

Abstract
BACKGROUND
To assess insulin secretion in pediatric cystic fibrosis (CF) patients with exocrine pancreatic sufficiency.
METHODS
Glucose and insulin responses during an oral glucose tolerance test (OGTT) were measured in 146 CF patients. Patients were divided into exocrine sufficient (CF-PS) and insufficient (CF-PI) groups based on pancreatic enzyme usage and fecal elastase. A reference group included healthy, non-diabetic subjects.
RESULTS
All CF groups showed reduced insulin secretion as measured by insulinogenic index. The CF-PS patients had normal glucose tolerance. There was direct correlation between BMI z-score and insulin area under the curve.
CONCLUSION
Patients with CF have reduced insulin secretion during an OGTT regardless of exocrine pancreatic status. The abnormal insulin secretion in all CF patients may predispose them for glucose intolerance, particularly when challenged by inflammation, infection, or nutritional deficiency. In addition, the diminished insulin secretion may contribute to increased catabolism. Lastly, the CF-related diabetes (CFRD) screening guidelines should be followed by all CF patients regardless of pancreatic status.
Keywords: Insulin deficiency, pancreatic cystic fibrosis, hyperglycemia, anabolism, insulin-secreting cells
INTRODUCTION
Pulmonary disease is the major cause of morbidity and mortality in patients with CF. However, pancreatic disease also significantly contributes to poor outcomes in patients with CF. The CF transmembrane regulator (CFTR) protein is expressed in the intralobular and intercalated duct epithelia of the exocrine pancreas (1). In the normal pancreas, chloride/bicarbonate exchangers and the CFTR protein create a bicarbonate-rich pancreatic secretion. This secretion aids in the digestion of protein and fat in the small intestine. In patients with CF, the impairment of epithelial chloride secretion caused by a disruption in CFTR results in the precipitation of proenzymes and the obstruction of pancreaticobiliary ducts (1). Pancreaticobiliary duct obstruction leads to inflammation, necrosis, and fibrosis of the exocrine pancreas resulting in exocrine pancreatic insufficiency. This process begins in utero and approximately 85% of CF patients have exocrine pancreatic insufficiency (CF-PI) with associated malabsorption within the first year of life (2).
The process of pancreaticobiliary duct obstruction places patients with CF at risk for developing endocrine pancreatic dysfunction and eventually CF-related diabetes (CFRD). The most common view of the etiology of CFRD is that inflammation, necrosis, and fibrosis associated with pancreatic duct obstruction cause fibrosis and fatty infiltration of the islets of Langerhans (3-5) leading to secondary β-cell dysfunction, reduced islet mass, and insulin deficiency. However, several groups have implicated decreased insulin sensitivity related to pulmonary disease and systemic inflammation in the pathogenesis of CFRD (6, 7). At present there is general agreement that insulin deficiency and insulin resistance contribute to hyperglycemia, and ultimately CFRD, in the CF-PI population, but this model is not well defined (8).
Previous reports suggest that the 15% of patients with CF who are exocrine pancreatic sufficient (CF-PS) are not at risk for secondary β-cell dysfunction and CFRD (9). These reports are based on the concept that if pancreatic ductal plugging and fibrosis have not progressed sufficiently to cause exocrine insufficiency, then β-cells will remain functional. Recent reports, however, challenge this concept, suggesting a primary mechanism of β-cell injury and insulin deficiency (10-12). Furthermore, in our own clinical experience, oral glucose tolerance testing (OGTT) in patients with CF-PS suggests abnormalities in insulin secretion. In view of our clinical observations and recent reports of primary β-cell dysfunction, we hypothesized that all CF patients regardless of exocrine pancreatic status would have insulin deficiency secondary to dysfunctional β-cell mass, even if they did not have prediabetes or CFRD. To test this hypothesis, we compared OGTT results from pediatric CF patients subgrouped by exocrine pancreatic status to OGTT results from lean, healthy patients.
METHODS
Patient population
The study population included all CF patients followed at Cincinnati Children’s Medical Hospital Center (CCHMC) who were 5 years of age and older and who underwent at least one OGTT between January 1, 2004 and June 30, 2009. These patients had undergone OGTT as part of the annual screening assessment and were consider baseline in health. We identified these patients using a clinical database established in 1998 that contains clinical and biochemical data on patients with CF who were followed by the pulmonary division at CCHMC. Nine percent of the patients included in this study population were also included in work previously published by the authors (13).
