 months (range: 14.7 to 50.2 months, n=12) and 13.8 months for ventilator-dependence (range: 8.2 to 33.6 months, n=11), similar to previously published data5. Only one patient from the HSAT group is currently alive; this patient received ITI treatment in the entrenched setting10.
Figure 2. Kaplan-Meier Curves* for (A) overall survival and (B) ventilator-free survival for HSAT, SIT, and LT groups.
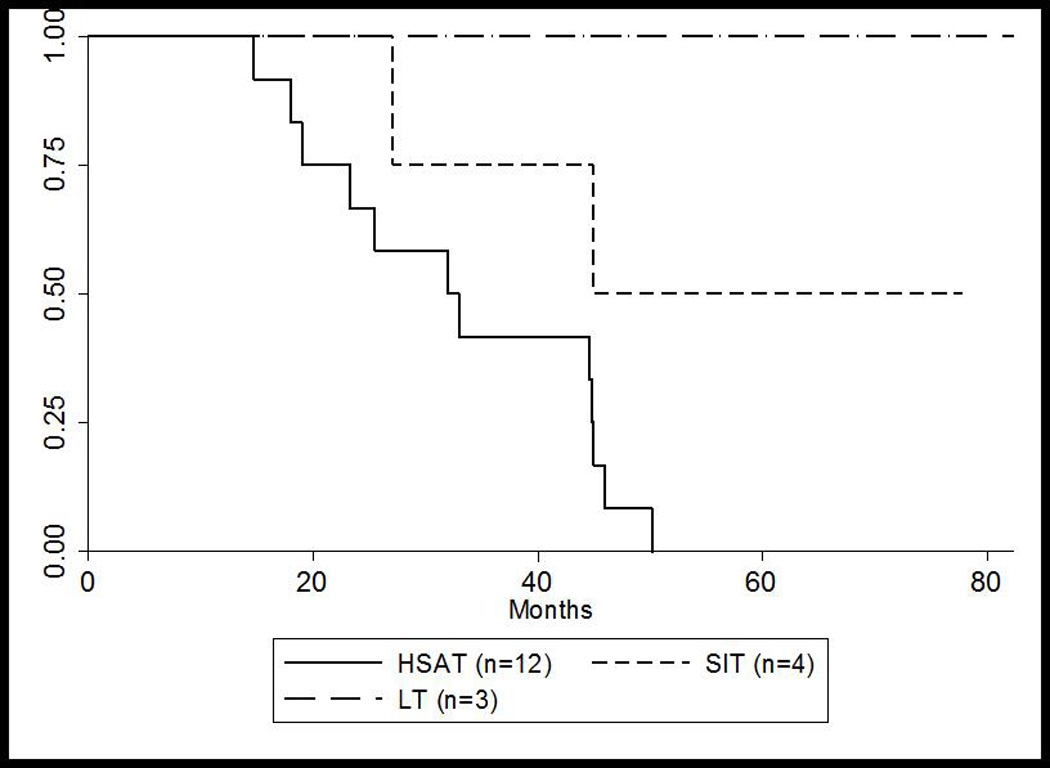
(A) Overall survival for HSAT, SIT, and LT groups
(B) Invasive ventilator-free survival for HSAT, SIT, and LT groups
* Patient 10 was successfully rescued using ITI at 86 weeks and was excluded in these calculations
HSAT, high sustained antibody titers; LT, low titers; SIT, sustained intermediate titers
Left Ventricular Mass Index
In this study, all groups had an elevated LVMI at baseline compared to the normal upper limit of 64 g/m2 for infants without Pompe disease18. The median LVMI for the HSAT group was 180.5 g/m2 at baseline (range: 89.0 to 253.0 g/m2; n=10). A net decrease was observed at 26 weeks on ERT (median=95.0 g/m2; range: 56.7 to 269.0 g/m2; n=11) followed by a net increase at 52 weeks on ERT (median=124.8 g/m2; range: 63.3 to 178.0 g/m2 n=8). For the HSAT group, the median LVMI continued to increase after week 52 of ERT (n=3; data not shown). Patient 18 of the LT group had an LVMI of 128.3 g/m2 and 50.8 g/m2 at baseline and week 111, respectively, not demonstrating a worsening in LVMI after week 52 as was seen in the HSAT group. Patient 20 in the LT group had an LVMI of 68 g/m2, 65 g/m2 and 58 g/m2 at baseline, week 30 and week 60, respectively. One patient with SIT (Patient 15) had LVMI measurements behaving similar to those seen in the HSAT group (203 g/m2 at baseline, 138 g/m2 at week 26 and 200 g/m2 at week 52), while another SIT patient’s baseline LVMI of 141.5 g/m2 normalized after 9 months of treatment (Patient 16)—consistent with those in the LT group.
