

Abstract
Impaired functionality of dendritic cells (DCs) significantly contributes to decreased adaptive immune responses in aged hosts. The expression of MHC-peptide on the DC surface is the critical first step in T cell priming, but few studies have addressed the effect of aging on antigen (Ag) acquisition, processing, and presentation by DCs. Here we show that aged murine DCs were less efficient in the cross-presentation of cell-associated Ag and subsequently in the cross-priming of CD8+ T cells than their young counterparts.
The decreased cross-presentation was associated with a reduction in the frequency of CD8αDCs and merocytic (CD8α−CD11b−)DCs that could endocytose cell-associated Ag, as well as the number and the size of the endocytosed particles in the DC that did internalize cell-associated materials.
Mechanistically, phagocytic capacity has been associated with mitochondrial activity and membrane potential (Δψm). Aged DCs exhibited profound signs of mitochondrial dysfunction, illustrated by lower Δψm, reduced ATP turnover and coupling efficiency, decreased baseline oxidative phosphorylation (OXPHOS), and greater proton leak and ROS production. Mimicking the aged metabolic phenotype in young DCs by pharmacologic manipulation indicated that the reductions in Δψm and ATP impeded the phagocytic capacity while ROS interfered with a later step in the cross-presentation process. Conversely, in vitro scavenging of ROS partially restored cross-presentation by aged DCs.
Together, these data suggest that improvement of aged DC functionality might be feasible in the elderly, by targeting metabolic dysfunction, or its downstream sequelae, thereby opening new avenues for enhancing vaccine efficiency in this population.
Keywords: aging, dendritic cells, metabolism, phagocytosis, antigen presentation, T cell priming
Introduction
Aging has a profound negative impact on the capacity of the immune system to develop efficient effector responses against a vast array of antigens (Ag). It is well established that vaccines are poorly immunogenic in older individuals compared to younger individuals (reviewed in (1)). Similarly, in pre-clinical models, cancer vaccination was less effective in old mice than in young mice, with short lasting and weak T cell responses to tumor-associated Ags (rev. in (2)).
Although age-related intrinsic defects in T cells and B cells, and notably the paucity of naïve T cells in old mice, are critical, it has become clear that alterations within the innate immune system significantly impact the development of adaptive immunity in the aging host. Recently impaired functionality of dendritic cells (DCs) has been identified as a significant contributor in this decreased response (3). Notably, young TCR-transgenic CD4+ and CD8+ T cells exhibited poor expansion in an aged environment in response to their cognate Ags (4, 5). Similarly, the transfer of Ag-pulsed aged DCs into young recipients resulted in the defective induction of Ag-specific endogenous CD8+ T cell responses compared to the transfer of young DCs (6–8).
DCs are a phenotypically and functionally heterogeneous population of leukocytes with distinct functions. Their capacity to activate T cells critically depends on the level of MHC-peptide complexes they display on their surface. High levels of MHC-peptide/TCR interaction have been proposed to lower the T cell’s need for additional costimulation to reach full activation. Several groups have studied the effect of aging on the expression of membrane-associated and soluble costimulatory molecules (5, 6, 9–18), but very few studies have addressed the effect of aging on Ag acquisition, processing, and presentation by DCs. Decreases in phagocytic capacity have been reported in aging murine macrophages and CD11c+ cells as well as DCs generated from mouse bone marrow or human monocytes (4, 9, 19–23). However, the molecular mechanisms that confer this defect in phagocytosis remain completely unknown.
Mechanistically, phagocytic capacity and endosomal trafficking have recently been associated with mitochondrial activity and mitochondrial membrane potential (Δψm) in fibroblasts and bone-marrow derived murine macrophages (24). Park et al showed that uptake of apoptotic cells increased Δψm and ATP, and that genetic or chemical reduction of Δψm and/or ATP levels resulted in accelerated trafficking of phagocytosed materials to low pH organelles (24). Importantly, aging is associated with a decline in mitochondrial function, including decreases in mitochondrial number, mitochondrial DNA, protein levels and protein activity (25, 26), as well as a reduced oxidative phosphorylation capacity (OXPHOS) resulting in decreased ATP levels and increased Reactive Oxygen Species (ROS) production (27, 28). Exploring how the mitochondrial function is affected, and its relationship with decreased DC functionality in aging, is thus of particular interest.
To address these important and unresolved questions, we used a well-established model of cross-presentation (the presentation of exogenous Ag on MHC class I) to determine the capacity of the aged DCs to cross-present cell-associated Ags in vitro and in vivo. We also studied whether age-related changes in mitochondrial functions are linked with DC impaired functionality.