We analyzed the weight, height, and spirometry values of these patients, as recorded during clinic appointments closest to the time of the OGTT testing. The mean number of days between recording height, weight, and spirometry data and the OGTT data was 17. Additionally, all study subjects had measures of hemoglobin A1C at the time of the OGTT. In patients who had undergone OGTT testing more than once, the data from the most recent OGTT were used for this study.
A reference group included 12 lean subjects enrolled in a previous study of glucose metabolism (14). These subjects were non-diabetic, free of chronic medical conditions, not taking medication, and healthy at the time of evaluation. These patients were similar to other previously published control groups used for measures of beta-cell function (15).
Nutritional status
Weight and height measurements were performed as part of a standard clinic visit. Body mass index (BMI) was calculated as weight (kg)/ height (m) 2. Z-scores for BMI were derived based on sex- and age-specific BMI charts developed by the Centers for Disease Control and Prevention (16).
Spirometry
We performed spirometry in the pulmonary function laboratory according to the American Thoracic Guidelines (17) and recorded the forced expiratory volume in 1 second (FEV1). We then calculated percent predicted FEV1 (FEV1%) using Wang et al (18) for study subjects ages 5-16 years and Hankinson et al (19) for subjects older than age 16.
Fecal elastase analysis
Patients not demonstrating symptoms of malabsorption underwent fecal elastase testing. Quest Diagnostics® laboratories (San Juan Capistrano, CA) performed the fecal elastase measurements using a quantitative enzyme linked immunosorbent assay for measuring concentrations of elastase-1 in feces. We defined CF-PS patients as those not taking pancreatic enzyme supplements and having a fecal elastase value of ≥246. Patients who did not meet these criteria were considered CF-PI.
Oral glucose tolerance testing
Following an overnight fast, patients ingested an oral glucose solution containing 1.75 mg/kg of glucose (maximum of 75 grams). Venous blood samples were obtained through an intravenous catheter before glucose ingestion and 30, 60, and 120 minutes after glucose ingestion. Samples were placed on ice and centrifuged within 1 hour. Plasma was then collected and stored for measurement of glucose and insulin.
We categorized patients as having normal or abnormal glucose metabolism using criteria recommended by the American Diabetes Association (ADA) (20), and divided our patients into normal glucose tolerance (CF-NGT) and abnormal glucose metabolism (CF-AGM) as we have done previously (13). The CF-AGM group included patients who had impaired fasting glucose (IFG), impaired glucose tolerance (IGT), IFG/IGT and CFRD.
Biochemical analysis
Plasma glucose was measured using a glucose oxidase-based-method run on an Ortho Clinical Diagnostics Vitro 950 instrument, and plasma insulin by radioimmunoassay (14) in the General Clinical Research Center laboratory at CCHMC. Hemoglobin A1C was determined with a modified high-performance-liquid-chromatography method using an Alliance 2690/2695 HPLC (Walters Corporation, Milford, MA) and a PolyCAT A (PolyLC, Inc, Columbia, MD) column to separate the hemoglobin fractions by cation-exchange chromatography. The hemoglobin A1C was then quantified using a dual wavelength detector (model 2487, Walters Corporation) and Empower Software (Walters Corporation); results were reported as percentages.
Biochemical calculations
The glycemic and insulin responses of each subject during the OGTT were integrated as the 3-hour areas above fasting levels (area under the curve-AUC) using the trapezoidal rule. To estimate insulin secretion, the insulinogenic index was calculated as insulin30min-insulin0min [pmol/L]/glucose30min-glucose0min[mmol/L] (21) (22).
Statistical analysis
In preliminary analyses, we used likelihood-ratio chi-square tests to assess differences between the reference group of lean subjects and each of the CF groups (CF PI-NGT, CF-PI-AGM, and CF-PS) with regard to gender and race. We tested the differences between groups for continuous variables using the Wilcoxon nonparametric test.
