

Abstract
Background
Protein kinase C epsilon (PKCε) is emerging as a potential target for the development of pharmacotherapies to treat alcohol use disorders, yet little is known regarding how a history of a highly prevalent form of drinking, binge alcohol intake, influences enzyme priming or the functional relevance of kinase activity for excessive alcohol intake.
Methods
Immunoblotting was employed on tissue from subregions of the nucleus accumbens (NAC) and the amygdala to examine both idiopathic and binge drinking-induced changes in constitutive PKCε priming. The functional relevance of PKCε translocation for binge drinking and determination of potential upstream signaling pathways involved were investigated using neuropharmacological approaches within the context of 2 distinct binge drinking procedures, Drinking in the Dark (DID) and Scheduled High Alcohol Consumption (SHAC).
Results
Binge alcohol drinking elevated p(Ser729)-PKCε levels in both the NAC and the central nucleus of the amygdala (CeA). Moreover, immunoblotting studies of selectively bred and transgenic mouse lines revealed a positive correlation between the propensity to binge drink alcohol and constitutive p(Ser729)-PKCε levels in the NAC and CeA. Finally, neuropharmacological inhibition of PKCε translocation within both regions reduced binge alcohol consumption in a manner requiring intact Group1 metabotropic glutamate receptors, Homer2, phospholipase C (PLC) and/or phosphotidylinositide-3 kinase (PI3K) function.
Conclusions
Taken together, these data indicate that PKCε signaling in both the NAC and CeA is a major contributor to binge alcohol drinking and to the genetic propensity to consume excessive amounts of alcohol.
Keywords: Homer, glutamate, Scheduled High Alcohol Consumption, Drinking in the Dark, alcohol use disorders, HDID-1 mice
Introduction
Globally, alcohol use is the leading cause of premature death and disability in persons aged 15–49 (1), with upwards of 50% of alcohol-drinking individuals reporting consuming 4+ alcoholic drinks per occasion (a.k.a. binge drinking) (2,3). Despite the high prevalence of binge alcohol drinking world-wide (2), a knowledge gap exists regarding the neurobiological consequences of this pattern of excessive alcohol intake relevant for understanding the etiology and treatment of this form of alcohol use disorder (AUD). In two different murine models for binge-like drinking, Drinking in the Dark (DID) and Scheduled High Alcohol Consumption (SHAC), a binge history sensitizes both pre- and post-synaptic aspects of glutamate signaling within the nucleus accumbens (NAC) and central nucleus of the amygdala (CeA) (4–7) – two mesocorticolimbic structures highly implicated in alcoholism psychopathology (8). Moreover, biochemical indices of a hyper-glutamate state in these structures are biochemical traits associated with an excessive alcohol drinking phenotype in mice (5–7,9,10). Thus, pharmacotherapeutic strategies aimed at reducing glutamate hyperactivity may well serve as a rational treatment option for curbing binge drinking behavior. In support of this assertion, neuropharmacological or transgenic manipulations that interfere with signaling through Group1 metabotropic glutamate receptors (mGluRs) in the NAC and/or CeA attenuate binge alcohol intake in murine models (6–7,11).
One emerging potential target for pharmacotherapeutic development for AUDs is protein kinase C epsilon (PKCε), a serine/threonine kinase that is diacylglycerol (DAG)-dependent (12,13). PKCε is activated by Gαq/11-coupled receptors, including Group1 mGluRs (14), following DAG formation from phosphatidylinositol 4,5-biphosphate after hydrolysis by phospholipase C (PLC) (15). Alcohol interacts directly with PKCε (16) and up-regulates indices of PKCε phosphorylation, priming, and activity in brain (10,17–19). Activated PKCε can then, in turn, phosphorylate Ser-327 on the γ2 subunits of the GABA type A (GABAA) receptor (20) to mediate acute tolerance to the intoxicating effects of alcohol. Additionally, null mutations of PKCε reduce alcohol reward-related behaviors in various paradigms as determined in studies of mice (13,21–27), and PKCε knock-down in the CeA blunts binge alcohol intake by mice (28). Thus, an understanding of the potential initiators of PKCε signaling will provide significant insight into the molecular pathways influenced by alcohol that lead to pathological drinking.
One potential intermediary may be Group1 mGluRs. These receptors are specifically coupled to PKCε in certain brain areas (14). Alcohol intake, including binge drinking, is partly mediated by mGlu5 activity (29,30) and the subsequent stimulation of PKCε- and/or phosphotidyl inositide-3 kinase (PI3K)-dependent transduction pathways (5–7,11,19,27), which were suggested much earlier to involve the Group1 mGluR-associated scaffolding protein Homer (19). However, no study to date has directly investigated the links between Group1 mGluRs, Homer2, PI3K, and PKCε for effects on alcohol consumption. Thus, we employed a combination of immunoblotting and neuropharmacological approaches to test the hypothesis that in both the NACs and the CeA, signaling from mGlu1/5-Homer2 to PKCε is critical for the manifestation of excessive alcohol drinking. Such findings further elucidate the intracellular signaling pathways influenced by alcohol that likely underpin anomalies in excitatory neurotransmission contributing to risky alcohol drinking behavior.
Materials and Methods
Overview of the Experiments
A history of binge alcohol drinking using modified DID (31) and SHAC (32) models up-regulates indices of Group1 mGluR/Homer2/PI3K signaling within the CeA and/or NACs of C57BL/6J (B6) mice (5–7,11). Therefore, we employed immunoblotting procedures (10) to determine the effects of a history of binge alcohol drinking upon PKCε levels and priming in both brain regions of the B6 mice assayed for the expression of putative up-stream signaling molecules in our prior work (5–7). Across inbred mice, strain differences exist regarding PKCε phosphorylation of Ser729 in the NACs suggestive of a relation between higher levels of constitutive PKCε priming and alcohol intake (10). Thus, we also examined the relation between basal p(Ser729)-PKCε levels in the CeA and NAC and genetic propensity to binge drink by immunoblotting tissue from alcohol-naïve S15 HDID-1 [1st replicate line of mice selected for high blood alcohol concentrations (BACs) under DID procedures] vs. non-selected mice from a genetically heterogeneous HS/Npt stock (33). We also compared S4 SHAC (selected for high BACs under SHAC procedures) vs. SLAC (selected for low BACs under SHAC procedures) mice (5) and mGlu5F1128R transgenic (TG) mice, with a point mutation in the Homer binding site on mGlu5. TG mice exhibit low binge drinking under SHAC procedures vs. their wild-type (WT) counterparts that binge drink with this paradigm (5).
