

Abstract
Lipoproteins are protein-lipid nanoparticles that transport lipids in circulation and are central in atherosclerosis and other disorders of lipid metabolism. Apolipoproteins form flexible structural scaffolds and important functional ligands on the particle surface and direct lipoprotein metabolism. Lipoproteins undergo multiple rounds of metabolic remodeling that is crucial to lipid transport. Important aspects of this remodeling, including apolipoprotein dissociation and particle fusion, are mimicked in thermal or chemical denaturation and are modulated by free energy barriers. Here we review our biophysical studies that revealed kinetic mechanism of lipoprotein stabilization and unraveled its structural basis. The main focus is on high-density lipoprotein (HDL). An inverse correlation between stability and functions of various HDLs in cholesterol transport suggests functional role of structural disorder. A mechanism for conformational adaptation of the major HDL proteins, apoA-I and apoA-II, to the increasing lipid load is proposed. Together, these studies help understand why HDL form discrete subclasses separated by kinetic barriers, which have distinct composition, conformation and functional properties. Understanding these properties may help improve HDL quality and develop novel therapies for cardiovascular disease.
Keywords: Lipid surface-binding proteins, thermodynamic and kinetic stability, cholesterol transport and metabolism, conformational ensemble
Introduction
Lipoproteins are dynamic vehicles for lipid transport
Lipids in the body are transported via lipoproteins that are non-covalent dynamic assemblies of specific proteins, termed apolipoproteins (apos), and lipids. Lipoproteins vary in size (101–102 nm), density, composition, structure and function. Plasma lipoproteins are central to cardiovascular health and disease [1, 2]; this disease remains the leading cause of death in the developed countries [3]. Lipoproteins are also central to other major disorders of lipid metabolism such as metabolic syndrome, obesity and diabetes II [1–2], whereas apoE, which is the main apolipoprotein in the brain [4], acts in an isoform-specific manner as a major genetic risk factor for Alzheimer’s disease [5]. Moreover, apolipoproteins are prone to misfolding and can form fibrils that are found in most amyloid deposits in vivo [6–8]. Apolipoprotein misfolding can cause systemic amyloidoses and has been implicated in atherosclerosis [6–8]. Both normal functions of apolipoproteins and their pathologic misfolding are critically hinged on their remarkable structural flexibility that enables these amphipathic proteins to adapt their conformations to lipoproteins of various sizes and to the aqueous environment such as plasma [9–11].
Apolipoproteins comprise up to 50% of the total lipoprotein mass and form flexible structural scaffolds and essential functional ligands on the lipoprotein surface [11, 12]. These proteins direct lipoprotein metabolism by activating lipophilic enzymes, interacting with lipid transfer proteins, and binding and activating lipoprotein receptors [11, 12]. Each lipoprotein particle contains several apolipoproteins and hundreds of lipids. The particle surface is comprised of apolipoproteins embedded into a monolayer of polar lipids, mainly phospholipids and cholesterol. The polar moieties of the proteins and lipids face solvent and thereby confer lipoprotein solubility, while the apolar lipids (mainly cholesterol esters and triacylglycerides, or fat) are sequestered in the core (Figure 1 below). This assembly helps solubilize lipids and transport them to and from peripheral cells in the aqueous environment of plasma, lymph and cerebrospinal fluid.
Figure 1.
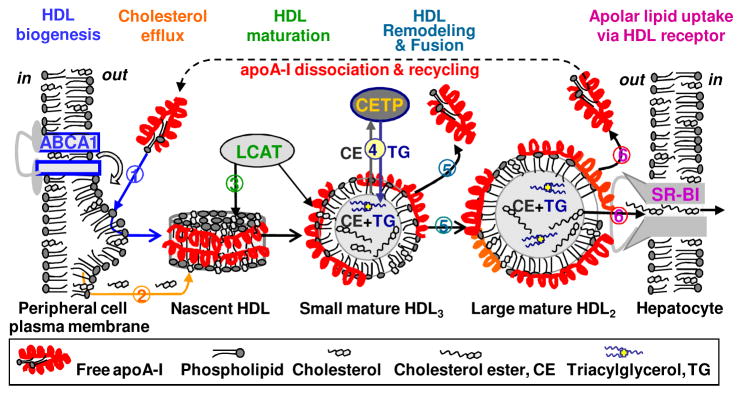
Reverse cholesterol transport in a nut shell. Cartoon illustrates selected steps in this complex process. ApoA-I interacts with the plasma membrane and ABCA1 transporter to generate nascent “discoidal” HDL (1). These HDL take up additional cholesterol from the membrane (2) which is esterified by LCAT. The hydrophobic cholesterol ester produced in this reaction moves from the HDL surface to its core, leading to HDL maturation and formation of small spherical particles termed HDL3 (3). These small HDL are further remodeled by LCAT and other plasma factors, such as cholesterol ester transfer protein (CETP) (4). This remodeling can cause imbalance between the polar surface and the apolar core of the particle, leading to partial protein dissociation and lipoprotein fusion to form larger HDL2 (5). Large lipid-loaded HDL2 form preferential substrates for the scavenger receptor, SR-BI, that mediates uptake of apolar core lipids and HDL disintegration (6). The dissociated apoA-I is either recycled or gets degraded or misfolded. Figure modified from [61].
Lipoproteins form major classes differing in the particle size, density, biochemical composition and function: high-, low-, intermediate-, and very low-density lipoproteins (HDL, LDL, IDL, VLDL) and chylomicrons [11–14]. Since proteins, which are heavier than lipids, are located on the particle surface, the particle diameter increases with decreasing density, from HDL (7–12 nm) and LDL (20–24 nm) to VLDL (40–100 nm). Each lipoprotein class has specific functions in lipid transport. HDLs remove excess cholesterol from peripheral cells, such as arterial macrophages, and have other beneficial properties. Plasma levels of HDL cholesterol (“good cholesterol”) and the major HDL protein, apoA-I (28 kDa), have long been known to correlate inversely with the risk of cardiovascular disease [14–16]. In contrast, LDL, which is the major plasma carrier of cholesterol in the form of cholesterol ester, delivers it to the peripheral tissues. The plasma levels of LDL cholesterol (“bad cholesterol”) and the major LDL protein, apoB (550 kDa), are the main causative risk factors of atherosclerosis [1, 2, 13, 16]. VLDL, which is the main plasma carrier of fat, is the metabolic precursor of LDL and a critical risk factor for metabolic syndrome and diabetes II [1, 2, 13, 17]. Each lipoprotein class is further subdivided into subclasses with distinct particle size, biochemical composition, and functional properties [18–20]. Analysis of these properties is the major thrust in the ongoing efforts to improve lipoprotein functionality and develop novel diagnostic tools and therapies for cardiovascular disease to complement statins, fibrates and other lipid-lowering drugs [20–22].
Lipoprotein metabolism is an extremely complex process during which individual particles exchange their proteins and lipids and undergo extensive remodeling by lipophilic enzymes, lipid transfer proteins and lipoprotein receptors. One example of such remodeling is HDL maturation and growth in reverse cholesterol transport [23, 24]. In this complex pathway, nascent HDLs, which can be envisioned as “discoidal” particles comprised of a cholesterol-containing phospholipid bilayer with proteins wrapped around the perimeter, are transformed into mature “spheroidal” HDLs that contain a core of apolar lipids, mainly cholesterol esters (Figure 1). This transformation is driven by lecithin:cholesterol acyltransferase (LCAT) that converts polar molecules of cholesterol and phosphatidylcholine into apolar cholesterol ester and free fatty acid. This and many other remodeling reactions shift the balance between the polar surface of a lipoprotein and its apolar core. Below we describe how this balance can be restored upon spontaneous lipoprotein fusion, fission and protein dissociation.
Although over 90% of all apolipoproteins circulate on the lipoprotein surface in a stable highly α-helical conformation, a small sub-population is found in a transient lipid-poor or lipid-free form [24–27], termed “free” for brevity. This structurally labile metabolically active form can be generated de novo or upon dissociation from the lipoprotein surface. Free apos are rapidly recruited for binding to plasma membrane or to other lipoproteins (Figure 1); alternatively, they are either degraded or misfolded and form amyloid [6–8]. Apolipoproteins undergo large conformational changes in these transitions, from the highly dynamic partially unfolded free state, to a more stable largely α-helical lipid-bound state, to intermolecular cross-β-sheet in amyloid.
Lipoprotein heterogeneity and ability to exchange their protein and lipid constituents raises several fundamental questions. First, what maintains the overall integrity of the lipoprotein assembly in the absence of unique specific packing of its proteins and lipids? Second, what structural stability is optimal for lipoprotein functions? A related question is: how do apolipoproteins adapt their conformation to the changing lipid load during lipid transport? What is the conformational ensemble of an apolipoprotein free in solution and on the surface of various lipoproteins? Finally, what makes apolipoproteins amyloidogenic? The answers are beginning to emerge as a result of decades of work by many research groups.
Here, we review our biophysical studies of lipoproteins, with the focus on HDLs which are the smallest particles whose structural and stability properties have been best characterized. First, we provide a brief overview of HDL metabolism. Next, we outline the thermodynamic stabilization of lipid-free apolipoproteins in solution, and contrast it with the kinetic stabilization of model and plasma HDL. This is followed by the description of the key determinants for HDL stability and their relevance to HDL functions at specific steps of cholesterol transport. Finally, we propose a structure-based mechanism for functional adaptation of HDL proteins to the increasing lipid load during reverse cholesterol transport. The structural basis for apolipoprotein misfolding, which can lead to human amyloidosis [8], and the structural stability and fusion of LDL, which is an early triggering event in atherosclerosis [28], have been recently reviewed elsewhere.
Functionality of “good cholesterol”: quality versus quantity
HDLs and their major protein, apoA-I (28 kDa) remove excess cholesterol from peripheral cells via the reverse cholesterol transport, which is the sole pathway for cholesterol removal from the body [23, 24, 29] (Figure 1). In addition, HDLs possess anti-inflammatory, anti-thrombolytic, anti-oxidant and other beneficial properties [14, 15, 19, 20]. Although the inverse correlation between the risk of cardiovascular disease and the plasma levels of “good cholesterol” and apoA-I has been known for over 40 years, recent populational studies and clinical trials revealed that raising the HDL levels does not necessarily improve cardiovascular health [30]. Thus, the current consensus is that not all HDLs are created equal, and that functionality of lipoproteins is no less important than their steady-state levels [19, 30]. This functionality depends on the protein and lipid composition and conformation, which determine the dynamic properties of HDL and the overall rate of cholesterol removal.