For each glucose and insulin response, an analysis of variance (ANOVA) model was used to assess difference in means between the reference and each of the CF groups, and the CF-PS group to the CF-PI-NGT and CF-PI-AGM groups. Fasting glucose, AUCGlu, and AUCIns, were each log-transformed to achieve normality. A cube-root transformation was used to achieve normality for the insulinogenic index. Within each model, simulation-based, step-down multiple comparison adjustments were applied to all least-square means (LSM) comparisons between groups. Estimates for ANOVA models are reported in terms of back-transformed point-estimates and 95% confidence intervals for each LSM (Table 3). To determine the association between BMI z-score and insulin secretion in patients with CF, an analysis of covariance (ANCOVA) model was applied to the insulin responses including age and FEV1 as a continuous covariates.
TABLE 3.
Least squares means and 95% confidence intervals based on ANOVA models for CF-PS, CF-PI-NGT, CF-PI-AGM, and reference groups. Results have been back-transformed to original measurement scale. Comparisons for 1-hour glucose and HbgA1C% were made on the original values, using the Wilcoxon nonparametric test, and reported as means ± standard deviations. Reference group compared to CF-PS and CF-PI groups
| GLUCOSE RESPONSES | CF-PS | CF-PI-NGT | CF-PI-AGM | Reference |
|---|---|---|---|---|
| Fasting Glucose (mg/dL) | 80.5 (70.2, 92.3) |
83.9 (82.1, 85.9) |
93.8 (89.1, 98.8) |
88.2 (84.8, 91.8) |
| AUCGlu (mg/dL) | 13554 (11808, 15570) |
15768 †, §
(15246, 16308) |
21132 ††, **
(20052, 22266) |
14309 (13521, 15142) |
| 1-hour glucose (mg/dL) | 111.7±37.8 | 162.2±41.4* | 210.8±43.2** | 127.9±18.0 |
| HgbA1C % | 5.3±0.4 | 5.7±0.5* | 6±0.6* | n/a |
| INSULIN RESPONSES | CF-PS | CF-PI-NGT | CF-PI-AGM | Reference |
|
| ||||
| AUCIns (μU/mL) | 5505 (4217, 7200) |
3220 ††,*
(2910, 3570) |
3733 †,*
(3347, 4167) |
5338 (4355, 6542) |
p<0.05,
p<0.001.
CF-PS group compared to CF-PI groups
p=0.053,
p<0.05,
p<0.001
Influence diagnostics were obtained for each subject and included likelihood distance, Cook’s D and PRESS statistics(23). Based on the results of these diagnostics, it was determined that one subject in each of the CF groups was unduly influencing the model fit, parameter estimates and predicted values (using likelihood distance, Cook’s D and PRESS statistics). These three subjects were removed from all models. Significance level for all tests was set a priori at α < 0.05. SAS software (SAS Institute, version 9.2, Cary, NC) was used for all analyses.
RESULTS
Patient characteristics
A search of the Cincinnati Children’s Hospital database identified 146 patients with CF who were 5 years of age or older and who had undergone at least one OGTT. Seventy-three (50%) of the 146 patients were homozygous for ΔF508. Sixty-one (42%) were heterozygous ΔF508. Nine (6%) patients had identified mutations not including the ΔF508 mutation. Three patients (2%) did not undergo genetic analysis. The male: female ratio was 8:7. The racial distribution was 143 Caucasians, 2 African-Americans, and 1 multiracial patient.
Of the 146 CF patients, 139 were receiving pancreatic enzyme supplementation and were thus classified as pancreatic insufficient (CF-PI). Seven patients were not taking pancreatic enzyme supplements and had a fecal elastase value of ≥246 microgram/g of stool (Table 1). These patients were classified as pancreatic sufficient (CF-PS). The genotypes of these patients are listed in Table 1.
TABLE 1.