DISCUSSION
Similar to what has been previously noted, approximately 32% of our IPD cohort were determined to be CN—a clinical factor predicting reduced survival and poor clinical outcomes compared to CP patients5, specifically related to the development of HSAT in response to rhGAA6. The use of ITI protocols has changed the natural history of CN IPD with a significant difference in survival compared to CN IPD treated with ERT monotherapy9. Interestingly, only three CN cases have been reported in the literature to date that have not developed a significant immune response against rhGAA without the use of ITI11,12, which suggests the presence of other modifying or protective factors influencing treatment outcomes in these cases.
Given the advent of newborn screening for Pompe disease, we sought to determine if early initiation of ERT prevents the development of an immune response. As previously noted, despite early initiation of treatment, the majority of CN IPD patients developed sustained antibodies against rhGAA, many of whom mounted HSAT associated with subsequent clinical decline and ultimately death, including some patients who started ERT before one month of age19. Therefore, in contrast to previous claims12, early initiation of ERT does not protect against the development of an immune response against rhGAA20.
While two CN patients with early initiation of ERT did not develop a clinically significant immune response, both carried splice site mutations. As such, the patients may have been exposed to GAA epitopes similar to rhGAA if these mutations allowed for residual GAA production, yet at a level below detection by traditional western blot analysis. A closer look at the types of GAA mutations in our cohort showed that most of the patients with HSAT had at least one nonsense GAA mutation. In contrast, CN patients in the LT group exclusively carried splicing or frameshift GAA mutations, none of which were nonsense mutations.
Leaky splice site mutations or premature stop codons which occur toward the end of the GAA coding region may possibly result in some residual enzyme activity, which in turn helps prevent the development of an immune response to ERT. The current understanding of CRIM status is based on results from western blot; however, without correlation of GAA mutations, western blot may not provide the entire picture. Utilization of additional technologies, such as an “omics” approach, may be warranted to identify potential modifying or protective factors and reach a more complete understanding of immune responses in IPD. Additionally, the definitive role of various GAA mutations in the protein expression as well as the possibility of variable tissue expression needs further exploration.
In conclusion, very few CN patients have been reported with no immune response; however, these patients have unique genotypes frequently with splicing mutations, whose various degrees of immunogenicity needs to be further explored. This study confirms that CN status yields an overall poor clinical response, as the majority of CN IPD patients treated with ERT monotherapy develop an immune response and experience subsequent clinical decline or death. On the contrary, development of anti-rhGAA antibody titers appears to be dictated by the presence of GAA mutations known to severely affect protein expression; thus, results of western blot analysis should be correlated with GAA genotype for the confirmation of CRIM status in IPD. While early diagnosis and treatment is significant in improving overall clinical outcomes in IPD, early diagnosis and initiation of treatment by itself does not prevent the development of antibodies against rhGAA. ITI in the naïve setting has proven to be successful in preventing the development of antibody titers in CN IPD, which validates the use of ITI therapies in the ERT-naïve setting. Because immunological responses accompanied with clinical decline poses a significant risk for the CN IPD population, implementation of ITI in the naïve setting should be considered standard of care in treating these patients at risk to develop HSAT. Further investigation surrounding immune responses and management of IPD, irrespective of CRIM status, is required and warrants attention, particularly in the setting of newborn screening for Pompe disease.
Supplementary Material
HSAT, high sustained antibody titers; LT, low titers; SIT, sustained intermediate titers
{kind=link}
ACKNOWLEDGEMENTS
This research was supported by a grant from the Genzyme Corporation, a Sanofi Company (Cambridge, MA), and in part by the Lysosomal Disease Network, a part of National Institutes of Health Rare Diseases Clinical Research Network (RDCRN). The Lysosomal Disease Network (U54NS065768) is a part of the National Institutes of Health (NIH) Rare Diseases Clinical Research Network (RDCRN), supported through collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS), the National Institute of Neurological Disorders and Stroke (NINDS) and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors are grateful to the patients and their families who participated in this study. We thank Jian Dai, BS for providing western blot results. We would as also like to thank the local physicians and clinical care team for their collaboration, including Stephanie DeArmey, MHS, PA-C, David Viskochil, MD, Jennifer Propst, MS, CGC, Virginia Proud, MD and Mary-Alice Abbott, MD.