We next employed established neuropharmacological approaches (5–7,11) to examine the influence of locally inhibiting PKCε translocation using a Tat-εV1-2 peptide (34), either alone or in tandem with inhibition of mGlu1, mGlu5 and PLC upon binge alcohol intake by B6 mice. Finally, as Homer2 is in position to mediate the effects of mGlu1/5-PKCε signaling (35,36), we examined whether or not local inhibition of PKCε translocation reduced binge alcohol consumption in Homer2 knock-out (KO) animals (37).
The details of the experimental procedures for these experiments are provided in the Supplemental Online Methods section.
Results
Drinking in the Dark up-regulates the relative expression of p(Ser729)-PKCε
The average daily alcohol intake under DID procedures exhibited by the B6 mice was 4.8 ± 0.37 g/kg/2 hr (6,7,11), which is predicted to result in a mean BAC ~ 100 mg% (31) that exceedes the NIAAA criterion for binge drinking (38). In the NACs, binge drinking elevated the relative expression of p(Ser729)-PKCε [PKCε ratio: t22=−3.984, p=0.001], and this reflected a non-significant reduction in total PKCε levels (t-test: p>0.05) and a significant 50% increase in p(Ser729)-PKCε (Figure 1A) [t22=−2.314, p=0.03]. These same B6 mice also exhibited an increase in the relative levels of p(Ser729)-PKCε in the CeA (Figure 1B) [t19=2.122, p=0.047]; this reflected a strong trend toward reduced total kinase levels in binge drinking mice (t-test: p>0.05), as p(Ser729)-PKCε levels were not different from water controls (t-test: p>0.05). In contrast, no changes in total or phosphorylated PKCε were apparent in the NAC core (NACc) or basolateral amygdala (BLA) (t-tests, p’s>0.05; Suppl. Table S1).
Figure 1. Thirty days of binge alcohol consumption under Drinking in the Dark (DID) procedures up-regulates Protein Kinase C epsilon (PKCε) activity in the nucleus accumbens shell (NACs) and central nucleus of the amygdala (CeA).

Summary of the change in protein levels of total PKCε, p-Ser729-PKCε and their relative levels (PKCε ratio) in the NACs (A) and CeA (B) following 24 hr withdrawal from 30 days of alcohol drinking under Drinking in the Dark (DID or D) procedures. Two representative immunoblots for PKCε and p-Ser729-PKCε for each group are included as insets. After normalization for loading, the data are expressed as a percent of the average protein levels of water-drinking controls (Water or W; n=5–6/gel). Data represent the mean ± SEM of the number of animals indicated in parentheses in the figure, *p<0.05 (t-tests).
Basal PKCε phosphorylation as a correlate of genetic propensity to binge drink
We next determined whether or not basal p(Ser729)-PKCε expression is a biochemical correlate of a high binge alcohol drinking phenotype through a comparison of tissue from selectively bred HDID-1 and genetically heterogenous HS/Npt control mice (33). We observed no genotypic differences in PKCε expression within either NAC subregion (t-tests: p’s>0.05; Figure 2A and Suppl. Table S2). While no genotypic differences in total PKCε levels were apparent in CeA (t-test: p>0.05), HDID-1 mice exhibited a robust increase in the absolute, as well as relative levels of p(Ser729)-PKCε in this region (Figure 2B) [total: t26=3.25, p=0.003; ratio: t26=3.306, p=0.003]. However, HDID-1 mice did not differ from HS/Npt controls regarding the basal levels of Homer proteins, mGlu1/5, GluN2A/B, PI3K or AKT phosphorylation (the latter serving to index PI3K activity) in this subregion (Suppl. Table S3). Moreover, there were no genotypic differences in PKCε levels in BLA (t-tests: p>0.05; Table 2), nor were differences observed for any other protein examined (t-tests; p’s>0.05; Suppl. Table S3).
Figure 2. Line differences in central nucleus of the amygdala (CeA) protein kinase C epsilon (PKCε) activity between selectively bred HDID-1 and genetically heterogeneous HS/Npt control mice.

Summary of the protein levels of total PKCε and p-Ser729-PKCε, as well as their relative levels (PKCε ratio) in the nucleus accumbens shell (NACs) (A) and CeA (B) of mice from S15 of selective breeding under DID procedures (HDID-1) and mice from their HS/Npt founder line (Control). The data are expressed as a percentage of average levels of HS/Npt animals (5–6/gel). Representative immunoblots of PKCε and p-Ser729-PKCε are included as insets (C= HS/Npt founder line control, H= HDID-1 selectively bred line). Data represent the mean ± SEM of the number of animals indicated in parentheses the figure, *p<0.05 (t-tests).
PKCε signaling within the NACs is necessary for binge drinking
We next assessed the role for PKCε in maintaining binge drinking under DID procedures via intra-NAC infusions of the PKCε translocation inhibitor Tat-εV1-2 and employed co-infusion procedures to address whether or not the effects of PKCε inhibition were shared with those produced by infusion of maximally effective doses of mGlu1, mGlu5, PLC, and PI3K inhibitors (Figure 3). Prior to any drug infusions, the B6 mice exhibited an average alcohol intake of 4.56 ± 0.39 g/kg during the 2-h drinking period, an intake correlated with BACs > 80 mg% (31). An intra-NACs infusion of Tat-εV1-2 alone, as well as the co-infusion of this inhibitor with 3 g/side MTEP, 15 pg/side JNJ-16259685, 5.8 fg/side U-73122, or 0.17 ng/side LY294002 reduced alcohol intake under DID procedures (Figure 3A) [F(5,104)=6.54, p<0.001]. LSD post-hoc analyses confirmed that all intra-NACs manipulations lowered alcohol intake below that exhibited by control mice infused with the scrambled Tat peptide (all p’s <0.05). Post-hoc comparisons were made also between mice infused with the PKCε inhibitor alone and those co-infused with the pharmacological agents. As illustrated in Figure 3A, co-infusion of 3 μg/side MTEP attenuated significantly the capacity of Tat-εV1-2 to inhibit binge drinking (p=0.037), while this effect was also significant (albeit less robust) when animals were co-infused with 15 pg/side JNJ-16259685 (p=0.049), 5.8 fg/side U-73122 (p=0.047) or 0.17 ng/side LY294002 (p=0.047). Representative placements of our microinjector tips in the NACs are presented in Figure 3C.