The genetic mutations of the CF-PS group. Fecal Elastase reported as microgram/gram of stool.
| Mutation #1 | Mutation #2 | Fecal Elastase |
|
|---|---|---|---|
| 1 | DELTA-F508 | R553X | 461 |
| 2 | P5L | A349V | 500 |
| 3 | DELTA-F508 | 3272-26 A to G | 500 |
| 4 | 1898+1G->A | 3272-26 A to G | 436 |
| 5 | DELTA-F508 | NOT IDENTIFIED | 500 |
| 6 | DELTA-F508 | Multiple: R668C & G576A | 500 |
| 7 | DELTA-F508 | G85E | 246 |
The characteristics of the CF groups as well as the reference group are shown in Table 1. No differences were found between the CF-PS group and the CF-PI group with regard to BMI, BMI z-score, FEV1 and FEV1%. The CF groups were significantly younger than the reference group (p<0.01). The BMI of the reference group was significantly higher than the CF-PI group (Table 1). However, there was no significant difference in the BMI z-score between the reference group and the CF groups (Table 1). Based on CF Foundation nutritional guidelines, 25% of the CF-PS group and 50% of the CF-PI groups were classified as nutritionally “at risk” with a BMI <50th percentile. (24).
Glucose tolerance
All CF-PS patients had normal glucose tolerance. Of the 139 CF-PI patients, 96 (69%) demonstrated normal glucose tolerance (CF-PI-NGT) and 43 (31%) demonstrated abnormal glucose metabolism (CF-PI-AGM). Of the CF-PI-AGM patients, 22 met the diagnostic criteria of CFRD. There was no significant difference in the fasting glucose among the groups. The AUCGlu was not significantly different in the CF-PS group compared to the reference group. However, the AUCGlu was increased in the both CF-PI groups compared to the CF-PS group and reference group (Table 3). The mean 1-hour glucose measures and hemoglobin A1C from the CF-PS group was significantly lower than the values for the CF-PI-NGT and CF-PI-AGM groups (Table 3).
Insulin secretion
The AUCIns of both CF-PI groups was significantly lower than the AUCIns in the CF-PS group and the reference group (Table 2). When insulin secretion was expressed as a function of ambient glucose, the insulinogenic index supported a significant impairment in all three CF groups compared to the reference group (Figure 1). In fact, the mean insulinogenic index of the CF-PS group was half of the mean index of the reference group. The insulinogenic index was significantly lower in the CF-PI groups compared to in the CF-PS group.
TABLE 2.
Demographic and clinical parameters for the group of lean, healthy adolescents (reference) compared to the CF-PS, CF-PI-NGT, and CF-PI-AGM groups. Means ± standard deviations for continuous variables. Chi-square test for association was used for sex; Wilcoxon nonparametric test was used for continuous variables.
| CF-PS | CF-PI-NGT | CF-PI-AGM | Reference | |
|---|---|---|---|---|
| No. of Subjects | 7 | 96 | 43 | 12 |
| Male | 57% | 51% | 56% | 25% |
| Age (years) | 12.6±5.2 † | 12.7±4.5 † | 15.5±4.2 † | 19.8±1 |
| Weight (kg) | 43.4±15.1† | 41.9±16.2 | 49.4±13.5 | 66±10.5 |
| BMI | 19.2±3.0 | 18.5±3.1 † | 19.1±2.9 † | 21.7±2.5 |
| BMI z-score | 0.2±1.2 | −0.1±0.8 | −0.4±1.2 | −0.1±0.7 |
| Nutritionally at risk (BMI <50th percentile) |
25% | 50% | 51% | n/a |
| FEV1% predicted | 93.5±14.8 | 88.5±20.8 | 73.7±28 | n/a |
Compared to reference group p<0.01
Figure 1.
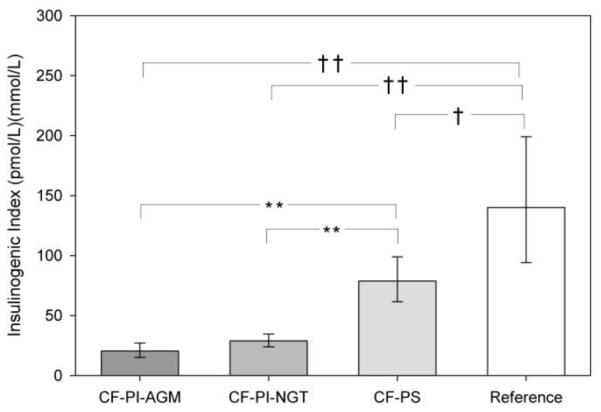
Insulinogenic index results in patients with CF-PS, CF-PI-NGT, CF-PI-AGM, and reference group during OGTT. Tests for differences between group means were performed in an ANOVA model. Reference group compared to CF-PS and CF-PI- † p<0.05, †† p<0.001. CF-PS group compared to reference and CF-PI groups- ** p<0.001
Correlations
In separate ANCOVA models (CF patients only) controlling for age and FEV1%, BMI z-score was significantly and positively correlated with AUCIns (p=0.001) (Figure 2). BMI z-score was not significantly correlated with insulinogenic index.