Materials and methods
Mice, cells, and peptides
Young (2–3 months) and aged (16–20 months) C57BL/6J mice were obtained from the NIA aging colony. Mice expressing OVA under the actin promoter (actmOVA) were crossed to the Kb−/− background in our facility. Mice were maintained under specific pathogen-free conditions in accordance with guidelines by the Association for Assessment and Accreditation of Laboratory Animal Care International.
DCs were isolated from spleens of naive mice as described before (29). Briefly, DCs were first enriched by negative selection using anti-biotin-beads and biotinylated Ab to TCR, CD19, IgM, IgD and NKp46, followed by positive selection with CD11c-beads (Miltenyi). Enriched DCs were either used directly or further (sub)sorted by flow cytometry based on their expression of CD11c, CD11b, and CD8α, using CD3, CD19, IgM, IgD, and NKp46 as lineage/dump markers (MoFlow, Coulter, X). Overall DC purity after flow cytometric sorting was ≥ 98% and after bead isolation ≥ 80%. In all studies viability was >95% (7-aminoactinomycin D [7-AAD] staining).
OVA257–264-specific B3Z hybridoma cells were cultured in IMDM supplemented with 10% FCS, 50 μM 2-ME, 2 mM L-glutamine, 20 U/ml penicillin, and 20μg/ml streptomycin (30). OVA257–264 (SIINFEKL) and control peptide LCMV gp33–41 (KAVYNFATC) were obtained from A&A Laboratories (San Diego, CA).
DC characterization, endocytosis, and pH studies
Enriched DCs were analyzed for the expression of CD4, CD8α, CD11b, CD11c, XCR1, CD40, CD80, CD86, CD273, CD274, CD275, Kb, Db, and I-A/E by flow cytometric analysis (Abs/isotype controls: eBioscience/BioLegend, San Diego, CA; using a LSRII, Becton-Dickinson, CA).
For endocytosis studies, enriched DCs were incubated with Cell-Trace Violet-labeled irradiated splenocytes (1:3 ratio) at 4°C and 37°C in the absence or presence of N-acetylL-cysteine (NAC, 0.5mM), Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, 1 nM), and Oligomycin (50nM). After 4–6 hrs DCs were stained with Abs to CD11c, MHCII, CD11b, CD8α, to identify DC populations together with a fixable live/dead staining and analyzed by flow cytometry or ImageStream (Amnis, Seattle, WA) (29, 31, 32). At least 5,000 events were acquired for each condition. Images of fixed cells were analyzed using IDEAS 6.0. The number and size of phagocytosed particles were determined using the spot counting and spot size features after tight masking on the brightfield image to exclude membrane-associated extracellular particles (29, 31, 32). The measure of phagosomal pH over time in enriched DCs was performed as described in Savina et al (33). Briefly, DCs were cultured with dually-labeled beads (pH sensitive FITC and pH insensitive FluoProbes 647) and analyzed at different time points by flow cytometry, using a gating FCS/SSC selective for cells containing one bead. The ratio of the mean fluorescence intensity (MFI) emission between the two dyes was determined and compared with a standard curve (ranging from pH 5.5 to 8).
Metabolism parameters
To determine mitochondrial mass, mitochondrial membrane potential (ΨΔm), and ROS production, enriched and subsorted DCs were stained with CD11c, CD11b, CD8α and combinations of Mitrotracker-Green, Mitotracker-Deep Red, JC1, and H2D-CFDA (Invitrogen). Cellular ATP was determined using an ATP Bioluminescent Assay Kit (Sigma).
For real-time analysis of oxidative phosphorylation, DCs were analyzed using an XF-24 Extracellular Flux Analyzer (Seahorse Bioscience) (34). 5×105 purified DCs were plated in XF Running Buffer (FX media, 10mM Dextrose, 2mM L-glutamax, 1mM sodium pyruvate) per the manufacturer’s instructions to obtain real-time measurements of the oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR). Where indicated, ECAR and/or OCR were analyzed in response to 10μM oligomycin, 4μM FCCP and 1μM rotenone plus 10μM antimycin A (all Sigma-Aldrich).