Figure 3. Inhibition of nucleus accumbens shell (NACs) and central nucleus of the amygdala (CeA) protein kinase C epsilon (PKCε) reduces binge alcohol drinking in C57BL/6J mice.
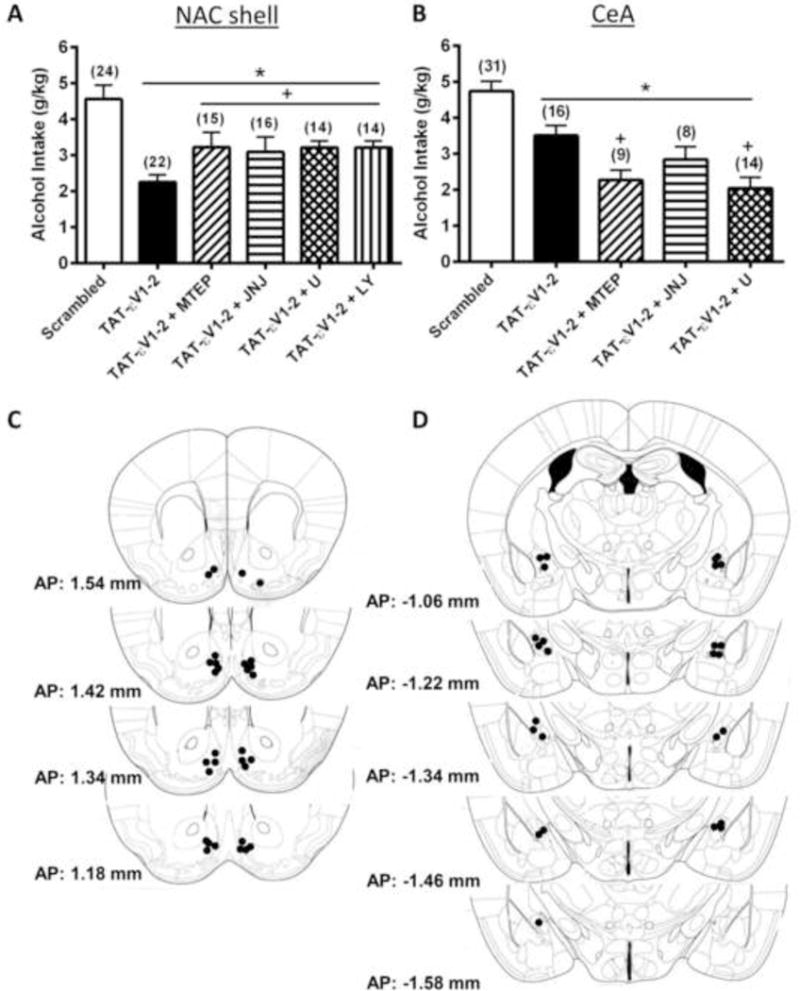
Summary of the effects of an intra-NACs (A) and intra-CeA (B) infusion of the PKCε inhibitor Tat-εV1-2 alone or in combination with the mGlu5 antagonist MTEP (3.0 μg/side), the mGlu1 antagonist JNJ-16259685 (JNJ; 15 pg/side), the PLC inhibitor U-73122 (U; 0.58 fg/side), and the PI3K antagonist LY-294002 (LY; 0.17 ng/side) upon 20% alcohol intake under DID procedures. Control animals were infused with a scrambled peptide (Scrambled). The data represent the mean ± SEM of the number of animals indicated in the figure, *p<0.05 vs. Scrambled, +p<0.05 vs. Tat-εV1-2 (LSD post-hoc tests). Pictograms depicting the location of microinjector tips in the NACs (C) and CeA (D) of mice from 1 replicate of the neuropharmacological studies. Numbers represent the anterior-posterior (AP) distance from Bregma (in mm) at which the microinjectors were localized.
PKCε signaling in the CeA is necessary for binge drinking
An intra-CeA infusion of Tat-εV1-2 or co-infusion of the PKCε inhibitor with 3 μg MTEP, 15 pg JNJ-16259685 or 5.8 fg U-73122 inhibited binge alcohol intake under our DID procedures in B6 mice (Figure 3B) [F(4,74)=14.612, p<0.001]. B6 mice infused intra-CeA with the scrambled control peptide consumed alcohol in excess of amounts predicted to result in BACs > 80 mg% (4.73 ± 0.28 g/kg) (31) and LSD post-hoc analyses confirmed that alcohol drinking was attenuated by infusion of Tat-εV1-2 alone (p=0.009), as well as by co-infusion of the inhibitor with MTEP (p<0.001), with JNJ-16259685 (p=0.002) or with U-73122 (p<0.001) (Figure 3B). However, in contrast to the NACs (Figure 3A), the magnitude of the reduction in alcohol intake produced by an intra-CeA infusion of Tat-εV1-2 was enhanced by co-infusion with MTEP (p=0.005) or U-73122 (p=0.001). These data suggest that, at least within the CeA, mGlu5 and PLC operate to influence alcohol drinking through a pathway independent of PKCε. In contrast, the reduction in drinking produced by Tat-εV1-2 co-infusion with JNJ-16259685 was similar in magnitude to that produced by the PKCε inhibitor alone (p>0.05), suggesting that mGlu1 and PKCε in the CeA operate to influence binge alcohol intake via a shared pathway. Representative placements of our microinjector tips in the CeA are presented in Figure 3D.
PKCε signaling and sucrose intake
To examine potential non-specific effects of our PKCε translocation inhibitor, we infused B6 mice with 0.25 μl/side Tat-εV1-2 into the NACs or CeA and measured 5% sucrose intake under our 2-hr DID procedures. As summarized in Suppl. Table S4, PKCε inhibition did not alter sucrose intake in this model (t-tests, p>0.05).
Homer2 is necessary for the “anti-drinking” effect of PKCε inhibition
To test the hypothesis that Homer proteins play a role in regulating the functional interaction between mGlu5 and PKCε (19), we next examined the effects of Tat-εV1-2 infusions upon alcohol intake by Homer2 WT and KO mice. As observed in previous reports (6,7,11), no differences were found in baseline alcohol intake between Homer2 WT and KO mice (Figure 4). The level of alcohol intake exhibited by the mice was 3.31±0.35 (NACs study) and 4.19±0.27 (CeA study) g/kg/2hr. The effects of infusing Tat-εV1-2 either into the NACs or into the CeA varied as a function of genotype, but not of region [Genotype × Pretreatment: F(1,31)=5.43, p=0.03; no main effect of, or interactions with, Region, p>0.05]. Post-hoc comparisons confirmed that Tat-εV1-2 inhibited alcohol intake by WT mice when infused into the NACs (Figure 4A) [t8=3.079, p=0.02] or CeA (Figure 4B) [t6=3.0, p=0.03]. In contrast, the inhibitor had no effect in KO animals (for NACs: p=0.97; for CeA: p=0.15).