To obtain key determinants for HDL function, it is important to understand the energetic basis for structural stability and remodeling of HDL at various steps of cholesterol transport. To this end, our laboratory has analyzed structural stability of free apos as well as of reconstituted “discoidal” and plasma “spherical” HDL and their individual subclasses differing in the particle size, composition, oxidative status, and other properties. The rationale for these studies and their representative results are outlined below.
Structural stability of HDL and its proteins
Structural flexibility of free apolipoproteins in solution
All HDL apos are water-soluble, or exchangeable. Exchangeable apos form a conserved family of proteins that have evolved from a single precursor gene via the duplication or deletion of 11-mer codon repeats. These repeats encode for tandem 11-residue sequence repeats with high propensity to form amphipathic α-helices [31]. Class-A α-helices that form the major structural and functional motif in the apolipoprotein family (A stands for apos) are distinct from the class-G helices found in globular proteins. Class-A helices contain a large fraction of apolar residues that confer lipid binding affinity, and a substantial fraction of charged residues that confer protein solubility. Characteristic radial distribution of these residues optimizes lipid surface binding. Large apolar faces in class-A helices, which comprise 30–50% of the total surface area (as compared to ~15% typical of globular proteins), form the lipid binding sites [31]. They are flanked by the basic residues that can contribute to lipid binding via the hydrophobic interactions with the phospholipid acyl chains and the electrostatic interactions with the phospholipid head groups. Acidic residues are located in the middle of the polar helical face and can form extensive salt links with the basic residues, which probably contribute to protein stability in solution and on HDL [32–34]. Thus, in contrast to 7-mer helical repeats found in globular proteins, apolipoprotein helices are comprised of 11-mer sequence repeats that are optimized for lipid surface binding.
In lipid-free exchangeable apos in solution, the large apolar helical faces are involved in protein-protein interactions. These interactions are apparently less extensive than those in globular proteins. As a result, the structural stability of most apolipoproteins is lower than that of typical globular proteins (under 3 kcal/mol for apoA-I and even lower for smaller apos), while their aggregating propensity is higher. These and other properties of free apos, including apoA-I, apoA-II, apoC-I and apoE, resemble the compact molten-globular folding intermediates [9, 35–38].
Larger apos such as apoA-I (28 kDa) [34] or apoE (43 Da) [5, 37] contain a globular helix-bundle domain in the N-terminal two-thirds of the protein, and an intrinsically disordered C-terminal domain that forms the primary lipid binding site. Smaller apos free in solution are even less well-ordered. For example, lipid-free apoA-II, a 9 kDa protein that forms a disulfide-linked homodimer in humans, has marginally stable α-helical structure at near-physiologic conditions, which unfolds upon either heating or cooling from 25 °C [35]. Another example is apoC-I (6 kDa), the smallest exchangeable apo that is ~30% α-helical in solution as a monomer, acquires additional helical structure upon self-association, and undergoes unfolding accompanied by oligomer dissociation upon either heating or cooling from 25 °C [36]. Other small apos such as apoC-II and apoC-III (9 kDa) lack stable secondary structure in solution [5, 8]. In sum, these and other free exchangeable apos are fully or partially intrinsically disordered.
Lipid binding induces helical structure in these proteins and stabilizes them against proteolyisis and misfolding [38–40]. What is the physical basis for this stabilization? With few exceptions, most published studies have been focused on thermodynamic stability of lipoproteins, perhaps because it is easier to access experimentally. In such studies, the samples are heated and cooled at a constant rate (in melting experiments), or denaturant concentration is increased incrementally (in titration experiments), and the resultant structural changes are monitored by calorimetry or spectroscopy as a function of temperature or denaturant concentration. Although the midpoint of the protein unfolding transition is often taken as a measure of relative stability, the ultimate goal is to determine the difference in free energy, ΔG=GD−GN, or enthalpy, ΔH=HD−HN, between the native (N) and the denatured (D) states. A key assumption is that these two states are in equilibrium throughout the transition, i. e. that the denaturation is thermodynamically reversible.
Application of the thermodynamic approach to lipoproteins has one major caveat: lipoproteins are irreversibly disrupted upon thermal or chemical denaturation. It is well-known that, except for simple model complexes described in the next section, lipoproteins do not spontaneously re-assemble into their native state upon denaturation. This irreversibility questions the application of the conventional thermodynamic approach to the analysis of lipoprotein stability and necessitates an alternative kinetic approach [41].
Model lipoproteins are stabilized by free energy barriers
To address the physical basis for HDL stability, we initially used complexes reconstituted from a model phospholipid dimyrisroyl phosphatidylcholine (DMPC) and apoC-I (57 a. a.), the smallest human apolipoprotein that modulates HDL and VLDL metabolism and is a structural and functional prototype of larger members of this protein family [36, 41]. DMPC (C14:0, C14:0) is a relatively short-chain fully saturated phospholipid that can spontaneously self-assemble with apolipoproteins upon incubation at 24 °C. The resultant lipoproteins are “discoidal” particles 10–15 nm in diameter, as suggested by non-denaturing gel electrophoresis and negative stain electron microscopy (Figure 2A, B). These lipoproteins provide a simple experimental model for nascent plasma HDL, which are comprised mainly of apoA-I, longer-chain PCs, and cholesterol.
Figure 2.
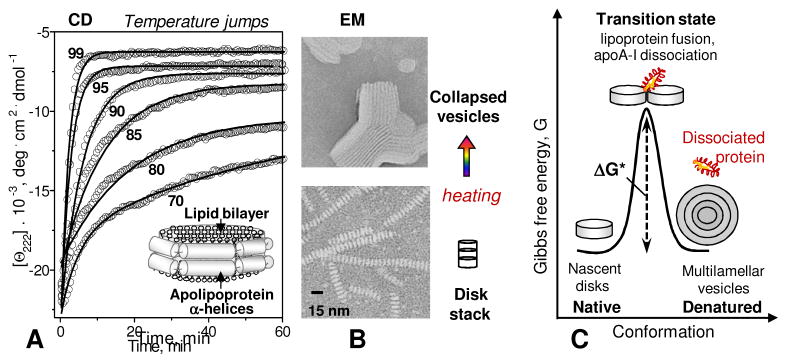
Model discoidal HDL are stabilized by free energy barriers. (A) Lipoproteins reconstituted from a model phospholipid and an apolipoprotein form bilayer “disks” with protein α-helices wrapped around the perimeter. Heat denaturation kinetics of these particles was monitored in T-jump experiments from 25 °C to higher constant temperatures by CD spectroscopy at 222 nm for α-helical unfolding. Solid lines show single-exponential data fitting that was used to determine k(T) for the Arrhenius analysis. (B) Negative stain electron microscopy of discoidal HDL before and after heat denaturation. Intact particles are seen as disk stacks; such stacking is characteristic of negative staining. (C) Cartoon showing intact discoidal lipoproteins, the products of their denaturation, and the proposed high-energy transition state of disk fusion and protein dissociation.
Arguably the easiest way to assess macromolecular stability is through the melting experiments. To this end, apoC-I:DMPC disks, which have ~60% α-helical content at 25 °C, were heated and cooled from 5 to 98 °C at a rate of 90 °C/h. Circular dichroism (CD) signal was recorded at 222 nm to monitor α-helical unfolding as a function of temperature, Θ222(T) (Figure 2A, B). Similar experiments were conducted with free monomeric apoC-I that is ~30% α-helical at 25 °C. Like other free apolipoproteins [9, 35, 37], apoC-I in solution undergoes a sigmoidal unfolding that is reversible upon cooling [36]. The heating and cooling data of free apos are independent of the scan rate and do not show hysteresis, indicating a thermodynamically reversible transition.
Compared to free apoC-I whose thermal unfolding was centered at Tm~55°C, the heat unfolding transition of apoC-I:DMPC disks shifted to higher temperatures (Tm,app =72°C at 90 °C/h) suggesting increased stability [41]. Moreover, the heating and cooling data showed a hysteresis, indicating thermodynamic irreversibility. To test its origin, we slowed down the scan rate to provide more time for the system to reach equilibrium at each temperature and thereby try and close the gap between the heating and cooling curves. CD melting data were collected at several scan rates from 90 to 3 °C/h, so the fastest experiment took a couple of hours while the slowest took a couple of days. Hysteresis was observed at any scan rate explored, indicating that heat denaturation of apoC-I:DMPC complexes is inherently thermodynamically irreversible [41].
Importantly, the apparent melting temperature, Tm,app, which was taken at the inflection point in the heating data, decreased from 72°C to 48°C upon slowing down the heating from 90 to 4 °C/h. Therefore, the melting temperature alone cannot be used as a reliable measure of lipoprotein stability. Moreover, large scan rate effect on Tm,app indicates high activation energy, Ea. The value estimated from the scan rate effects on the melting data of apoC-I:DMPC complexes was Ea=25±5 kcal/mol [41, 42].
To verify this value, we conducted kinetic temperature-jump (T-jump) experiments in which the transition was triggered by a rapid increase in temperature from 25 °C to a high constant value, and the time course of protein unfolding was monitored by far-UV CD at 222 nm, Θ222(t) (Figure 2A). By the time the first data point was recorded (~10 sec), free protein was fully unfolded. In contrast, the unfolding in apoC-I:DMPC complexes proceeded on a much slower time scale that could be monitored by CD. At each temperature, the time course of the unfolding was well-approximated by a single exponential, Θ222(t) = A exp (−kt), which was used to determine the reaction rate k(T) and obtain an Arrhenius plot, ln k(T) versus 1/T. The slope of this plot corresponded to an activation energy Ea=25±5 kcal/mol, verifying the value obtained from the melting experiments [41, 42].