In vitro cross-presentation by DCs
Flow cytometry sorted DC subtypes or total DCs were incubated with irradiated actmOVA-Kb−/− cells (1500 rad/3×105 cells/well) together with 1 × 105 OVA257–264-specific B3Z hybridoma cells in 96-well U-bottom plates in the presence or absence of NAC (0.5 mM). In parallel experiments DCs were incubated with irradiated actmOVA-Kb−/− in the absence or presence of FCCP (1 nM) or oligomycin (50 nM) for 4 hrs after which the DCs were repurified and cultured with B3Z cells in the absence or presence of NAC (0.5 mM). B3Z activation was determined 24 h later by chlorophenol red-β-D-galactopyrannoside conversion assay (30). OVA257–264-pulsed DCs were used as positive controls.
DC transfer studies
Total DCs from young and aged mice were incubated with irradiated actmOVA-Kb−/− cells as described above followed by sorting of CD8α−CD11b−DCs. 5×105 purified young or aged DCs were (i.v.) transferred into young and aged wt Bl/6 recipients. Seven days after the DC transfer, endogenous Ag-specific CD8+ T cell responses were assessed by intracellular cytokine production after a 5-hr stimulation with OVA257–264 peptide (cognate) or GP33–41 (control) in the presence of brefeldin A. Surface staining and intracellular cytokine staining for IFN-γ, IL-2, and TNF-α were performed using a Cytofix/Cytoperm kit (BD Pharmingen, San Diego, CA) according to the manufacturer’s directions. Capacity for secondary expansion in vitro was determined by stimulating the splenocytes on irradiated MEC.B7.SigOVA cells for 6 days and dividing the absolute number of endogenous Ag-specific CD8+ T cells at the beginning of the culture by the absolute number of endogenous Ag-specific CD8+ T cells at the end of the culture as described before (35, 36).
Statistical analysis
Unless stated otherwise, the data are expressed as means ± SEM. and evaluated using an ANOVA followed by a Dunnett test. A p value of <0.05 was considered statistically significant.
Results
Increased DC frequency but poorer T cell priming capacity in aged mice
To assess the effect of aging on the DCs’ capacity to cross-prime cell-associated Ags, we first analyzed the DC composition in young and aged C57Bl/6 mice. Aged mice contained higher frequencies as well as absolute numbers of total DCs (CD11c+ MHCII+ CD3−CD19−NKp46−) in the spleen, LN, and bone marrow. No differences were observed in the frequencies of splenic pDCs, CD11bDCs, and CD8αDCs. However, DCs with the CD8α−CD11b− (mcDCs) surface phenotype were significantly expanded (fig. 1a–c, and not shown).
Figure 1. Altered DC composition and cross-presenting capacity in aged mice.

Spleens were harvested from young (white bars) and aged (black bars) mice and stained for CD11c, MHCII, CD11b, CD8α, in combination with a lineage stain (CD3, CD19, IgM, IgD, and NKp46) and fixable live/dead stain. A. Representative image of the gating strategy to assess DC numbers and composition. B. Frequency of DC subpopulations within the total splenic DC population. C. Absolute number of DC subpopulations per spleen. Data expressed as mean ± s.e.m. with n=9–11/group.*, p<0.05.
D. Purified young (white bars) and aged (black bars) CD8αDCs, mcDCs, and CD11bDCs were incubated with irradiated actmOVA-Kb−/− cells, resorted and cultured with the OVA257–264-specific B3Z hybridoma. B3Z activation was determined 24 hrs later by CPRG conversion assay. Data from one experiment (of 3) are expressed as mean ± s.e.m. with n=3/group.*, p<0.05. E. Fluorescent intensity if Kb on young and aged DCs. Black circles, Kb; open circles, isotype control. N=4/group.
As these CD8α−CD11b− mcDCs, along with CD8αDCs, have recently been reported to induce strong CD8+ T cell responses to cell-associated Ags in both tumor and autoimmunity settings (29, 32), we assessed the effect of aging on their functionality. We first tested their capacity to cross-present cell-associated OVA in MHC class I, H-2Kb. Although we did not observe significant differences in costimulatory molecule expression between young and aged DCs (fig. S1a) as previously reported (18), we used the OVA257–264-specific B3Z hybridoma that does not require additional costimulation as reporter system for MHCI-peptide density on the DC surface. Young and aged CD8αDCs, mcDCs, and CD11bDCs were first incubated with irradiated actmOVA-Kb−/− cells, purified again and cultured with the OVA257–264-specific B3Z hybridoma. As shown before, CD8αDCs and mcDCs, but not CD11bDCs showed capacity for cross-presentation of cell-associated Ags. Aged CD8αDCs and mcDCs were both less efficient at B3Z activation than their young counterparts (fig. 1d). This was not a result of decreased MHCI expression or viability as both young and aged DC subsets showed comparable MHCI expression (fig. 1e, and S.1b) and in vitro survival for the duration of the experiment (>95%, data not shown).