Figure 4. Homer2 is necessary for the attenuating effects of nucleus accumbens shell (NACs) and central nucleus of the amygdala (CeA) protein kinase C epsilon (PKCε) inhibition.
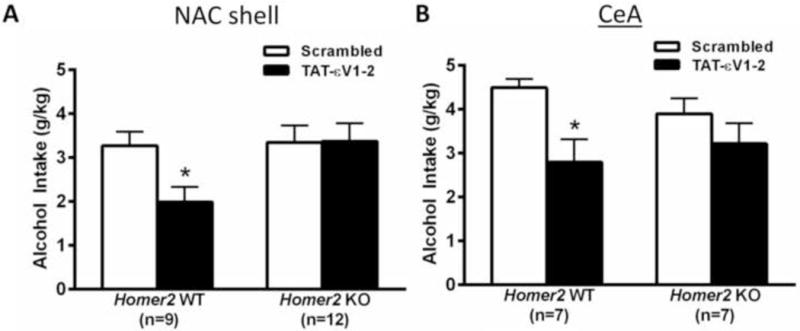
Summary of the effects of intra-NACs (A) and intra-CeA (B) infusions of the PKCε inhibitor Tat-εV1-2 or control peptide (Scrambled) upon 20% alcohol intake under Drinking in the Dark (DID) procedures by Homer2 wild-type (WT) and knock-out (KO) mice. The data represent the mean ± SEM of the number of animals indicated in the figure, *p<0.05 vs. Scrambled in WT mice (t-tests).
Interactions between PKCε and alcohol intake under SHAC procedures
A final series of experiments were designed to determine whether or not the interactions between NAC PKCε activity and alcohol drinking observed under DID procedures generalized to binge drinking under SHAC procedures. Thus, we examined relative p(Ser729)-PKCε levels in tissue from the entire NAC obtained from mice with a history of repeated bouts of binge alcohol drinking under SHAC procedures (average daily intake = 1.6 ± 0.2 g/kg/30 min; resultant average BAC=109.5 ± 37.3 mg%), which had been processed previously for binge drinking-induced changes in putative up-stream signaling molecules (5). Although binge drinking under SHAC procedures did not significantly affect either total or p(Ser729)-PKCε levels in the entire NAC (t-tests, p’s>0.05), there was an increase in the relative expression of p(Ser729)-PKCε in this study (Figure 5A) [t24=2.44, p=0.02]. We also immunoblotted for the relative levels of p(Ser279)-PKCε in the entire NAC of mice selectively bred for high versus low alcohol intake under SHAC procedures (SHAC vs. SLAC lines, respectively), which had been processed previously for line differences in glutamate receptor/Homer/PI3K expression (5). As indicated in Figure 5B, SHAC line mice exhibited higher relative levels of p(Ser729)-PKCε versus their low-drinking SLAC counterparts [t11=2.966, p=0.013], and this effect reflected both an increase in p(Ser729)-PKCε expression [t11=2.029, p=0.06] and a modest, albeit non-significant, reduction in total kinase levels [t-test: p>0.05].
Figure 5. Role for protein kinase C epsilon (PKCε) signaling in the nucleus accumbens (NAC) in binge alcohol consumption under Scheduled High Alcohol Consumption (SHAC) procedures.

A, Summary of the change in protein levels of total PKCε p-Ser729-PKCε and their relative levels (PKCε ratio) in the entire NAC following 24 hr withdrawal from 6 bouts of alcohol drinking under Scheduled High Alcohol Consumption (SHAC or S) procedures and water drinking controls (Water or W; 5–6/gel). Representative immunoblots are included as insets. B, Summary of the line differences in protein levels in the entire NAC of S4 mice selectively bred for high versus low alcohol intake under SHAC procedures (SLAC line or L = Scheduled Low Alcohol Consumption line; SHAC line or H = Scheduled High Alcohol Consumption line), at 3 months after their final 30 min alcohol-drinking session. The data are expressed as a percent of average levels of the SLAC line (5–6 SLAC mice/gel). Representative immunoblots are included as insets. C, Summary of the line differences in protein levels in the NAC of mGlu5F1128R transgenic mice (mGlu5 TG or T) and wild-type littermates (mGlu5 WT or W), expressed as a percent of average levels of WT animals (5–6 WT mice/gel). Representative immunoblots are included as insets. D, Summary of the effects of an intra-NAC shell infusion of the PKCε inhibitor Tat-εV1-2 or control peptide (Scrambled) upon 5% alcohol intake exhibited by B6 mice under SHAC procedures. Data represent the mean ± SEM of the number of animals indicated in the figure, *p<0.05 (t-test
We next asked whether or not the converse might be true and assayed for genotypic differences in basal NAC PKCε phosphorylation between low SHAC-intake mGlu5F1128R transgenic (TG) mice, with a point mutation in the Homer binding site on mGlu5 versus their WT littermates (5). While total PKCε levels did not differ between genotypes (p>0.05), TG mice exhibited 40% lower p(Ser729)-PKCε levels [t17=3.747, p=0.002], as well as significantly lower relative levels of phosphorylated versus non-phosphorylated kinase [PKCε ratio; t17=2.513, p=0.022], compared to WT animals (Figure 5C).
We also examined also the ability of the Tat-εV1-2 translocation inhibitor to reduce alcohol intake under SHAC procedures when infused into the NACs of B6 mice. Indeed, Tat-εV1-2 infusion significantly reduced consumption of 5% alcohol, relative to mice infused with a scrambled control peptide (Figure 5D) [t10=4.28, p=0.002). Importantly, the alcohol intake of the control mice in this study correlated with BACs > 80 mg%, while that of Tat-εV1-2-infused animals was lower than that predicted to meet the criterion for binge drinking (5). Thus, PKCε inhibition in the NACs is sufficient to prevent binge alcohol drinking under SHAC procedures. Finally, intra-NAC inhibition of PKCε did not influence the consumption of 5% sucrose under SHAC procedures (p>0.05; Suppl. Table S4), providing further evidence that interfering with PKCε activity selectively affects alcohol intake.