What is the meaning of this value? Arrhenius activation energy, Ea ≅ΔHv*, is the enthalpic component of the free energy barrier that defines kinetic stability, ΔG*=ΔH*−TΔS*; here * denotes the high-energy transition state that is the bottleneck of the reaction, and Δ refers to the difference between the transition and the native states, ΔG*=G*−GN (Figure 2C). The free energy barrier, ΔG*, determines the rate k of the lipoprotein denaturation. According to the transition state theory, ΔG*=−RT ln(k/K), where k is the observed rate constant and K is the rate constant in the absence of the barrier. The heat- or denaturant-induced structural transition in lipoproteins observed in our kinetic experiments occurred on a time scale of hours (k~10−3 sec−1), much slower than the typical unfolding rate in small helical proteins in solution (K~108–1010 sec−1) which, in turn, is slower than the theoretical “speed limit” in Eyring equation (kBT/h~ 6*1012 sec−1). Thus, apoC-I binding to DMPC decelerates the unfolding by more than 10 orders of magnitude, which corresponds to ΔG*=18±2 kcal/mol [41].
This value of kinetic stability is comparable or higher than the typical thermodynamic stability of globular proteins, and is much higher than the thermodynamic stability of free apos in solution under ambient conditions, ΔG≤3 kcal/mol. This kinetic barrier helps explain why lipoprotein formation stabilizes apos against unfolding, misfolding and proteolysis despite the lack of unique packing interactions in these dynamic assemblies.
Physical origin of free energy barriers
What is the physical basis for the kinetic stabilization of lipoproteins? A related question is: what happens to the lipids as the proteins unfold? The answer requires basic physical understanding of the native and the denatured lipoprotein state, as well as the high-energy transition state between them (Figure 2C). To this end, we analyzed lipoproteins at various stages of thermal or chemical denaturation by using negative stain electron microscopy (Figure 2B) and non-denaturing gel electrophoresis. We also recorded changes in turbidity or in 90° light scattering in CD experiments [41, 43]. This enabled us to simultaneously monitor micro- and macroscopic changes during lipoprotein heating and cooling, such as the α-helical unfolding (by far-UV CD) and the changes in the particle size (by turbidity or light scattering). The results revealed that lipoprotein denaturation involves α-helical unfolding coupled to apolipoprotein dissociation and the increase in the particle size due to fusion [41–44]. The observations that the activation energy Ea and the free energy barrier ΔG* increased nearly linearly with increasing protein size and acyl chain length suggested strongly that the rate-limiting step in lipoprotein disruption involves dissociation of a lipid-poor protein molecule [44, 45]. Other kinetic studies of apoA-I exchange support this conclusion [27, 46, 47].
Together, our studies of model lipoproteins have revealed a molecular basis for kinetic stabilization of discoidal HDL, which can be summarized as follows. The heat- or denaturant-induced protein unfolding disrupts the apolar lipid-binding surfaces of the amphipathic apolipoprotein α-helices and thereby promotes their dissociation from the lipid [41, 44]. Protein dissociation produces transient hydrophobic packing defects, or sticky patches, on the lipoprotein surface, which initiate lipoprotein aggregation and fusion. Lipoprotein fusion reduces surface-to-volume ratio and thereby compensates for the loss of the polar surface moiety. Such fusion involves transient disruption of multiple protein-lipid and lipid-lipid packing interactions [41, 44, 45], providing a large favorable enthalpic contribution ΔH*≅Ea to the free energy barrier ΔG*=ΔH*−TΔS*. This barrier defines the kinetic stability of discoidal lipoproteins and determines the rate of their remodeling (Figure 2C).
Human plasma HDL is stabilized by free energy barriers
Is structural stability of plasma HDL also modulated by kinetic barriers? If so, what is the physical origin of these barriers? Compared to lipoprotein “nanodisks”, mature plasma HDL have more complex structure and biochemical composition: in addition to the proteins and polar lipids on the surface, they also contain apolar lipids in the core. Therefore, the free energy landscape of mature HDL is expected to be more complex, potentially featuring more than one barrier separating more than two distinct structural states [48]. This idea is supported by multiple lines of evidence obtained in calorimetric, electron microscopic, spectroscopic and biochemical studies [48–51].
For example, differential scanning calorimetry of mature human plasma HDL shows two high-temperature peaks in the heat capacity function, Cp(T), which correspond to two irreversible transitions (Figure 3A) [49, 50]. In comparison, just one high-temperature calorimetric peak corresponding to fusion is observed in discoidal lipoproteins [50]. All these transitions shift to lower temperatures at slower heating rates, indicating kinetic control.
Figure 3.

Mature plasma HDL are stabilized by free energy barriers. Thermal or chemical denaturation of human plasma HDL was monitored by differential scanning calorimetry (A), negative stain electron microscopy (B), circular dichroism spectroscopy (C) and non-denaturing gel electrophoresis (D). The results revealed two morphological transitions, one involving HDL fusion and another HDL rupture and release of apolar core lipids. Both transitions involve apolipoprotein dissociation. (E) Cartoon illustrating the proposed high-energy transition state of HDL fusion.
Electron microscopic analysis of HDL at various stages of thermal denaturation reveals that the first high-temperature calorimetric peak corresponds to fusion whereby two-to-three HDL merge to form a larger HDL-like particle (Figure 3A, B). The second calorimetric peak corresponds to HDL rupture, i. e. collapse of the lipoprotein morphology and release of the apolar core lipids that coalesce into droplets (Figure 3A, B). Irreversible increase in the particle size upon heat-induced HDL fusion and rupture and the concomitant apolipoprotein unfolding was also observed in the melting experiments monitored by CD spectroscopy and turbidity or light scattering [32, 48–51]. Finally, kinetic experiments such as temperature- or denaturant-jumps showed a slow two-phase unfolding kinetics that could be approximated with a double-exponential function (Figure 3C). We proposed that the first kinetic phase reflects HDL fusion, while the second phase corresponds to HDL rupture and release of core lipids [32, 48]. Both phases involve dissociation and unfolding of HDL proteins [32, 46, 50] (Figure 3D, E).
This idea agrees with other studies reporting two distinct populations of apoA-I on HDL: one readily exchangeable and the other more strongly associated with the particle [52–54]. Studies using non-denaturing gel electrophoresis and Western blotting suggest that HDL fusion involves the dissociation of the loosely bound population of apoA-I and minor apos, while the more strongly bound population of apoA-I, as well as apoA-II that inserts deeper into the lipid, stay on the particle until its rupture [32, 44, 51].
The activation energy of HDL fusion, which was estimated by using an Arrhenius analysis of the kinetic data recorded at near-physiologic salt concentrations and pH, was Ea~50 kcal/mol [32]. We proposed that this high activation energy arises from transient disruption of multiple protein-lipid and lipid-lipid interactions. Such disruption occurs during dissociation of apoA-I molecule and re-packing of the remaining proteins and lipids from two-to-three HDL that fuse to form a single particle [48]. Similarly, fusion in other lipid systems such as cell membranes involves transient disruption of many packing interactions, leading to high activation energy of tens of kcal/mol. In HDL, this activation energy provides an enthalpic contribution to the free energy barrier that determines the rate of HDL fusion in vitro and in vivo.
Physiological role of kinetic HDL stability
HDL fusion, rupture and apolipoprotein dissociation observed upon thermal or chemical denaturation mimic important aspects of the metabolic HDL remodeling by plasma factors [10, 48, 52, 54]. For example, electron microscopic analysis suggests that heat- or denaturant-induced HDL fusion resembles metabolic conversion of smaller denser HDL3 into larger lipid-loaded HDL2 particles [48, 52] (Figure 3B, D; Figure 4) Such a conversion is thought to occur during reverse cholesterol transport upon increasing lipid load in HDL. In fact, HDL remodeling by several plasma factors, such as LCAT or cholesterol ester and phospholipid transfer proteins, shifts the balance between the core and surface of HDL, which induces apoA-I dissociation and HDL fusion [55–58]. Another example is HDL rupture and release of the apolar core lipids observed in our denaturation studies. This process mimics aspects of HDL interactions with its receptor, scavenger receptor SR-BI, which mediates selective uptake of apolar lipids from HDL core at the last step of reverse cholesterol transport [59, 60] (Figures 1, 4). Thus, heat- or denaturant-induced HDL fusion and rupture, accompanied by apoA-I dissociation, mimic key aspects of metabolic transformations of plasma HDL during reverse cholesterol transport.
Figure 4.

Cartoon illustrating free energy barriers between HDL subclasses. The figure is not meant to represent the relative energies of the stable HDL states or the barriers between them. This dynamic energy landscape depends on the biochemical composition of HDL that constantly changes, and on the environmental conditions. Factors that help traverse these barriers by lowering them during reverse cholesterol transport are indicated: ABCA1 transporter, LCAT, cholesterol ester and phospholipid transport proteins (CETP and PLTP), hepatic lipase and SR-BI receptor. Remarkably, thermal, chemical and other perturbations (e. g. by detergents, bacterial factors etc. 46, 47, 52, 53]) induces very similar HDL fusion, rupture and apolipoprotein dissociation, mimicking key aspects of the morphologic HDL transitions in vivo. This similarity suggests that HDL remodeling in vivo and in vitro is modulated by similar kinetic barriers. Figure modified from [10].
We propose that HDL perturbations by various factors (such as thermal or chemical denaturation, hydrolysis of various lipids, cholesterol esterification, lipid transfer, receptor interactions, etc.) shift the balance between the apolar core and the polar surface. To restore the balance, the lipoprotein assembly has a limited repertoire of spontaneous responses: to release excess polar moieties from the surface (via protein dissociation) or to compensate for lack of such moieties (via lipoprotein fusion and, ultimately, rupture). HDL fusion and rupture involve concomitant repacking of many protein and lipid molecules and are thermodynamically irreversible. The rates of these transitions are modulated by kinetic barriers (Figure 4) that separate HDL subclasses and decelerate their spontaneous inter-conversions in vivo [61].
Other lipoproteins are stabilized by free energy barriers
Our stability studies revealed that, similar to HDL, other lipoprotein classes such as LDL and VLDL are also stabilized by free energy barriers [62–65]; however, the height of these barriers and the activation energy is different for different lipoprotein classes. The underlying morphological transitions are also different, reflecting distinct metabolic transformations that occur in these lipoproteins in vivo. For example, heat-induced LDL aggregation and fusion observed in our biophysical studies mimic closely LDL aggregation and fusion that occur upon LDL retention in the arterial wall [62, 63]. These LDL reactions are thought to provide an early triggering event in atherosclerosis [28].