Footnotes
Supplementary information is available at the Genetics in Medicine website.
REFERENCES
- 1.Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11:210–219. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- 2.Kishnani PS, Nicolino M, Voit T, et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 4.Kishnani PS, Corzo D, Leslie ND, et al. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009;66:329–335. doi: 10.1203/PDR.0b013e3181b24e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banugaria SG, Prater SN, Ng YK, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med. 2011;13:729–736. doi: 10.1097/GIM.0b013e3182174703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendelsohn NJ, Messinger YH, Rosenberg AS, et al. Elimination of antibodies to recombinant enzyme in Pompe’s disease. N Engl J Med. 2009;360:194–195. doi: 10.1056/NEJMc0806809. [DOI] [PubMed] [Google Scholar]
- 8.Messinger YH, Mendelsohn NJ, Rhead W, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. 2012;14:135–142. doi: 10.1038/gim.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banugaria SG, Prater SN, Patel TT, et al. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-Negative classic Infantile Pompe disease: A step towards improving the efficacy of ERT. PLoS ONE. 2013;8:e67052. doi: 10.1371/journal.pone.0067052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banugaria SG, Prater SN, McGann JK, et al. Bortezomib in the rapid reduction of high sustained antibody titers in disorders treated with therapeutic protein: lessons learned from Pompe disease. Genet Med. 2013;15:123–131. doi: 10.1038/gim.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohrbach M, Klein A, Kohli-Wiesner A, et al. CRIM-negative infantile Pompe disease: 42-month treatment outcome. J Inherit Metab Dis. 2010;33:751–757. doi: 10.1007/s10545-010-9209-0. [DOI] [PubMed] [Google Scholar]
- 12.Al Khallaf HH, Propst J, Geffrard S, et al. CRIM-Negative Pompe Disease Patients with Satisfactory Clinical Outcomes on Enzyme Replacement Therapy. JIMD Reports. 2013;9:133–137. doi: 10.1007/8904_2012_192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bali DS, Goldstein JL, Banugaria SG, et al. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet. 2012;160C:40–49. doi: 10.1002/ajmg.c.31319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abbott MA, Prater SN, Banugaria SG, et al. Atypical immunologic response in a patient with CRIM-negative Pompe disease. Mol Genet Metab. 2011;104:583–586. doi: 10.1016/j.ymgme.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 16.Hunley TE, Corzo D, Dudek M, et al. Nephrotic syndrome complicating alpha-glucosidase replacement therapy for Pompe disease. Pediatrics. 2004;114:e532–e535. doi: 10.1542/peds.2003-0988-L. [DOI] [PubMed] [Google Scholar]
- 17.Banugaria SG, Patel TT, Mackey J, et al. Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: need for agents to target antibody-secreting plasma cells. Mol Genet Metab. 2012;105:677–680. doi: 10.1016/j.ymgme.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogel M, Staller W, Buhlmeyer K. Left ventricular myocardial mass determined by cross-sectional echocardiography in normal newborns, infants, and children. Pediatr Cardiol. 1991;12:143–149. doi: 10.1007/BF02238520. [DOI] [PubMed] [Google Scholar]
- 19.Prater SN, Banugaria SG, Morgan C, et al. Letter to the Editors: Concerning “CRIM-negative Pompe disease patients with satisfactory clinical outcomes on enzyme replacement therapy” by Al Khallaf et al. J Inherit Metab Dis. 2014;37:141–143. doi: 10.1007/s10545-013-9637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Gelder CM, Hoogeveen-Westerveld M, Kroos MA, et al. Enzyme therapy and immune response in relation to CRIM status: the Dutch experience in classic infantile Pompe disease. J Inherit Metab Dis. 2014 doi: 10.1007/s10545-014-9707-6. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HSAT, high sustained antibody titers; LT, low titers; SIT, sustained intermediate titers