Discussion
Herein, alcohol consumption up-regulated PKCε phosphorylation on Ser729 in both the NACs and the CeA and basal p(Ser729)-PKCε levels in these regions were a correlate of the genetic propensity to binge drink. These data implicate either idiopathic or alcohol-induced increases in PKCε priming in NACs and CeA in the etiology of excessive alcohol drinking. Furthering this hypothesis, inhibition of PKCε translocation in the NACs or CeA reduced alcohol intake under our DID and SHAC procedures, and experiments conducted in Homer2 KO mice provided evidence that this scaffolding molecule is integrally involved. The neuropharmacological studies suggested that PKCε in the NACs regulates binge alcohol drinking via signaling pathways involving mGlu1, mGlu5, PLC and PI3K, while PKCε in the CeA operates in a signaling pathway involving mGlu1. These results extend current knowledge, derived from studies using constitutive PKCε KO mice (13) and shRNA-PKCε approaches (28), by demonstrating that PKCε operates in both the NACs and CeA to facilitate voluntary alcohol consumption through distinct Group1 mGluR-associated signaling pathways.
In vivo regulation of PKCε by alcohol and relation to alcoholism vulnerability
In vitro, alcohol stimulates PKCε translocation (39), and chronic alcohol exposure increases PKCε immunoreactivity and PKCε-mediated phosphorylation (17). The precise neuroanatomical loci of alcohol-PKCε interactions are less well-described and were examined herein using phosphorylation of PKCε at Ser729 as an indicator of isozyme priming (40). Binge alcohol drinking increased p(Ser729)-PKCε specifically in NAC and amygdala subregions that are components of the extended amygdala circuit highly implicated in alcoholism neurobiology (41,42). Moreover, this effect was observed within the same tissue reported previously by our group to exhibit increases in Group1 mGluR, Homer2a/b levels and/or PI3K activation (5–7). Such observations indicate that elevated PKCε priming in these brain regions, presumably via pathways involving Group1 mGluRs and their Homer2 scaffolds, may be central to the manifestation of excessive alcohol intake.
In support of this assertion, a susceptibility locus for alcohol dependence is indicated on human chromosome 2p21, where the gene for PKCε is mapped (43,44), and genotypic differences exist for basal p(Ser729)-PKCε between inbred mouse strains with divergent alcohol drinking phenotypes (10). Herein, we extend these prior data to selectively bred and transgenic lines of mice varying in binge drinking behavior. When assayed in the entire NAC, SHAC line mice selectively bred for high BACs under SHAC procedures exhibited higher p(Ser729)-PKCε levels, compared to SLAC line animals selected for low BACs under SHAC procedures. Conversely, mGlu5F1128R TG mice that manifest low binge alcohol drinking (5) exhibit lower constitutive p(Ser279)-PKCε levels in the NAC than WT littermates that exhibit binge drinking behavior (5). As the mGlu5F1128R TG mice employed in the present study exhibit reduced Homer binding to mGlu5 (5), these later results provide novel evidence in support of an important role for mGlu5-Homer interactions in regulating constitutive PKCε priming in vivo, as suggested previously (19). However, while HDID-1 mice, selected for high BAC under 4-h DID procedures, exhibited higher indices of CeA p(Ser729)-PKCε than their genetically heterogeneous HS/Npt controls, no differences were observed in putative up-stream signaling molecules in amygdala, nor were differences present for p(Ser729)-PKCε levels in the NAC. As strain differences exist regarding basal NAC levels of p(Ser729)-PKCε (10; Figure 5A), the failure to detect differences in PKCε priming between HDID-1 and HS/Npt controls could simply relate to the genetic heterogeneity of the control line employed in this study. Alternatively, the use of more sensitive mass spectrometry procedures to examine the phosphorylation of Ser729, or of other sites more reflective of enzyme activation [e.g., Thr566 or Ser710 (40)], might provide more information pertaining to the link between idiopathic or alcohol-induced changes in PKCε activity and alcohol drinking. Further preclinical exploration of this potential link is warranted, particularly considering evidence indicating the functional relevance for NAC Group1 mGluR/Homer2/PI3K/PKCε signaling in excessive alcohol drinking (present study; 5–7,11).
PKCε translocation and binge alcohol consumption
The correlational nature of the aforementioned immunoblotting studies cannot inform as to cause-effect relations between PKCε activity and binge drinking. However, earlier data from studies of constitutive PKCε KO mice argued an active and necessary role for this isozyme in alcoholism-related behaviors (13, 25) and herein, the inhibition of PKCε translocation in either the NACs or the CeA by the local infusion of a Tat-εV1-2 peptide (34) was sufficient to reduce binge alcohol consumption in 2 distinct murine models. To the best of our knowledge, these data are the first demonstration that PKCε translocation specifically in the NACs modulates binge alcohol intake, and further support the CeA as an anatomical site of action of PKCε for excessive alcohol consumption (28). This demonstration of a causal relation between PKCε translocation in at least two AUD-relevant brain regions and binge alcohol intake not only highlights the significance of both our immunoblotting results (10; present study) and those of the genetic mapping studies in humans (43,44), but also provides neuroanatomical insight into the facilitatory role for PKCε in excessive alcohol drinking. Potential up-stream modulators of PKCε translocation and binge alcohol consumption. Previously, a signaling pathway initiated upon mGlu5 stimulation, that involved Homers and PI3K activity, was proposed to play an important role in the activation of PKCε underpinning voluntary alcohol intake (19). In partial validation of this hypothesis, we demonstrated previously a critical role for Group1 mGluR/Homer2-mediated activation of both Gαq/11 and Gβγ signaling in the NACs and the CeA in maintaining binge alcohol drinking (5–7,11). Herein, an intra-NAC infusion of Tat-εV1-2 while effective at reducing alcohol intake in WT mice, did not inhibit alcohol intake in Homer2 KO mice, implicating Homer2 as an important regulator of both PKCε priming and the facilitation of binge alcohol drinking elicited by PKCε translocation. In the NACs, the inhibitory effects of co-infusion of the Tat-εV1-2 inhibitor with mGlu1, mGlu5, PLC or PI3K antagonists were not different from those produced by PKCε inhibition alone, indicating that at least in this striatal subregion, mGlu1/5, PLC, PI3K and PKCε operate within shared intracellular signaling pathways, perhaps scaffolded by Homer2. While there is no evidence that Homer proteins interact directly with any PKC, our prior work indicated that intact Homer2 expression or Homer coupling to mGlu5 is required in order to observe an attenuation of binge drinking by mGlu1/5 receptor antagonists and inhibitors of PLC and PI3K [6,7,11]. Moreover, the present observation of decreased constitutive PKCε priming in the NAC of mGlu5F1128R mice with reduced Homer binding supports some functional relation between mGluR-Homer complexes and PKCε. Thus, the coincident up-regulation in mGlu1/5, Homer2, and PLC expression, the activational state of PI3K and PKCε priming observed in the NACs of binge drinking animals or in animals with a high genetic propensity to binge drink [5,6,11; present study] likely reflects a shared signaling pathway that is permissive to excessive alcohol consumption.