Figure 2.

Scatter plots of insulin AUCIns by BMI z-score in all CF patients, with line of best fit. In ANCOVA models of AUCIns, BMi z-score was significant and positive predictor of parameters of insulin secretion (p=0.0017).
DISCUSSION
This study found abnormalities of insulin secretion in all CF patients regardless of exocrine pancreatic function, with the CF-PI patients more pronounced compared to the CF-PS patients. The insulin secretion abnormalities in the CF-PS have not been previously reported. Our findings support a model of intrinsic β-cell dysfunction in CF rather than secondary dysfunction caused by progressive pancreatic plugging and fibrosis producing exocrine pancreatic insufficiency and eventual endocrine insufficiency. Concordant with our findings, previous autopsy studies demonstrated no difference in the loss of pancreatic islet cells in patients with CFRD compared to patients with NGT (3) (5). Additionally, some investigators have proposed an intrinsic mechanism within the β-cell of CF patients causing dysfunction. In the rat endocrine pancreas, CFTR mRNA and protein have been localized to the β-cell and β-cell in the islet of Langerhans (25). CFTR knockout mice have been found to be more susceptible to β-cell damage than control mice (12). Finally, a mechanism of abnormal apoptosis in the β-cell caused by chronic stress in the endoplasmic reticulum due to the presence of misfolded CFTR has been reported (10).
Previous reports are discordant regarding the presence of hyperglycemia in the exocrine pancreatic sufficient CF population. Specifically, two studies focusing on CFTR genetic mutations and associated co-morbidities report the incidental finding that adults with CF and pancreatic sufficiency develop abnormal glucose metabolism including CFRD (26) (27). In contrast, Moran et al (9) reported that adult CF patients with pancreatic sufficiency had normal glucose metabolism. All of these studies involved small patient numbers, and there may be some variability among CF-PS patients that confounds discrimination from control subjects. Resolution of this issue will require larger samples and additional assessments of β-cell function.
Based on our findings, we recommend that all CF patients undergo glucose screening with illness and annual OGTT regardless of pancreatic status. The abnormal insulin secretion in all CF patients generates a risk of glucose intolerance. Decreased insulin sensitivity related to pulmonary disease and systemic inflammation (6, 7) further contributes to that risk of hyperglycemia. The CF-related diabetes (CFRD) screening guidelines recommend that CF patients with acute pulmonary exacerbation requiring intravenous antibiotics and/or systemic glucocorticoids should be screened for CFRD by monitoring fasting and 2-h postprandial plasma glucose levels for the first 48 hours (28). However, previous reports of adult CF-PS patients not having β-cell dysfunction (9) may cause clinicians to forego this screening in pancreatic sufficiency CF patients and risk unrecognized hyperglycemia.
Additionally, we found that BMI z-score was a significant predictor of AUCIns in all CF patients. Insulin demonstrates multiple actions within the body including preventing protein and fatty acid breakdown. Increased protein breakdown is reported in insulin deficient CF patients (29) (30). Our findings suggest that the loss of insulin-mediated anabolism contributes to malnutrition and poor outcomes in CF. Development of interventions such as insulin replacement in non-diabetic CF patients to offset insulin secretion dysfunction may improve the nutritional status of these patients.