Another example is thermal denaturation of human plasma VLDL. Intriguingly, this process involves not only lipoprotein fusion, rupture and dissociation of exchangeable apos, but also fission of small apoE-containing HDL-like particles [64, 65]. Very similar apoE-containing HDL are released from VLDL upon hydrolysis of core lipids during VLDL-to-LDL conversion in vivo [64].
Together, these examples support the idea that various biophysical or biochemical perturbations lead to a finite number of structural responses in lipoproteins that help restore the balance between their polar surface and the apolar core. Therefore, we postulate that lipoprotein remodeling in vitro and in vivo involves similar structural responses and is modulated by similar kinetic barriers. Such barriers help explain why lipoprotein classes and subclasses co-exist in plasma and do not spontaneously converge into the lowest free-energy state, or the proverbial primordial soup. At the same time, such barriers help ensure that potentially deleterious modifications (such as the protein and lipid oxidation, hydrolysis, dissociation of individual protein and lipid molecules, etc.) are not detrimental to the overall structural integrity of the lipoprotein assembly.
Kinetic barriers have been proposed to optimize structural stability and functions of other macromolecules and their assemblies and protect them against potentially deleterious effects of oxidation, hydrolysis and other post-translational modifications [66]. Our research has added lipoproteins to the growing list of macromolecular complexes whose structural stability and functional remodeling are modulated by free energy barriers, and has established experimental approaches for measuring these barriers [41, 42, 44].
Functional role of structural disorder in HDL
What height of the free energy barriers is optimal for HDL function? Since HDL perform various functions and undergo multiple rounds of metabolic remodeling, these dynamic barriers might differ at different metabolic steps and the balance among them probably modulates the overall rate of reverse cholesterol transport. Studies of model and plasma HDL varying in protein and lipid composition suggest a general trend: less stable particles tend to be functionally more efficient [61]. Examples are outlined below, starting with HDL biogenesis at the first step of reverse cholesterol transport, and ending with the uptake of apolar lipids by HDL receptor at the last step (Figure 1).
-
HDL biogenesis involves remodeling of plasma membrane by apoA-I and formation of small discoidal HDL (Figure 1). Such remodeling can occur spontaneously in vitro in model liposomes comprised of relatively short-chain phospholipids (C14 or shorter), which form relatively thin bilayers whose packing defects, which are maximal at the chain melting temperature, facilitate protein insertion. Longer-chain phospholipids such as those found in the plasma membrane (C18 or longer) form thicker more ordered bilayers. The remodeling of such bilayers by apoA-I does not occur spontaneously at an appreciable rate, and is facilitated by the lipid transporter ABCA1 in vivo. This ATP-powered transporter pumps lipids from the inner to the outer leaflet of the plasma membrane and thereby perturbs lipid packing and forms protrusions on the membrane surface [67]. Direct and lipid-mediated ABCA1-apoA-I interactions also contribute to HDL biogenesis [68]. Although the complex interactions between apoA-I, ABCA1 and the plasma membrane are far from clear, the perturbed lipid packing probably contributes to apoA-I insertion into the membrane. Thus, packing disorder in the lipid bilayer probably augments apolipoprotein insertion into the membrane and HDL biogenesis.
Moreover, in vitro studies using various apolipoproteins clearly showed that reduced structural stability of free protein accelerates lipoprotein formation [61]. For example, the rate of lipoprotein formation by the three major isoforms of apoE correlates inversely with the structural stability of free protein, apoE4<apoE3<apoE2 [5, 69]. This inverse correlation probably reflects the competition between the tertiary interactions among the protein α-helices and the protein-lipid interactions. In sum, structural disorder in the protein and in the lipid bilayer augments formation of discoidal lipoproteins.
HDL maturation is the next metabolic step in reverse cholesterol transport. At this step, discoidal HDL take up additional cholesterol from the cells, which is then esterified via the LCAT reaction. Cholesterol ester, which is highly hydrophobic, moves inside the particle forming its apolar core and converting it into a mature spherical HDL. Stability and functional studies showed that larger proteins and longer more fully saturated phospholipids form more stable discoidal lipoproteins [44, 45, 61] that are less efficient acceptors of cell cholesterol and less efficient activators of LCAT [70, 71]. These studies suggest an inverse correlation between structural stability of discoidal HDL and their functions in cell cholesterol efflux and esterification [61].
-
Functions and stability of mature spherical HDL show a similar trend observed in the oxidation studies of plasma HDL [72]. Interestingly, mild oxidation by various reagents was shown to destabilize human HDL (i.e. promote apoA-I dissociation and HDL fusion and rupture) as well as improve HDL function in promoting cholesterol efflux from cells. In contrast, more extensive oxidation had opposite effects: it stabilized HDL against fusion and impaired HDL ability to stimulate cholesterol efflux from cells [72]. Thus, the inverse correlation between structural stability and function is not limited to model discoidal but also extends to plasma spherical HDL.
One of the important functional determinants of spherical HDL is the particle size [19]. Epidemiologic studies suggest that large lipid-loaded HDL2 are more cardioprotective than their smaller HDL3 counterparts. Stability studies show that larger HDLs undergo rupture and release of their apolar lipids more readily than smaller particles [47] (Figure 3A). This suggests that reduced stability of HDL2 may contribute to the preferential receptor-mediated uptake of apolar lipids from these larger lipid-loaded particles at the final step of reverse cholesterol transport (Figure 1).
Taken together, these studies illustrate that inverse correlation between HDL stability and function extends from the first to the last step of reverse cholesterol transport. Since reduced macromolecular stability generally promotes local structural flexibility, we propose that local flexibility of HDL facilitates insertion of various plasma factors (LCAT, hepatic lipase, phospholipid and cholesterol ester transfer proteins etc.) whose action displaces a considerable portion of apoA-I and promotes HDL fusion [38, 55–58, 73–75], which is crucial reverse cholesterol transport [61].
Does this imply that low structural stability of HDL necessarily benefits cholesterol removal? We think this is unlikely, since drastic destabilization will adversely affect HDL interactions with its functional partners (e.g. LCAT, CETP, etc.) and promote HDL degradation. Moreover, the rate of HDL turnover depends upon concerted action of many factors which does not necessarily correlate with HDL stability. Thus, structural stability is just one of the important determinants of HDL functionality. We propose that HDL stability must be delicately balanced to maintain the structural integrity of the lipoprotein assembly yet enable its rapid remodeling and turnover at key junctures of reverse cholesterol transport.
Structural adaptation of HDL proteins to the increasing lipid load
Detailed HDL structure remains unknown
Nascent HDL, which are generated in vivo through the interactions of free apoA-I with the plasma membrane and ABCA1 transporter, are generally thought to form discoidal particles ~8 nm in diameter, each containing two copies of apoA-I, although particles of other sizes containing more copies of apoA-I have also been reported [76]. In vivo, these particles are converted into mature spherical HDL with incrementally increasing diameters approaching 12 nm, each containing up to four or five copies of apoA-I [19, 77]. Mid-size HDL can also contain several copies of apoA-II, the second-major HDL protein whose structure and function remain unclear [78]. ApoA-II is particularly strongly associated with mature HDL and can hamper their functional remodeling via the unknown mechanism [51]. As illustrated in Figure 1, HDL maturation and growth can be accompanied by HDL fusion and dissociation of free apoA-I. Finally, selective uptake of core lipids by the SR-BI receptor leads to HDL disruption and dissociation of the remaining apos.
What conformational changes facilitate apoA-I conversion from lipid-free to HDL-bound state? How does apoA-I confer curvature to the otherwise flat phospholipid monolayer on the surface of HDL, which is necessary to encapsulate apolar lipids in its core? What is the structural ensemble of apoA-I on HDLs of various shapes and sizes? These questions are important since apoA-I forms the structural scaffold and the functional ligand on HDL surface, which confers HDL shape and interacts with many HDL-associated proteins [11, 34].
The answers to these questions are unclear because spatial and temporal heterogeneity of HDLs makes them resilient to high-resolution structural studies. However, the wealth of low-resolution information on model and plasma HDL, combined with the two available x-ray crystal structures of free N- or C-terminally truncated apoA-I, allow educated guessing.
Low- and high-resolution structural studies of apoA-I
ApoA-I has long been known to contain a globular α-helical N-terminal domain that opens up on the lipid, and a flexible C-terminal tail that initiates lipid binding and aggregation. Low thermodynamic stability and high aggregating propensity of free apoA-I have complicated its structural studies. Low-resolution x-ray crystal structure of the N-terminally truncated protein, Δ(1-43)apoA-I, solved by Borhani, Brouillette and colleagues, was proposed to represent an HDL-bound protein conformation [79]. This structure revealed an unusual “horseshoe-shape” conformation in which two highly α-helical protein molecules formed an antiparallel dimer. In this dimer, the central 22-mer helical repeat 5 (resides 121-142), as well as the C-terminal repeats 10 (residues 220-241) were in register. This “double-belt” dimer, with its antiparallel helical arrangement and 5/5 and 10/10 repeat register, provided an important starting model for understanding apoA-I conformation on HDL of various shapes and sizes [80].
Protein cross-linking and mass spectrometry studies by Davidson’s team suggested overall conformational similarity for apoA-I on HDL disks and spheres. This lead to the development of the trefoil-tertafoil models proposing how apoA-I can adapt to the surface of discoidal and spherical HDL via minimal structural rearrangements [77, 81].
Despite extensive studies by many teams, several important aspects of the apoA-I structure remained unknown. These included the solution conformation of free apoA-I, as well as the detailed conformations of apoA-I, particularly its N-terminal part, on various HDL. Also, despite significant advances in low-resolution structural studies of apoA-II on HDL [82], the structure-function relationship in apoA-II remained unclear. Thus, low-resolution structural and biophysical studies yielded invaluable information on the overall HDL architecture but fell short of providing a complete picture of the structural ensemble of HDL proteins.
Two recent developments greatly advanced our understanding of HDL structure-function. First, Phillips, Englander and colleagues used hydrogen-deuterium mass spectrometry (HX MS) to characterize full-length human apoA-I in solution and on model discoidal HDL [83, 84]. Second, Mei and Atkinson determined the first atomic-resolution x-ray crystal structure of free human C-terminally truncated apoA-I, Δ(185-243)apoA-I [34]. The hydrophobic flexible C-terminal tail was truncated to facilitate crystallization. This truncation had little effect on the the rest of the protein structure, as evident from the agreement between the crystal structure of Δ(185-243)apoA-I and the HX MS analysis of full-length apoA-I in solution [34, 83].