Although binge alcohol drinking up-regulates p(Ser729)-PKCε expression in the CeA and higher constitutive PKCε priming in the CeA is a genetic correlate of excessive alcohol drinking, the specific signaling pathway(s) operating in the CeA to regulate binge alcohol intake appear to be distinct from those operating within the NACs. For one, PI3K inhibition within the CeA does not affect binge alcohol drinking (5,6 vs. 7), arguing that Akt/PI3K hyperactivity in the CeA is not a major mediator of binge alcohol intake and likely not a major contributor to alcohol-induced increases in PKCε priming observed within this region. Moreover, mGlu1 and PLC operate independently within the CeA to influence binge drinking (7) and herein, the effect of an intra-CeA co-infusion of Tat-εV1-2 with an mGlu5 antagonist was greater than that produced by the antagonist alone. Such a result argues against a specific role for CeA mGlu5 in PKC translocation facilitating alcohol intake. However, the inhibitory effect of an intra-CeA co-infusion of Tat-εV1-2 and an mGlu1 antagonist was not different from that produced by the mGlu1 alone, arguing that PKCε operates in the CeA, via some mGluR1-dependent pathway, to influence binge alcohol intake. This mGlu1/PKCε pathway does not involve Gαq/11-mediated stimulation of PLC, as the attenuation of binge drinking by co-infusion of PLC and PKCε inhibitors was greater than that produced by PKCε inhibition alone. Thus, the capacity of PKCε priming to regulate binge drinking may be entirely independent of the generation of DAG by PLC. Although evidence indicates that PI3K can regulate PKCε priming (19), our failure to observe alcohol-induced increased PI3K activity in the CeA, coupled with no effect of intra-CeA infusions of different PI3K inhibitors upon binge drinking (7), argue that alcohol-induced or idiopathic increases in PKCε priming observed in this region likely do not involve βγ-mediated activation of PI3K upon mGlu1 stimulation (7).
What then is the pathway involved in mGlu1/PKCε-dependent signaling that regulate binge drinking in the CeA? mGlu1 activation can elicit cAMP formation in vitro (45), raising the possibility that mGlu1-PKCε signaling in the CeA may involve this second messenger. Indeed PKCε activation can be regulated by Gαi-coupled corticotrophin-releasing factor receptor 1 (CRF1) receptors (34) and there is cross-talk between PKA and PKC in opioid-withdrawn animals (46), supporting this possibility. Alternatively, mammalian target of rapamycin complex 2 (mTORC2) is an important upstream regulator of PKCε priming (47). While not yet assessed in brain, mTORC2 activity can be up-regulated by alcohol when assayed in myocytes (48), Group1 mGluR-mediated inhibition of tuberous sclerosis complexes can increase mTORC activation (49), and the capacity of Group1 mGluRs to augment mTORC activity depend upon constitutively expressed Homer proteins (50). Thus, either alcohol-induced or idiopathic increases in mGluR/Homer2-PKCε signaling in CeA may involve mTORC1 and/or mTORC2 as an intermediary, and this possibility requires systematic investigation. Alternatively, both mGlu1 and PKCε can regulate the function of N-type calcium channels, and Homer proteins have been implicated in mGlu1-regulation of channel function (51). While this latter mechanism cannot account for how mGlu1 stimulation elicits PKCε translocation in the CeA, genetic or pharmacological interruption of mGlu1, Homer2, N-type calcium channels or PKCε all produce an alcohol-avoiding phenotype in preclinical animal models (5–7,11,23,52). Thus, N-type calcium channels may be an important point of signaling convergence between mGlu1 and PKC to be explored in future studies.
Conclusion
Using a combination of immunoblotting and neuropharmacological approaches, the results of the present study support idiopathic or alcohol-induced increases in PKCε priming/translocation in the NACs and CeA as an important signaling event in the etiology of alcoholism and provide experimental support for the potential utility of targeting PKCε for the pharmacotherapeutic intervention of AUDs.
Supplementary Material
Acknowledgments
This work was funded by NIH grants AA016650 to KKS, AA013478 and AA016981 to DAF, AA13519 and AA10760 to JCC, as well as DA00266 and DA011742 (PFW). This work was also funded by in part by VA Merit Review grants from the US Department of Veterans Affairs to DAF and JCC, and by resources and the use of facilities at VA Portland Healthcare System (DAF and JCC). DKC was supported by T32 AA007468. We would like to thank Dr. Robert O. Messing (University of Texas, Austin) for his generous donation of the Tat-εV1-2 peptide for our studies. We thank Ms. Burgundy Tyrrel for her assistance with some of the behavioral studies. Previously presented in part as posters at the Research Society on Alcoholism Meeting (2009 and 2012) and the Alcoholism and Stress: Framework for Future Treatment Strategies Conference (2011).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
FINANCIAL DISCLOSURES
The authors report no biomedical financial interests or potential conflicts of interest. The contents do not represent the views of the US Department of Veterans Affairs or the United States Government.