The present study has several limitations. First, the age difference between the reference and study groups with the mean age of the reference group indicating they were post-pubertal. Whereas the mean age of the CF patients would indicate these patients were pubertal or pre-pubertal. Second, it is likely that we underestimated the number of subjects who were pancreatic sufficient. We report on 5.5% of participants being pancreatic sufficient compared to a report of 15% in the CF national registry (2). In addition, fecal elastase testing was performed only on patients with uncertain pancreatic status. However, because the CF-PI groups demonstrated significantly reduced insulin secretion in all measures compared to the CF-PS group, it is likely that the correct classification of all CF-PS patients would have strengthened our results. Third, with our results, we do not have an accurate measure of insulin sensitivity. The Homeostatic model assessment measure, calculated from fasting insulin and glucose values, is an acceptable measure of insulin sensitivity for the analysis of a large cohort. However, HOMA-IR in an insulin deficient population falsely underestimates the true values associated with insulin sensitivity. Thus, we can accurately report on insulin secretion only. Due to the limitations of this study, we recommend a future larger prospective study including assessment pubertal status of all patients, measurement of fecal elastase on all CF patients, and a higher number of CF-PS patients.
In summary, this study demonstrates that insulin secretion is impaired in CF patients regardless of exocrine pancreatic function status. It is well documented that CF patients with exocrine pancreatic insufficiency have greater degrees of insulin deficiency as judged by glucose tolerance and insulin responses to an OGTT. This insulin deficiency places them at risk for poor outcomes. However, it seems that CF patients with pancreatic sufficiency may also be at risk for complications related to insulin deficiency. All CF patients should complete glucose screening with illness and annual OGTT regardless of pancreatic status. Additionally, interventions to offset insulin deficiency may markedly improve health outcomes in patients with CF.
Highlights.
Insulin secretion is impaired in CF patients regardless of pancreatic exocrine function status.
Insulin secretion deficiency correlates with poor nutritional status in CF patients.
Pancreatic sufficient CF patients may be at risk for complications related to insulin deficiency.
All CF patients should complete glucose screening with illness and annual OGTT.
ACKNOWLEDGEMENTS
The authors would like to thank Aliza Cohen, MA, for her invaluable writing and editorial expertise in helping to clearly deliver our thoughts in this manuscript.
This work was supported by The Research Foundation and the Division of Pulmonary Medicine, Cincinnati Children’s Hospital Medical Center and in part by USPHS Grant # UL1 RR026314 from the National Center for Research Resources, NIH.
ABBREVIATIONS
- AGM
Abnormal glucose metabolism
- ANOVA
Analysis of variance
- ANCOVA
Analysis of covariance
- AUC
Area under the curve
- BMI
Body mass index
- CCHMC
Cincinnati Children’s Medical Hospital Center
- CF
Cystic fibrosis
- CFRD
Cystic fibrosis-related diabetes
- CFTR
Cystic fibrosis transmembrane conductance regulator
- DM
Diabetes mellitus
- FEV1
Forced expiratory volume in 1 second
- GLU
Glucose
- HgbA1C
Hemoglobin A1C
- IAPP
islet amyloid polypeptide
- IFG
Impaired fasting glucose
- IGT
Impaired glucose tolerance with normal fasting glucose
- INS
Insulin
- LSM
Least-square means
- NGT
Normal glucose tolerance
- OGTT
Oral glucose tolerance test
- PI
Exocrine Pancreatic insufficient
- PS
Exocrine Pancreatic sufficient
- REF
Reference group
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
No conflict of interest or disclosures to be submitted.
No reprints requested.