The crystal structure of Δ(185-243)apoA-I was a true eye-opener that clarified some existing concepts and revealed new important features. The crystallographic dimer forms a semicircular arc punctuated by prolines, with diameter d~11 nm commensurate with that of HDL (Figure 5A). This structure is consistent with the horseshoe-shaped conformation of the N-terminally truncated protein [34, 79], making it crystal-clear how the protein confers curvature to the lipid monolayer on HDL surface. Importantly, the antiparallel helical dimers observed in the crystal structures of the N- and the C-terminally truncated proteins showed many similarities, including 5/5 repeat register (Figure 5), supporting the idea that this register is an inherent property of apoA-I dimer. The structure of Δ(185-243)apoA-I also provided a detailed view of the central opening between the juxtaposed repeats 5, which probably facilitates LCAT insertion during cholesterol esterification [34]. Moreover, this structure helped understand the action of two putative gain-of-function mutants, apoA-I Milano and Paris [85], provided the first structural basis for understanding the misfolding of nearly 20 other mutants that cause amyloid disease in humans [86, 87], and helped understand the structural and functional role of apoA-II on HDL [51].
Figure 5.
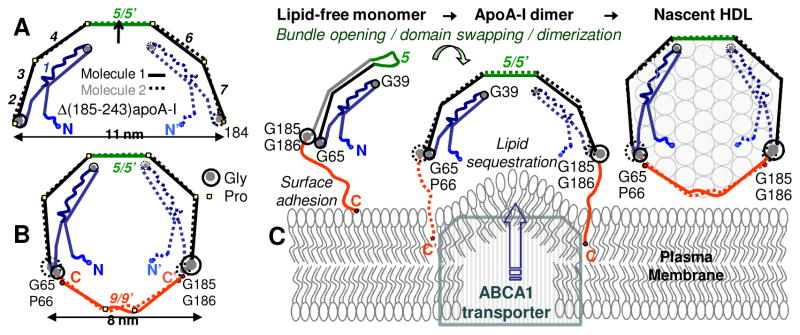
Crystal structure of the C-terminally truncated free apoA-I provides new insights into HDL biogenesis. (A) Cartoon drawn to scale shows x-ray crystal structure of Δ(185-243)apoA-I [34] (PDB ID: 3R2P). Two antiparallel dimer-forming molecules are related via the 2-fold symmetry axis passing through the middle of repeat 5. Proline-induced helical kinks in these two molecules are in register, conferring the semi-circular dimer shape whose curvature is commensurate with that of HDL. Fragment 1-184 contains seven 11/22-mer sequence repeats (numbers in italics). Straight segments represent α-helices. Critical Gly residues are indicated. (B) Proposed conformation of full-length apoA-I on small HDL [88]. The model was derived from the crystal structure in (A) by tightening the Pro-induced kinks, which allows the double-belt closure via the 185-243 fragments (red). (C) Proposed interaction mechanism of full-length apoA-I with the plasma membrane and generation of nascent HDL. Protein dimerization and insertion of the hydrophobic flexible C-terminal tails into the membrane are crucial in this process [34], which is also proposed to involve apoA-I interactions with ABCA1 transporter [68]. Figure modified from [88].
Adaptation of apoA-I structure to HDL of various shapes and sizes
The 2.2Å resolution crystal structure of free Δ(185-243)apoA-I, together with the biophysical information on apoA-I obtained over the years by many groups, provided the starting model for understanding the conformations of the full-length protein in solution and on HDL [34, 88]. For example, the globular helix-bundle conformation of full-length free apoA-I monomer can be obtained from the crystallographic dimer by swapping the polypeptide chain around the flexible repeat 5 [34], with the flexible C-terminal tail loosely folded against the helix bundle [25, 89]. Further, the crystal structure suggests that the flexible C-terminal tails from the two dimer-forming molecules act in concert to insert into the plasma membrane and sequester lipids to form nascent HDL [34, 88]. In the resulting small nascent HDL, two copies of apoA-I probably completely encircle the particle perimeter in an antiparallel double belt (Figure 5C).
This and other structural and biophysical information enabled us to propose the conformational ensemble of apoA-I on various HDLs (Figure 6) [88]. To derive it, we used two basic principles. First, HDL undergoes minimal structural re-arrangement at each step of metabolic remodeling to minimize kinetic barriers modulating these steps. Second, such rearrangements involve conformational changes facilitated by Gly-containing flexible regions: the G56-P66 andG184-G185 hinges, the G39-containing turn, and repeat 5. Further, we assumed that HDL fusion involves incremental elongation of the apoA-I double belt, while HDL rupture and release of core lipids involves disruption of the belt-closing contacts between the N- and the C-termini that probably form the weakest link in the belt [88].
Figure 6.

Hypothetical molecular mechanism of structural adaptation of apoA-I to the increasing lipid load in HDL during reverse cholesterol transport. Constant (66-184, in black) and variable regions (1-66, in blue, and 185-243, in red) are indicated. (A) Small nascent HDL (d~8 nm, 2 copies of apoA-I); (B) small mature HDL (d~8 nm, 2 copies of apoA-I); (C) mid-size mature HDL (d~9.6 nm, 3 copies of apoA-I); (D) large mature HDL (d~11.5 nm, 4 copies of apoA-I). Swing axis that, we propose, connects G65, P66 and G185, G186 hinges is in yellow. Odd copies of apoA-I are proposed to adopt a “double hairpin” conformation (C, bottom). Mid-size human HDL can also carry several copies of apoA-II as their second-major protein [51] (not shown). Figure modified from [88].
Figure 6 illustrates the hypothetical molecular mechanism for apoA-I adaptation to the increasing lipid load. To elongate the belt, the N-terminal helix bundle is proposed to sequentially open around two flexible hinges containing G39 and G65-P66 [34]. We postulate that, to fully close the double belt, this unhinging must be accompanied by incremental change in resister of the belt-closing segments near the molecular termini [88]. One way to accomplish it is via the change in register of the helical repeats, from 9/9 on the small, to 10/10 on the midsize, to G/G on the large HDL (the G-repeat encompasses N-terminal residues) (Figure 6). Currently, only the 10/10 repeat register on midsize HDL has been experimentally established, while the others remain hypothetical [51, 80, 88].
Another important aspect of the proposed mechanism is the conformational adaptation of apoA-I to the surface of discoidal and spherical HDL, which is curved in 1D or 2D, respectively. In our model, this adaptation occurs via the swing motion of the double belt around two hinges, G65-P66 and G185-G186 (Figure 6). These hinges split the molecule neatly into two ~120-residue halves, a constant region containing central residues 66-185 (black) and a variable region containing the N- and C-terminal residues, 1-65 (blue) and 185-243 (red). This swing motion of the two halves of the double-belt is reminiscent of the trefoil/tetrafoil model proposed by Davidson’s team [77]. Although their proposed swing axis was different from ours, the key concept underlying these two independently derived models is very similar. The arguments in favor of the G65, P66 – G185, G186 swing axis and other experimental evidence supporting our model has been outlined elsewhere [88].
An interesting idea proposed in the original trefoil/tertrafoil model is the domain swapping among the double belts [77] whereby a polypeptide chain segment from one apoA-I molecule rotates around the central axis to pair with another copy of apoA-I (Figure 6D, circular arrow). Such a rotation, which is akin to flipping book pages, integrates multiple copies of apoA-I on HDL surface (e.g. 1 and 2, 2 and 3, 3 and 4, 4 and 1) into a single framework.
Despite its esthetic appeal, this idea has several caveats. First, domain swapping can only involve segments of similar shape and size. Our model suggests that this is only possible on the large HDL where apoA-I adopts a fully extended helical conformation, but not on the small or midsize particles where the N-terminal part is folded back (Figure 6). Second, domain swapping of a protein bound to the lipid surface is expected to involve transient disruption of multiple protein and lipid interactions, which is energetically costly. Third, experimental studies showed the presence of two comparable-size populations of apoA-I on HDL, one easily exchangeable and the other tightly bound to the particle [51–54, 90]. The presence of the exchangeable population argues against integration of multiple copies of apoA-I into a single framework via the domain swapping. Thus, we believe that such a domain swapping is unlikely.
We propose that the strongly bound apoA-I is probably the primary double belt that formed the nascent HDL, whereas the loosely bound population contains additional protein that adsorbed to the growing HDL particle at later stages. This “easy come – easy go” population is probably comprised of the monomeric apoA-I that may adopt a double-hairpin conformation (Figure 6C). In fact, compared to the dimer, the apolipoprotein monomer has lower activation energy of dissociation from the lipid surface and hence, is more readily exchangeable than the dimer [44].
The presence of two distinct populations of apoA-I on HDL is probably important for HDL function in normal state and in disease. For example, in acute inflammation, nearly 50% of total apoA-I can be displaced from HDL by serum amyloid A [90]. Intriguingly, this displacement does not greatly alter several functions of acute-phase HDL [90], suggesting that these functions are determined mainly by the non-exchangeable apoA-I. Thus, we posit that the non-exchangeable apoA-I, which is the major determinant of HDL structure and function, represents the original double-belt dimer, whereas the exchangeable apoA-I serves as an additional modulator of HDL metabolism.
The models in Figures 5 and 6 represent useful structure-based starting points for deriving the conformational ensemble of apoA-I on HDL, but do not represent a complete ensemble. For example, they can accommodate but do not explicitly account for the “looped belt” model in which apoA-I segment around residue 140 adopts a dynamic conformation that comes on and off the lipid [91, 92]. Also, our models can accommodate other HDL stoichiometries, including nascent HDL containing three, four or more copies of apoA-I. Figure 7 shows a hypothetical model of one such particle containing four copies of apoA-I organized as two head-to-tail dimers.
Figure 7.

Hypothetical model of a large nascent HDL that contains more than two copies of apoA-I. Protein arrangement around the perimeter of a particle containing four copies of apoA-I in two antiparallel dimers with 5/5′ helix register and head-to-tail dimer packing is illustrated (top view). Other dimer-based models, with N-terminal segments partially folded back, or monomer-based models with apoA-I in a double-hairpin conformation (Fig. 6C, bottom) can also be envisioned.