References
- 1.Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–60. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark DB, Bukstein O, Cornelius J. Alcohol use disorders in adolescents: epidemiology, diagnosis, psychosocial interventions, and pharmacological treatment. Paediatr Drugs. 2002;4:493–502. doi: 10.2165/00128072-200204080-00002. [DOI] [PubMed] [Google Scholar]
- 3.Substance Abuse and Mental Health Services Administration. (Office of Applied Studies, NSDUH Series H-38A, HHS Publication No. SMA 10-4586 Findings).Results from the 2009 National Survey on Drug Use and Health: Volume I Summary of National Findings. 2010 [Google Scholar]
- 4.Szumlinski KK, Diab ME, Friedman R, Henze LM, Lominac KD, Bowers MS. Accumbens neurochemical adaptations produced by binge-like alcohol consumption. Psychopharmacology. 2007;190:415–431. doi: 10.1007/s00213-006-0641-7. [DOI] [PubMed] [Google Scholar]
- 5.Cozzoli DK, Goulding SP, Zhang PW, Xiao B, Hu JH, Ary AW, et al. Binge drinking upregulates accumbens mGluR5-Homer2-PI3K signaling: functional implications for alcoholism. J Neurosci. 2009;29:8655–8668. doi: 10.1523/JNEUROSCI.5900-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cozzoli DK, Courson J, Caruana AL, Miller BW, Greentree DI, Thompson AB, et al. Nucleus accumbens mGluR5-associated signaling regulates binge alcohol drinking under drinking-in-the-dark procedures. Alcohol Clin Exp Res. 2012;36:1623–1633. doi: 10.1111/j.1530-0277.2012.01776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cozzoli DK, Courson J, Wroten MG, Greentree DI, Lum EN, Campbell RR, et al. Binge alcohol drinking by mice requires intact group1 metabotropic glutamate receptor signaling within the central nucleus of the amygdala. Neuropsychopharmacology. 2014;39:435–444. doi: 10.1038/npp.2013.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koob GF, Roberts AJ, Schulteis G, Parsons LH, Heyser CJ, Hyytia P, et al. Neurocircuitry targets in ethanol reward and dependence. Alcohol Clin Exp Res. 1998;22:3–9. [PubMed] [Google Scholar]
- 9.Kapasova Z, Szumlinski KK. Strain differences in alcohol-induced neurochemical plasticity: a role for accumbens glutamate in alcohol intake. Alcohol Clin Exp Res. 2008;32:617–631. doi: 10.1111/j.1530-0277.2008.00620.x. [DOI] [PubMed] [Google Scholar]
- 10.Goulding SP, Obara I, Lominac KD, Gould AT, Miller BW, Klugmann M, Szumlinski KK. Accumbens Homer2-mediated signaling: A factor contributing to mouse strain differences in alcohol drinking? Genes Brain Behav. 2011;10:111–126. doi: 10.1111/j.1601-183X.2010.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lum EN, Campbell RR, Rostock C, Szumlinski KK. mGluR1 receptors within the nucleus accumbens regulate alcohol intake in mice under limited-access conditions. Neuropharmacology. 2014;79:679–87. doi: 10.1016/j.neuropharm.2014.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newton PM, Messing RO. Increased sensitivity to the aversive effects of ethanol in PKCepsilon null mice revealed by place conditioning. Behav Neurosci. 2007;121:439–442. doi: 10.1037/0735-7044.121.2.439. [DOI] [PubMed] [Google Scholar]
- 13.Newton PM, Ron D. Protein kinase C and alcohol addiction. Pharmacol Res. 2007;55:570–577. doi: 10.1016/j.phrs.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Newton PM, Messing RO. The substrates and binding partners of protein kinase Cepsilon. Biochem J. 2010;427:189–196. doi: 10.1042/BJ20091302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akita Y. Protein kinase C-epsilon (PKC-epsilon): its unique structure and function. J Biochem. 2002;132:847–852. doi: 10.1093/oxfordjournals.jbchem.a003296. [DOI] [PubMed] [Google Scholar]
- 16.Das J, Pany S, Rahman GM, Slater SJ. PKC epsilon has an alcohol-binding site in its second cysteine-rich regulatory domain. Biochem J. 2009;421:405–413. doi: 10.1042/BJ20082271. [DOI] [PubMed] [Google Scholar]
- 17.Messing RO, Peterson PJ, Henrich CJ. Chronic ethanol exposure increases levels of protein kinase C δ and ε and protein kinase C-mediated phosphorylation in cultured neural cells. J Biol Chem. 1991;266:23428–23432. [PubMed] [Google Scholar]
- 18.Roivainen R, Hundle B, Messing RO. Protein kinase C and adaptation to ethanol. In: Jansson B, Jorvall H, Rydberg U, Terenius L, Vallee BL, editors. Toward a Molecular Basis of Alcohol Use and Abuse. Birkhauser Verlag; Basel: 1994. pp. 29–38. [Google Scholar]
- 19.Olive MF, McGeehan AJ, Kinder JR, McMahon T, Hodge CW, Janak PH, Messing RO. The mGluR5 antagonist 6-methyl-2-(phenylethynyl)pyridine decreases ethanol consumption via a protein kinase Cε-dependent mechanism. Mol Pharmacol. 2005;67:349–355. doi: 10.1124/mol.104.003319. [DOI] [PubMed] [Google Scholar]
- 20.Qi ZH, Song M, Wallace MJ, Wang D, Newton PM, McMahon T, et al. Protein kinase C epsilon regulates gamma-aminobutyrate type A receptor sensitivity to ethanol and benzodiazepines through phosphorylation of gamma2 subunits. J Biol Chem. 2007;282:33052–33063. doi: 10.1074/jbc.M707233200. [DOI] [PubMed] [Google Scholar]
- 21.Hodge CW, Mehmert KK, Kelley SP, McMahon T, Haywood A, Olive MF, et al. Supersensitivity to allosteric GABAA receptor modulators and alcohol in mice lacking PKCε. Nat Neurosci. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- 22.Hodge CW, Raber J, McMahon T, Walter H, Sanchez-Perez AM, Olive MF, et al. Decreased anxiety-like behavior, reduced stress hormones, and neurosteroid supersensitivity in mice lacking protein kinase Cε. J Clin Invest. 2002;110:1003–1010. doi: 10.1172/JCI15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olive MF, Mehmert KK, Messing RO, Hodge CW. Reduced operant ethanol self-administration and in vivo mesolimbic dopamine responses to ethanol in PKCε deficient mice. Eur J Neurosci. 2000;12:4131–4140. doi: 10.1046/j.1460-9568.2000.00297.x. [DOI] [PubMed] [Google Scholar]
- 24.Olive MF, Mehmert KK, Nannini MA, Camarini R, Messing RO, Hodge CW. Reduced ethanol withdrawal severity and altered withdrawal-induced c-fos expression in various brain regions of mice lacking protein kinase Cε. Neuroscience. 2001;103:171–179. doi: 10.1016/s0306-4522(00)00566-2. [DOI] [PubMed] [Google Scholar]