REFERENCES
- 1.Kopelman H, Corey M, Gaskin K, Durie P, Weizman Z, Forstner G. Impaired chloride secretion, as well as bicarbonate secretion, underlies the fluid secretory defect in the cystic fibrosis pancreas. Gastroenterology. 1988 Aug;95(2):349–55. doi: 10.1016/0016-5085(88)90490-8. [DOI] [PubMed] [Google Scholar]
- 2.Foundation CF. Cystic Fibrosis Foundation Patient Registry: 2007 Annual Data Report Bethesda, Maryland. 2007 [Google Scholar]
- 3.Iannucci A, Mukai K, Johnson D, Burke B. Endocrine pancreas in cystic fibrosis: an immunohistochemical study. Hum Pathol. 1984 Mar;15(3):278–84. doi: 10.1016/s0046-8177(84)80191-4. [DOI] [PubMed] [Google Scholar]
- 4.Handwerger S, Roth J, Gorden P, Agnese PDS, Carpenter DF, Peter G. Glucose Intolerance in Cystic Fibrosis. The New England Journal of Medicine. 1969;281(9):11. doi: 10.1056/NEJM196908282810901. [DOI] [PubMed] [Google Scholar]
- 5.Lohr M, Goertchen P, Nizze H, Gould NS, Gould VE, Oberholzer M, Heitz PU, Kloppel G. Cystic fibrosis associated islet changes may provide a basis for diabetes. An immunocytochemical and morphometrical study. Virchows Arch A Pathol Anat Histopathol. 1989;414(2):179–85. doi: 10.1007/BF00718598. [DOI] [PubMed] [Google Scholar]
- 6.Hardin DS, Moran A. Diabetes mellitus in cystic fibrosis. Endocrinol Metab Clin North Am. 1999 Dec;28(4):787–800. doi: 10.1016/s0889-8529(05)70102-x. ix. [DOI] [PubMed] [Google Scholar]
- 7.Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, Adams KS, Seaquist ER. Insulin sensitivity in cystic fibrosis. Diabetes. 1994 Aug;43(8):1020–6. doi: 10.2337/diab.43.8.1020. [DOI] [PubMed] [Google Scholar]
- 8.Kelly A, Moran A. Update on cystic fibrosis-related diabetes. J Cyst Fibros. 2013 Jul;12(4):318–31. doi: 10.1016/j.jcf.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Moran A, Diem P, Klein DJ, Levitt MD, Robertson RP. Pancreatic endocrine function in cystic fibrosis. J Pediatr. 1991 May;118(5):715–23. doi: 10.1016/s0022-3476(05)80032-0. [DOI] [PubMed] [Google Scholar]
- 10.Ali BR. Is cystic fibrosis-related diabetes an apoptotic consequence of ER stress in pancreatic cells? Med Hypotheses. 2009 Jan;72(1):55–7. doi: 10.1016/j.mehy.2008.07.058. [DOI] [PubMed] [Google Scholar]
- 11.Couce M, O'Brien TD, Moran A, Roche PC, Butler PC. Diabetes mellitus in cystic fibrosis is characterized by islet amyloidosis. J Clin Endocrinol Metab. 1996 Mar;81(3):1267–72. doi: 10.1210/jcem.81.3.8772610. [DOI] [PubMed] [Google Scholar]
- 12.Stalvey MS, Muller C, Schatz DA, Wasserfall CH, Campbell-Thompson ML, Theriaque DW, Flotte TR, Atkinson MA. Cystic fibrosis transmembrane conductance regulator deficiency exacerbates islet cell dysfunction after beta-cell injury. Diabetes. 2006 Jul;55(7):1939–45. doi: 10.2337/db05-1647. [DOI] [PubMed] [Google Scholar]
- 13.Elder DA, Wooldridge JL, Dolan LM, D'Alessio DA. Glucose tolerance, insulin secretion, and insulin sensitivity in children and adolescents with cystic fibrosis and no prior history of diabetes. J Pediatr. 2007 Dec;151(6):653–8. doi: 10.1016/j.jpeds.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 14.Elder DA, Prigeon RL, Wadwa RP, Dolan LM, D'Alessio DA. Beta-cell function, insulin sensitivity, and glucose tolerance in obese diabetic and nondiabetic adolescents and young adults. J Clin Endocrinol Metab. 2006 Jan;91(1):185–91. doi: 10.1210/jc.2005-0853. [DOI] [PubMed] [Google Scholar]
- 15.Casazza K, Phadke RP, Fernandez JR, Watanabe RM, Goran MI, Gower BA. Obesity attenuates the contribution of African admixture to the insulin secretory profile in peripubertal children: a longitudinal analysis. Obesity (Silver Spring) 2009 Jul;17(7):1318–25. doi: 10.1038/oby.2008.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuczmarski RJ, Ogden CL, Grummer-Strawn LM, Flegal KM, Guo SS, Wei R, Mei Z, Curtin LR, Roche AF, Johnson CL. CDC growth charts: United States. Adv Data. 2000 Jun;8(314):1–27. [PubMed] [Google Scholar]