Possible role of apoA-II in HDL structure and dynamics
ApoA-I and apoA-II comprise 70% and 20% of HDL protein content, respectively, and are the major determinants of HDL structure and function. ApoA-II is an enigmatic protein that modulates reverse cholesterol transport via the unclear mechanisms. Human apoA-II forms a disulfide-linked homodimer that is strongly bound to mature HDL [49, 78, 82]. These HDL are termed HDL(A-I/A-II) as they contain both apoA-I and apoA-II. Although the structural and functional role of apoA-II is subject of debate, the consensus is that apoA-II can hamper metabolic remodeling of HDLs and modulate their metabolism indirectly by influencing the apoA-I conformation [49, 78].
We proposed a structural model explaining why human apoA-II circulates almost exclusively on the mid-size HDL and why it hampers HDL remodeling in vivo [51]. Briefly, in our model of small spherical HDL (Figure 6B), the variable region of the apoA-I double belt provides the 2D curvature and the adequate surface coverage, leaving no room for apoA-II. On midsize HDL (Figure 6C), the variable region of apoA-I swings out of plane of the double belt, vacating the surface for binding additional protein molecules such as apoA-I monomer (Figure 6E) or apoA-II homodimer. The latter consists of two 77-residue molecules disulfide-linked via Cys6, which are probably arranged in an antiparallel double hairpin [82]. The length of the apoA-II homodimer matches best the variable region of apoA-I in the midsize double belt (~85 residues, blue and red segments in Figure 6C)). This explains the preference of apoA-II for the midsize human plasma HDL. Notably, apoA-II is too short to match the fully expanded variable or constant half of apoA-I on large HDL (~120 residues, Figure 6D), which helps explain why apoA-II is not found on large plasma HDL(A-I/A-II). Moreover, this structural mismatch suggests that apoA-II on HDL(A-I/A-II) hampers conformational change in apoA-I, from the relatively compact midsize to the large fully expanded double belt [49].
In sum, our structural model suggests that apoA-II decelerates the remodeling of the midsize HDL(A-I/A-II) into large lipid-loaded particles, which are the preferred substrates for HDL receptor. Therefore, we posit that apoA-II generates a structurally and functionally inert population of plasma HDL(A-I/A-II) with increased lifetime but reduced rate of cholesterol transport [51]. This inert population can be viewed as a buffer that maintains HDL quantity but probably not its quality. This hypothesis is consistent with the observation that humans lacking apoA-II show normal lipoprotein profile and susceptibility to atherosclerosis [93]. The presence of such inert populations of human plasma HDL may contribute to the complex relationship between the plasma levels of “good cholesterol“ and cardioprotection.
Prediction of intrinsic disorder in HDL proteins
Large secondary and/or tertiary structural changes induced in the exchangeable apolipoproteins upon lipid binding, combined with the molten globule-like properties of these proteins in solution [9, 35–37] suggest that free apos are fully or partially intrinsically disordered. Computational analysis supported this idea. Amino acid sequences of selected human apos were analyzed by using two disorder prediction metaservers, D2P2 [94] and Gensilico [95]. The detailed results will be reported elsewhere. Briefly, the results of the two methods were in excellent agreement with each other and indicated increased disorder in apos as compared to typical globular proteins. Thus, human apoA-I and apoA-II were predicted to have global disorder tendency circa 0.4 by GenSilico and have over 50% of disordered residues by PONDR VSL2 [96], which clearly classifies them as intrinsically disordered. Importantly, the disorder prediction results agreed with the structural studies of apos by x-ray crystallography, HX MS and other methods. For example, regions of apoA-I that form the helix bundle in solution were predicted to be largely ordered, while the predicted disorder was concentrated largely in the central part encompassing flexible repeat 5, and in the C-terminal part whose flexibility has been established experimentally [34, 83]. Notably, the amyloidogenic segments in apoA-I were predicted to be ordered, in agreement with our recently proposed molecular mechanism of apoA-I misfolding in amyloidosis [87]. In sum, the disorder prediction methods classify apoA-I and other exchangeable apos as intrinsically disordered, which agrees with high structural flexibility characteristic of this protein family.
Concluding remarks
This review illustrates how combined biophysical and structural approaches help elucidate functional properties of dynamic macromolecular assemblies such as HDL. The kinetic origin of HDL stability, which is critical for maintaining discrete HDL subclasses (Figure 4) and thereby modulating reverse cholesterol transport (Figure 1), is ultimately rooted in HDL structure (Figures 5, 6). We propose that large discrete structural changes in HDL assembly (such as the incremental opening of the apoA-I double belt upon increasing lipid load (Figure 6C, D), or the dissociation of apoA-I monomer and lipoprotein fusion (Figure 3E)) involve large activation energy, Ea, and large free energy barriers, ΔG*. In contrast, continuous structural changes (such as the gradual swing motion of a double-belt segment to accommodate apolar lipids accumulating in HDL core during maturation (Figure 6B)) are expected to proceed smoothly, unencumbered by high kinetic barriers. The combination of the continuous and the discrete conformational changes probably accounts for HDL heterogeneity, with multiple kinetic barriers separating distinct subclasses of plasma HDL varying in their composition and functional properties (Figure 4).
Highlights.
Plasma apolipoproteins are protein constituents of “good” and “bad cholesterol”
Structural flexibility of apolipoproteins is key to their functions in lipid binding and transport
Kinetic barriers modulate lipoprotein stability and functions in vitro and in vivo
Structure-based mechanism of apolipoprotein adaptation to increasing lipid load is proposed
Water-soluble apolipoproteins are predicted and observed to be intrinsically disordered
Acknowledgments
Many studies reviewed here have been carried out by the former or current members of the Gursky laboratory, including Drs. Ranjana Mehta, Sangeeta Benjwal, Madhumita Guha, Xuan Gao, Mengxio Lu, Madhurima Das and, particularly, Dr. Shobini Jayaraman who has been the major driving force behind this research. Special thanks are due to our long-term collaborators, Mr. Donald Gantz and Drs. Xiaohu Mei, David Atkinson, and Haya Herscovitz. The author is indebted to Dr. Vladimir Uversky for useful suggestions and help with computational analysis of the predicted disorder. This work was supported by the National Institutes of Health grant GM067260.
Abbreviations
- apo
apolipoprotein
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
- VLDL
very low-density lipoprotein
- LCAT, lecithin
cholesterol acyltransferase
- CETP
cholesterol ester transfer protein
- PLTP
phospholipid transfer protein
- DMPC
dimyristoyl phosphatidylcholine
- CD
circular dichroism
- HX MS
hydrogen deuterium exchange mass spectrometry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References cited
- 1.Nesto RW. Beyond low-density lipoprotein: addressing the atherogenic lipid triad in type 2 diabetes mellitus and the metabolic syndrome. Am J Cardiovasc Drugs. 2005;5(6):379–387. doi: 10.2165/00129784-200505060-00005. [DOI] [PubMed] [Google Scholar]
- 2.Hegele RA. Plasma lipoproteins: genetic influences and clinical implications. Nat Rev Genet. 2009;10(2):109–121. doi: 10.1038/nrg2481. [DOI] [PubMed] [Google Scholar]
- 3.Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ladu MJ, Reardon C, Van Eldik L, Fagan AM, Bu G, Holtzman D, Getz GS. Lipoproteins in the central nervous system. Ann N Y Acad Sci. 2000;903:167–175. doi: 10.1111/j.1749-6632.2000.tb06365.x. [DOI] [PubMed] [Google Scholar]
- 5.Hauser PS, Ryan RO. Impact of apolipoprotein E on Alzheimer’s disease. Curr Alzheimer Res. 2013;10(8):809–817. doi: 10.2174/15672050113109990156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hatters DM, Howlett GJ. The structural basis for amyloid formation by plasma apolipoproteins: a review. Eur Biophys J. 2002;31(1):2–8. doi: 10.1007/s002490100172. [DOI] [PubMed] [Google Scholar]
- 7.Teoh CL, Griffin MD, Howlett GJ. Apolipoproteins and amyloid fibril formation in atherosclerosis. Protein Cell. 2011;2(2):116–127. doi: 10.1007/s13238-011-1013-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das M, Gursky O. Amyloid-forming properties of human apolipoproteins: sequence analyses and structural insights. Subcell Biochemistry. 2015 doi: 10.1007/978-3-319-17344-3_8. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gursky O, Atkinson D. Thermal unfolding of human high-density apolipoprotein A-1: Implications for a lipid-free molten globular state. Proc Natl Acad Sci USA. 1996;93:2991–2995. doi: 10.1073/pnas.93.7.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gursky O. Apolipoprotein structure and dynamics. Curr Opin Lipidol. 2005;16(3):287–294. doi: 10.1097/01.mol.0000169348.61191.ac. [DOI] [PubMed] [Google Scholar]
- 11.Phillips MC. New insights into the determination of HDL structure by apolipoproteins: Thematic review series: high density lipoprotein structure, function, and metabolism. J Lipid Res. 2013;54(8):2034–2048. doi: 10.1194/jlr.R034025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hevonoja T, Pentikainen MO, Hyvonen MT, Kovanen PT, Ala-Korpela M. Structure of low density lipoprotein (LDL) particles: basis for understanding molecular changes in modified LDL. Biochim Biophys Acta. 2000;1488:189–210. doi: 10.1016/s1388-1981(00)00123-2. [DOI] [PubMed] [Google Scholar]
- 13.Krauss RM. Lipoprotein subfractions and cardiovascular disease risk. Curr Opin Lipidol. 2010;21(4):305–311. doi: 10.1097/MOL.0b013e32833b7756. [DOI] [PubMed] [Google Scholar]