- 25.Choi DS, Wang D, Dadgar J, Chang WS, Messing RO. Conditional rescue of protein kinase C ε regulates ethanol preference and hypnotic sensitivity in adult mice. J Neurosci. 2002;22:9905–9911. doi: 10.1523/JNEUROSCI.22-22-09905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Besheer J, Lepoutre V, Mole B, Hodge CW. GABA(A) receptor regulation of voluntary ethanol drinking requires PKC epsilon. Synapse. 2006;60:411–419. doi: 10.1002/syn.20314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gass JT, Olive MF. Role of protein kinase C epsilon (PKCε) in the reduction of ethanol reinforcement due to mGluR5 antagonism in the nucleus accumbens shell. Psychopharmacology (Berl) 2009;204:587–597. doi: 10.1007/s00213-009-1490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lesscher HMB, Wallace MJ, Zeng L, Wang V, Deitchman JK, McMahon T, et al. Amygdala protein kinase C epsilon controls alcohol consumption. Genes Brain Behav. 2009;8:493–499. doi: 10.1111/j.1601-183X.2009.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gass JT, Olive MF. Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol. 2008;75:218–265. doi: 10.1016/j.bcp.2007.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bird MK, Lawrence AJ. Group 1 metabotropic glutamate receptors: involvement in drug-seeking and drug-induced plasticity. Curr Mol Pharmacol. 2009;2:83–94. doi: 10.2174/1874467210902010083. [DOI] [PubMed] [Google Scholar]
- 31.Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 32.Finn DA, Belknap JK, Cronise K, Yoneyama N, Murillo A, Crabbe JC. A procedure to produce high alcohol intake in mice. Psychopharmacology. 2005;178:471–480. doi: 10.1007/s00213-004-2039-8. [DOI] [PubMed] [Google Scholar]
- 33.Crabbe JC, Metten P, Rhodes JS, Yu CH, Brown LL, Phillips TJ, Finn DA. A line of mice selected for high blood ethanol concentrations shows drinking in the dark to intoxication. Biol Psychiatry. 2009;65:662–670. doi: 10.1016/j.biopsych.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bajo M, Cruz MT, Siggins GR, Messing R, Roberto M. Protein kinase c epsilon mediation of CRF- and ethanol-induced GABA release in central amygdala. Proc Natl Acad Sci U S A. 2008;105:8410–8415. doi: 10.1073/pnas.0802302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, et al. PI3 kinase enhancer-Homer complex couples mGluR1 to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6:1153–1161. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- 36.Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, et al. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717–726. doi: 10.1016/s0896-6273(00)80589-9. [DOI] [PubMed] [Google Scholar]
- 37.Yuan JP, Kiselyov K, Shin DM, Chen J, Shcheynikov N, Kang SH, et al. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell. 2003;114:777–789. doi: 10.1016/s0092-8674(03)00716-5. [DOI] [PubMed] [Google Scholar]
- 38.National Institute on Alcohol Abuse and Alcoholism. NIAAA Newsletter. Vol. 3. National Institute on Alcohol Abuse and Alcoholism; Bethesda, MD: 2004. NIAAA council approves definition of binge drinking; p. 3. [Google Scholar]
- 39.Gordon AS, Yao L, Wu ZL, Coe IR, Diamond I. Ethanol alters the subcellular localization of delta- and epsilon protein kinase C in NG108-15 cells. Mol Pharmacol. 1997;52:554–559. doi: 10.1124/mol.52.4.554. [DOI] [PubMed] [Google Scholar]
- 40.Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. EMBO J. 2000;19:496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koob GF. Brain stress systems in the amygdala and addiction. Brain Res. 2009;1293:61–75. doi: 10.1016/j.brainres.2009.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vilpoux C, Warnault V, Pierrefiche O, Daoust M, Naassila M. Ethanol-sensitive brain regions in rat and mouse: a cartographic review, using immediate early gene expression. Alcohol Clin Exp Res. 2009;33:945–969. doi: 10.1111/j.1530-0277.2009.00916.x. [DOI] [PubMed] [Google Scholar]
- 43.Chen X-N, Knauf JA, Gonsky R, Wang M, Lai EH, Chissoe S, et al. From amplification to gene in thyroid cancer: a high-resolution mapped bacterial-articial-chromosome resource for cancer chromosome aberrations guides gene discovery after comparative genome hybridization. Am J Hum Genet. 1998;63:625–637. doi: 10.1086/301973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reich T, Edenberg HJ, Goate A, Williams JT, Rice JP, Van Eerdewegh P, et al. Genome-wide search for genes affecting the risk for alcohol dependence. Am J Med Genet. 1998;81:207–215. [PubMed] [Google Scholar]
- 45.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 46.Fan P, Jiang Z, Diamond I, Yao L. Up-regulation of AGS3 during morphine withdrawal promotes cAMP superactivation via adenylyl cyclase 5 and 7 in rat nucleus accumbens/striatal neurons. Mol Pharmacol. 2009;76:526–533. doi: 10.1124/mol.109.057802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cameron AJ, Linch MD, Saurin AT, Escribano C, Parker PJ. mTORC2 targets AGC kinases through Sin1-dependent recruitment. Biochem J. 2011;439:287–297. doi: 10.1042/BJ20110678. [DOI] [PubMed] [Google Scholar]
- 48.Hong-Brown LQ, Kazi AA, Lang CH. Mechanisms mediating the effects of alcohol and HIV anti-retroviral agents on mTORC1, mTORC2 and protein synthesis in myocytes. World J Biol Chem. 2012;3:110–120. doi: 10.4331/wjbc.v3.i6.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feliciano DM, Lin TV, Hartman NW, Bartley CM, Kubera C, Hsieh L, et al. A circuitry and biochemical basis for tuberous sclerosis symptoms: from epilepsy to neurocognitive deficits. Int J Dev Neurosci. 2013;31:667–678. doi: 10.1016/j.ijdevneu.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Riordan K, Gerstein H, Hullinger R, Burger C. The role of Homer1c in metabotropic glutamate receptor-dependent long-term potentiation. Hippocampus. 2014;24:1–6. doi: 10.1002/hipo.22222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kammermeier PJ, Xiao B, Tu JC, Worley PF, Ikeda SR. Homer proteins regulate coupling of group 1 metabotropic glutamate receptors to N-type calcium and M-type potassium channels. J Neurosci. 2000;20:7238–7245. doi: 10.1523/JNEUROSCI.20-19-07238.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Newton PM, Orr CJ, Wallace MJ, Kim C, Shin HS, Messing RO. Deletion of N-type calcium channels alters ethanol reward and reduces ethanol consumption in mice. J Neurosci. 2004;24:9862–9869. doi: 10.1523/JNEUROSCI.3446-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.