- 17.Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP, Gustafsson P, Jensen R, Johnson DC, MacIntyre N, McKay R, Navajas D, Pedersen OF, Pellegrino R, Viegi G, Wanger J. Standardisation of spirometry. Eur Respir J. 2005 Aug;26(2):319–38. doi: 10.1183/09031936.05.00034805. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Dockery DW, Wypij D, Fay ME, Ferris BG., Jr. Pulmonary function between 6 and 18 years of age. Pediatr Pulmonol. 1993 Feb;15(2):75–88. doi: 10.1002/ppul.1950150204. [DOI] [PubMed] [Google Scholar]
- 19.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999 Jan;159(1):179–87. doi: 10.1164/ajrccm.159.1.9712108. [DOI] [PubMed] [Google Scholar]
- 20.Standards of medical care in diabetes--2007. Diabetes Care. 2007 Jan;30(Suppl 1):S4–S41. doi: 10.2337/dc07-S004. [DOI] [PubMed] [Google Scholar]
- 21.Phillips DI, Clark PM, Hales CN, Osmond C. Understanding oral glucose tolerance: comparison of glucose or insulin measurements during the oral glucose tolerance test with specific measurements of insulin resistance and insulin secretion. Diabet Med. 1994 Apr;11(3):286–92. doi: 10.1111/j.1464-5491.1994.tb00273.x. [DOI] [PubMed] [Google Scholar]
- 22.Wareham NJ, Phillips DI, Byrne CD, Hales CN. The 30 minute insulin incremental response in an oral glucose tolerance test as a measure of insulin secretion. Diabet Med. 1995 Oct;12(10):931. doi: 10.1111/j.1464-5491.1995.tb00399.x. [DOI] [PubMed] [Google Scholar]
- 23.Allen DM. The Relationship between Variable Selection and Data Augmentation and a Method of Prediction. Technometrics. 1974;16:125–7. [Google Scholar]
- 24.Stallings VA, Stark LJ, Robinson KA, Feranchak AP, Quinton H. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc. 2008 May;108(5):832–9. doi: 10.1016/j.jada.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 25.Boom A, Lybaert P, Pollet JF, Jacobs P, Jijakli H, Golstein PE, Sener A, Malaisse WJ, Beauwens R. Expression and localization of cystic fibrosis transmembrane conductance regulator in the rat endocrine pancreas. Endocrine. 2007 Oct;32(2):197–205. doi: 10.1007/s12020-007-9026-x. [DOI] [PubMed] [Google Scholar]
- 26.Preumont V, Hermans MP, Lebecque P, Buysschaert M. Glucose homeostasis and genotype-phenotype interplay in cystic fibrosis patients with CFTR gene deltaF508 mutation. Diabetes Care. 2007 May;30(5):1187–92. doi: 10.2337/dc06-1915. [DOI] [PubMed] [Google Scholar]
- 27.Koch C, Cuppens H, Rainisio M, Madessani U, Harms H, Hodson M, Mastella G, Navarro J, Strandvik B, McKenzie S. European Epidemiologic Registry of Cystic Fibrosis (ERCF): comparison of major disease manifestations between patients with different classes of mutations. Pediatr Pulmonol. 2001 Jan;31(1):1–12. doi: 10.1002/1099-0496(200101)31:1<1::aid-ppul1000>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 28.Moran A, Brunzell C, Cohen RC, Katz M, Marshall BC, Onady G, Robinson KA, Sabadosa KA, Stecenko A, Slovis B. Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care. 2010 Dec;33(12):2697–708. doi: 10.2337/dc10-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardin DS, LeBlanc A, Lukenbaugh S, Para L, Seilheimer DK. Proteolysis associated with insulin resistance in cystic fibrosis. Pediatrics. 1998 Mar;101(3):433–7. doi: 10.1542/peds.101.3.433. Pt 1. [DOI] [PubMed] [Google Scholar]
- 30.Moran A, Milla C, Ducret R, Nair KS. Protein metabolism in clinically stable adult cystic fibrosis patients with abnormal glucose tolerance. Diabetes. 2001 Jun;50(6):1336–43. doi: 10.2337/diabetes.50.6.1336. [DOI] [PubMed] [Google Scholar]