- 14.Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol. 2011;8:222–232. doi: 10.1038/nrcardio.2010.222. [DOI] [PubMed] [Google Scholar]
- 15.Rader DJ, Hovingh GK. HDL and cardiovascular disease. Lancet. 2014;384(9943):618–625. doi: 10.1016/S0140-6736(14)61217-4. [DOI] [PubMed] [Google Scholar]
- 16.Castelli WP, Anderson K, Wilson PW, Levy D. Lipids and risk of coronary heart disease. The Framingham Study. Ann Epidemiol. 1992;2:23–28. doi: 10.1016/1047-2797(92)90033-m. [DOI] [PubMed] [Google Scholar]
- 17.Havel RJ. Triglyceride-rich lipoproteins and plasma lipid transport. Arterioscler Thromb Vasc Biol. 2010;30(1):9–19. doi: 10.1161/ATVBAHA.108.178756. [DOI] [PubMed] [Google Scholar]
- 18.Diffenderfer MR, Schaefer EJ. The composition and metabolism of large and small LDL. Curr Opin Lipidol. 2014;25(3):221–226. doi: 10.1097/MOL.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 19.Rothblat GH, Phillips MC. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol. 2010;21:229–238. doi: 10.1097/mol.0b013e328338472d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenson RS, Brewer HB, Jr, Ansell B, Barter P, Chapman MJ, Heinecke JW, Kontush A, Tall AR, Webb NR. Translation of high-density lipoprotein function into clinical practice: current prospects and future challenges. Circulation. 2013;128(11):1256–1267. doi: 10.1161/CIRCULATIONAHA.113.000962. [DOI] [PubMed] [Google Scholar]
- 21.Gao X, Yuan S. High density lipoproteins-based therapies for cardiovascular disease. J Cardiovasc Dis Res. 2010;1(3):99–103. doi: 10.4103/0975-3583.70898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kingwell BA, Chapman MJ, Kontush A, Miller NE. HDL-targeted therapies: progress, failures and future. Nat Rev Drug Discov. 2014;13(6):445–464. doi: 10.1038/nrd4279. [DOI] [PubMed] [Google Scholar]
- 23.Rosenson RS, Brewer HB, Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR, Yvan-Charvet L. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curtiss LK, Valenta DT, Hime NJ, Rye KA. What is so special about apolipoprotein AI in reverse cholesterol transport? Arterioscler Thromb Vasc Biol. 2006;26:12–19. doi: 10.1161/01.ATV.0000194291.94269.5a. [DOI] [PubMed] [Google Scholar]
- 25.Jayaraman S, Cavigiolio G, Gursky O. Folded functional lipid-poor apolipoprotein A-I obtained by heating of high-density lipoproteins: Relevance to HDL biogenesis. Biochem J. 2012;442(3):703–712. doi: 10.1042/BJ20111831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyazaki O, Ogihara J, Fukamachi I, Kasumi T. Evidence for the presence of lipid-free monomolecular apolipoprotein A-1 in plasma. J Lipid Res. 2014;55(2):214–225. doi: 10.1194/jlr.M041038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Handa D, Kimura H, Oka T, Takechi Y, Okuhira K, Phillips MC, Saito H. Kinetic and thermodynamic analyses for spontaneous exchange between high density lipoprotein-bound and lipid-free apolipoprotein A-I. Biochemistry. 2015;54(4):1123–1131. doi: 10.1021/bi501345j. [DOI] [PubMed] [Google Scholar]
- 28.Lu M, Gursky O. Aggregation and fusion of low-density lipoproteins in vivo and in vitro. (2013) J BioMol Concepts. 2013;4:501–518. doi: 10.1515/bmc-2013-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289(35):24020–24029. doi: 10.1074/jbc.R114.583658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toth PP, Barter PJ, Rosenson RS, Boden WE, Chapman MJ, Cuchel M, D’Agostino RB, Sr, Davidson MH, Davidson WS, Heinecke JW, Karas RH, Kontush A, Krauss RM, Miller M, Rader DJ. High-density lipoproteins: a consensus statement from the National Lipid Association. J Clin Lipidol. 2013;7(5):484–525. doi: 10.1016/j.jacl.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Segrest JP, Garber DW, Brouillette CG, Harvey SC, Anantharamaiah GM. The amphipathic alpha helix: a multifunctional structural motif in plasma apolipoproteins. Adv Protein Chem. 1994;45:303–69. doi: 10.1016/s0065-3233(08)60643-9. [DOI] [PubMed] [Google Scholar]
- 32.Jayaraman S, Gantz DL, Gursky O. Effects of salt on thermal stability of human plasma high-density lipoproteins. Biochemistry. 2006;45:4620–4628. doi: 10.1021/bi0524565. [DOI] [PubMed] [Google Scholar]
- 33.Benjwal S, Jayaraman S, Gursky O. Electrostatic effects on the kinetic stability of model discoidal high-density lipoproteins. Biochemistry. 2005;44:10218–10226. doi: 10.1021/bi050781m. [DOI] [PubMed] [Google Scholar]
- 34.Mei X, Atkinson D. Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem. 2011;286(44):38570–38582. doi: 10.1074/jbc.M111.260422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gursky O, Atkinson D. High- and low-temperature unfolding of human high-density apolipoprotein A-2. Protein Sci. 1996;5 (9):1874–1882. doi: 10.1002/pro.5560050913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gursky O, Atkinson D. Thermodynamic analysis of human plasma apolipoprotein C-1: High-temperature unfolding and low-temperature oligomer dissociation. Biochemistry. 1998;37(5):1283–1291. doi: 10.1021/bi971801q. [DOI] [PubMed] [Google Scholar]
- 37.Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, Weisgraber KH. Apolipoprotein E4 forms a molten globule. A potential basis for its association with disease. J Biol Chem. 2002;277(52):50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- 38.Massey JB, Hickson-Bick DL, Gotto AM, Jr, Pownall HJ. Kinetics of tryptic hydrolysis as a probe of the structure of human plasma apolipoprotein A-II. Biochim Biophys Acta. 1989;999(2):121–127. doi: 10.1016/0167-4838(89)90208-2. [DOI] [PubMed] [Google Scholar]
- 39.Lins L, Piron S, Conrath K, Vanloo B, Brasseur R, Rosseneu M, Baert J, Ruysschaert JM. Enzymatic hydrolysis of reconstituted dimyristoyl phosphatidylcholine - apoA-I complexes. Biochim Biophys Acta. 1993;1151(2):137–142. doi: 10.1016/0005-2736(93)90096-i. [DOI] [PubMed] [Google Scholar]
- 40.Cavigiolio G, Jayaraman S. Proteolysis of apolipoprotein A-I by secretory phospholipase A : A new link between inflammation and atherosclerosis. J Biol Chem. 2014;289(14):10011–10023. doi: 10.1074/jbc.M113.525717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gursky O, Ranjana, Gantz DL. Complex of human apolipoprotein C-1 with phospholipid: Thermodynamic or kinetic stability? Biochemistry. 2002;41:7373–7384. doi: 10.1021/bi025588w. [DOI] [PubMed] [Google Scholar]
- 42.Mehta R, Gantz DL, Gursky O. Effects of mutations on the reconstitution and kinetic stability of discoidal lipoproteins. Biochemistry. 2003;42:4751–4758. doi: 10.1021/bi0341253. [DOI] [PubMed] [Google Scholar]
- 43.Benjwal S, Verma S, Röhm KH, Gursky O. Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Sci. 2006;15:635–639. doi: 10.1110/ps.051917406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jayaraman S, Gantz DL, Gursky O. Kinetic stabilization and fusion of discoidal lipoproteins containing human apoA-2 and DMPC: comparison with apoA-1 and apoC-1. Biophys J. 2005;88:2907–2918. doi: 10.1529/biophysj.104.055921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guha M, Gantz DL, Gursky O. Effect of fatty acyl chain length, unsaturation and pH on the stability of discoidal high-density lipoproteins. J Lipid Res. 2008;49(8):1752–1761. doi: 10.1194/jlr.M800106-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gillard BK, Courtney HS, Massey JB, Pownall HJ. Serum opacity factor unmasks human plasma high-density lipoprotein instability via selective delipidation and apolipoprotein A-I desorption. Biochemistry. 2007;46(45):12968–12978. doi: 10.1021/bi701525w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pownall HJ, Hosken BD, Gillard BK, Higgins CL, Lin HY, Massey JB. Speciation of human plasma high-density lipoprotein (HDL): HDL stability and apolipoprotein A-I partitioning. Biochemistry. 2007;46(25):7449–7459. doi: 10.1021/bi700496w. [DOI] [PubMed] [Google Scholar]
- 48.Mehta R, Gantz DL, Gursky O. Human plasma high-density lipoproteins are stabilized by kinetic factors. J Mol Biol. 2003;328(1):183–192. doi: 10.1016/s0022-2836(03)00155-4. [DOI] [PubMed] [Google Scholar]
- 49.Gao X, Yuan S, Jayaraman S, Gursky O. Differential stability of high-density lipoprotein subclasses: Effects of particle size and protein composition. J Mol Biol. 2009;387(3):628–638. doi: 10.1016/j.jmb.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jayaraman S, Jasuja R, Zakharov M, Gursky O. Pressure perturbation calorimetry of lipoproteins reveals an endothermic transition without detectable volume changes: Implications for apolipoprotein adsorption to phospholipid surface. Biochemistry. 2011;50(19):3919–3927. doi: 10.1021/bi200090y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao X, Yuan S, Jayaraman S, Gursky O. Effect of apolipoprotein A-II on the structure and stability of human high-density lipoprotein: Implications for the role of apoA-II in HDL metabolism. Biochemistry. 2012;51(23):4633–4641. doi: 10.1021/bi300555d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pownall HJ. Remodeling of human plasma lipoproteins by detergent perturbation. Biochemistry. 2005;44(28):9714–9722. doi: 10.1021/bi050729q. [DOI] [PubMed] [Google Scholar]
- 53.Han M, Gillard BK, Courtney HS, Ward K, Rosales C, Khant H, Ludtke SJ, Pownall HJ. Disruption of human plasma high-density lipoproteins by streptococcal serum opacity factor requires labile apolipoprotein A-I. Biochemistry. 2009;48(7):1481–1487. doi: 10.1021/bi802287q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lund-Katz S, Nguyen D, Dhanasekaran P, Kono M, Nickel M, Saito H, Phillips MC. Surface plasmon resonance analysis of the mechanism of binding of apoA-I to high density lipoprotein particles. J Lipid Res. 2010;51(3):606–617. doi: 10.1194/jlr.M002055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korhonen A, Jauhiainen M, Ehnholm C, Kovanen PT, Ala-Korpela M. Remodeling of HDL by phospholipid transfer protein: demonstration of particle fusion by 1H NMR spectroscopy. Biochem Biophys Res Commun. 1998;249(3):910–916. doi: 10.1006/bbrc.1998.9162. [DOI] [PubMed] [Google Scholar]
- 56.Settasatian N, Duong M, Curtiss LK, Ehnholm C, Jauhiainen M, Huuskonen J, Rye KA. The mechanism of the remodeling of high density lipoproteins by phospholipid transfer protein. J Biol Chem. 2001;276(29):26898–26905. doi: 10.1074/jbc.M010708200. [DOI] [PubMed] [Google Scholar]
- 57.Rye KA, Hime NJ, Barter PJ. Evidence that cholesteryl ester transfer protein-mediated reductions in reconstituted high density lipoprotein size involve particle fusion. J Biol Chem. 1997;272(7):3953–3960. doi: 10.1074/jbc.272.7.3953. [DOI] [PubMed] [Google Scholar]
- 58.Clay MA, Pylem DH, Ryem KA, Barter PJ. Formation of spherical, reconstituted high density lipoproteins containing both apolipoproteins A-I and A-II is mediated by lecithin:cholesterol acyltransferase. J Biol Chem. 2000;275(12):9019–9025. doi: 10.1074/jbc.275.12.9019. [DOI] [PubMed] [Google Scholar]
- 59.Pownall HJ. Detergent-mediated phospholipidation of plasma lipoproteins increases HDL cholesterophilicity and cholesterol efflux via SR-BI. Biochemistry. 2006;45(38):11514–11522. doi: 10.1021/bi0608717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84(4):276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 61.Guha M, Gao X, Jayaraman S, Gursky O. Correlation of structural stability with functional remodeling of high-density lipoproteins: The importance of being disordered. Biochemistry. 2008;47:11393–97. doi: 10.1021/bi8014746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jayaraman S, Gantz DL, Gursky O. Structural basis for thermal stability of human low-density lipoprotein. Biochemistry. 2005;44(10):3965–3971. doi: 10.1021/bi047493v. [DOI] [PubMed] [Google Scholar]
- 63.Lu M, Gantz DL, Herscovitz H, Gursky O. Kinetic analysis of thermal stability of human low-density lipoproteins: model for LDL fusion in atherogenesis. J Lipid Res. 2012;53:2175–2185. doi: 10.1194/jlr.M029629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guha M, England CO, Herscovitz H, Gursky O. Thermal transitions in human very low-density lipoprotein: Fusion, rupture and dissociation of HDL-like particles. Biochemistry. 2007;46(20):6043–6049. doi: 10.1021/bi7001532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guha M, Gursky O. Effects of oxidation on structural stability and remodeling of human very low density lipoprotein. Biochemistry. 2010;49(44):9584–9593. doi: 10.1021/bi101391z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanchez-Ruiz JM. Protein kinetic stability. Biophys Chem. 2010;148(1–3):1–15. doi: 10.1016/j.bpc.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 67.Tang C, Oram JF. The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochim Biophys Acta. 2009;1791(7):563–572. doi: 10.1016/j.bbalip.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 68.Vedhachalam C, Ghering AB, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC. ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler. Thromb Vasc Biol. 2007;27(7):1603–1609. doi: 10.1161/ATVBAHA.107.145789. [DOI] [PubMed] [Google Scholar]
- 69.Acharya P, Segall ML, Zaiou M, Morrow J, Weisgraber KH, Phillips MC, Lund-Katz S, Snow J. Comparison of the stabilities and unfolding pathways of human apolipoprotein E isoforms by differential scanning calorimetry and circular dichroism. Biochim Biophys Acta. 2002;1584(1):9–19. doi: 10.1016/s1388-1981(02)00263-9. [DOI] [PubMed] [Google Scholar]
- 70.Davidson WS, Gillotte KL, Lund-Katz S, Johnson WJ, Rothblat GH, Phillips MC. The effect of high density lipoprotein phospholipid acyl chain composition on the efflux of cellular free cholesterol. J Biol Chem. 1995;270(11):5882–5890. doi: 10.1074/jbc.270.11.5882. [DOI] [PubMed] [Google Scholar]
- 71.Davidson WS, Lund-Katz S, Johnson WJ, Anantharamaiah GM, Palgunachari MN, Segrest JP, Rothblat GH, Phillips MC. The influence of apolipoprotein structure on the efflux of cellular free cholesterol to high density lipoprotein. J Biol Chem. 1994;269(37):22975–22982. [PubMed] [Google Scholar]
- 72.Gao X, Jayaraman S, Gursky O. Mild oxidation promotes and advanced oxidation prevents protein dissociation and remodeling of human plasma high-density lipoprotein in vitro. J Mol Biol. 2008;376:997–1007. doi: 10.1016/j.jmb.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Silver ET, Scraba DG, Ryan RO. Lipid transfer particle-induced transformation of human high density lipoprotein into apolipoprotein A-I-deficient low density particles. J Biol Chem. 1990;265(36):22487–22492. [PubMed] [Google Scholar]
- 74.Lusa S, Jauhiainen M, Metso J, Somerharju P, Ehnholm C. The mechanism of human plasma phospholipid transfer protein-induced enlargement of high-density lipoprotein particles: evidence for particle fusion. Biochem J. 1996;313(1):275–282. doi: 10.1042/bj3130275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liang HQ, Rye KA, Barter PJ. Remodelling of reconstituted high density lipoproteins by lecithin: cholesterol acyltransferase. J Lipid Res. 1996;37(9):1962–1970. [PubMed] [Google Scholar]
- 76.Sorci-Thomas MG, Thomas MJ. High density lipoprotein biogenesis, cholesterol efflux, and immune cell function. Arterioscler Thromb Vasc Biol. 2012;32(11):2561–2565. doi: 10.1161/ATVBAHA.112.300135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Silva RA, Huang R, Morris J, Fang J, Gracheva EO, Ren G, Kontush A, Jerome WG, Rye KA, Davidson WS. Structure of apolipoprotein A-I in spherical high density lipoproteins of different sizes. Proc Natl Acad Sci USA. 2008;105:12176–12181. doi: 10.1073/pnas.0803626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gauthamadasa K, Rosales C, Pownall HJ, Macha S, Jerome WG, Huang R, Silva RA. Speciated human high-density lipoprotein protein proximity profiles. Biochemistry. 2010;49(50):10656–10665. doi: 10.1021/bi1015452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-1 suggests a lipid-bound conformation. Proc Natl Acad Sci USA. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural models of human apolipoprotein A-I: A critical analysis and review. Biochim Biophys Acta. 2001;1531:4–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- 81.Huang R, Silva RA, Jerome WG, Kontush A, Chapman MJ, Curtiss LK, Hodges TJ, Davidson WS. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat Struct Mol Biol. 2011;18(4):416–422. doi: 10.1038/nsmb.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Silva RA, Schneeweis LA, Krishnan SC, Zhang X, Axelsen PH, Davidson WS. The structure of apolipoprotein A-II in discoidal high density lipoproteins. J Biol Chem. 2007;282(13):9713–9121. doi: 10.1074/jbc.M610380200. [DOI] [PubMed] [Google Scholar]
- 83.Chetty PS, Mayne L, Lund-Katz S, Stranz D, Englander SW, Phillips MC. Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2009;106:19005–19010. doi: 10.1073/pnas.0909708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sevugan Chetty P, Mayne L, Kan ZY, Lund-Katz S, Englander SW, Phillips MC. Apolipoprotein A-I helical structure and stability in discoidal high-density lipoprotein (HDL) particles by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2012;109(29):11687–1192. doi: 10.1073/pnas.1209305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gursky O, Jones M, Mei X, Segrest J, Atkinson D. Structural basis for distinct functions of the naturally occurring Cys-containing mutants of human apolipoprotein A-I. J Lipid Res. 2013;54(12):3244–3257. doi: 10.1194/jlr.R037911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gursky O, Mei X, Atkinson D. Crystal structure of the C-terminal truncated apolipoprotein A-I sheds new light on the amyloid formation by the N-terminal segment. Biochemistry. 2012;51(1):10–18. doi: 10.1021/bi2017014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Das M, Jayaraman S, Mei X, Atkinson D, Gursky O. Amyloidogenic mutations in human apolipoprotein A-I are not necessarily destabilizing: a common mechanism of apoA-I misfolding in familial amyloidosis and atherosclerosis. FEBS J. 2014;281(11):2525–2542. doi: 10.1111/febs.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gursky O. Crystal structure of Δ(185-243)apoA-I suggests a mechanistic framework for the protein adaptation to the changing lipid load in good cholesterol. From Flatland to Sphereland via Double Belt, Belt-Buckle, Double Hairpin and Trefoil/Tetrafoil. J Mol Biol. 2013;425:1–16. doi: 10.1016/j.jmb.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Segrest JP, Jones MK, Shao B, Heinecke JW. An experimentally robust model of monomeric apolipoprotein A-I created from a chimera of two X-ray structures and molecular dynamics simulations. Biochemistry. 2014;53(48):7625–7640. doi: 10.1021/bi501111j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jahangiri A, de Beer MC, Noffsinger V, Tannock LR, Ramaiah C, Webb NR, van der Westhuyzen DR, de Beer FC. HDL remodeling during the acute phase response. Arterioscler Thromb Vasc Biol. 2009;29(2):261–267. doi: 10.1161/ATVBAHA.108.178681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maiorano JN, Jandacek RJ, Horace EM, Davidson WS. Identification and structural ramifications of a hinge domain in apolipoprotein A-I discoidal high-density lipoproteins of different size. Biochemistry. 2004;43:11717–11726. doi: 10.1021/bi0496642. [DOI] [PubMed] [Google Scholar]
- 92.Martin DD, Budamagunta MS, Ryan RO, Voss JC, Oda MN. Apolipoprotein A-I assumes a “looped belt” conformation on reconstituted high density lipoprotein. J Biol Chem. 2006;281:20418–20426. doi: 10.1074/jbc.M602077200. [DOI] [PubMed] [Google Scholar]
- 93.Deeb SS, Takata K, Peng RL, Kajiyama G, Albers JJ. A splice-junction mutation responsible for familial apolipoprotein A-II deficiency Am. J Hum Genet. 1990;46:822–827. [PMC free article] [PubMed] [Google Scholar]
- 94.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker AK, Gough J. D2P2: database of disordered protein predictions. Nucleic Acids Res. 2013;41:D508–D516. doi: 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kozlowski LP, Bujnicki JM. MetaDisorder: a meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinformatics. 2012;13(1):111. doi: 10.1186/1471-2105-13-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, Obradovic Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol. 2005;3(1):35–60. doi: 10.1142/s0219720005000886. [DOI] [PubMed] [Google Scholar]