We next tested whether aging affected the DCs potential for cross-priming in vivo. As mcDC were the most dysregulated in number in old animals, we focused on this subset. McDCs from young and aged mice were purified and cultured with irradiated actmOVA-Kb−/− cells, repurified and transferred into young and aged wt Bl/6 recipients. Transfer of young mcDCs induced a ~4 fold higher frequency and absolute number of endogenous OVA257–264-specific CD8+ T cells than transfer of aged mcDCs into young hosts (fig. 2.a–c). Moreover, young endogenous OVA257–264-specific CD8+ T cells induced by young mcDCs underwent significant expansion upon secondary encounter with Ag in vitro (fig. 2.d). In contrast, young endogenous OVA-specific CD8+ T cells induced by aged mcDCs failed to undergo secondary expansion (fig. 2.d). Similar results were obtained when aged mice received young or aged mcDCs. While overall priming in aged mice was significantly lower than in young mice, transfer of young mcDCs into aged mice induced significantly more endogenous OVA257–264-specific CD8+ T cells than transfer of aged mcDCs (fig. 2.a–c). Importantly, aged endogenous CD8+ T cells primed by young mcDCs retained some of their potential for secondary expansion, while aged endogenous OVA257–264-specific CD8+ T cells primed by aged mcDCs contracted upon secondary encounter with Ag (fig. 2.d). Together these data reveal an intrinsic defect in the aged DCs’ capacity to prime CD8+ T cells to cell-associated Ags.
Figure 2. Decreased cross-priming to cell-associated Ags by aged mcDCs in vivo.
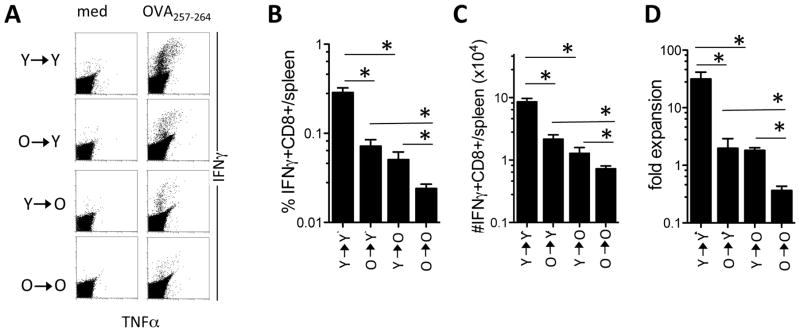
CD11c+CD11b−CD8α− mcDCs from young (Y) and aged (O) mice were isolated and cultured with irradiated actmOVA-Kb−/− cells, repurified and transferred into young and aged recipients. Seven days later the spleens were harvested and CD8+ T cell responses to OVA were examined. A. Representative flow cytometric dot-plot of CD8+ T cells producing IFNγ and TNFα upon stimulation with medium or OVA257–264 peptide. B. The frequency of OVA257–264 specific CD8+ T cells per spleen. C. The absolute number of OVA257–264 specific CD8+ T cells per spleen. D. Capacity for secondary expansion in vitro of CD8+ T cells primed by young or aged mcDC in young and aged recipients. CD8+ T cells were restimulated on irradiated MEC.B7.SigOVA cells and the fold expansion was determined by dividing the absolute number of Ag-specific CD8+ T cells at the beginning of the culture by the absolute number of Ag-specific CD8+ T cells at the end of the culture. Data from one experiment (of 2) are expressed as mean ± s.e.m. with n=4–5/group.*, p<0.05.
Altered endocytosis of cell-associated Ags in aged DCs
To identify the underlying mechanisms for this decreased cross-priming, we first determined whether aging affected uptake of cell-associated Ags. Young and aged DCs were cultured with Cell-Trace Violet labeled irradiated splenocytes and the endocytic process for each DC population was analyzed using classic and imaging flow cytometry (29).
Aged CD8αDCs and mcDCs were less efficient at endocytosing irradiated cells than their young counterparts, as determined by flow cytometry (fig. 3a). Detailed analysis by imaging flow cytometry showed that aging also reduced the number and the size of the endocytosed particles in the DC (fig. 3b–d). Indeed, the majority (>75%) of endocytosing young CD8αDCs and mcDCs contained more than 2 particles, with approximately 45% of the DCs containing more than 4 particles (fig. 3c). In contrast, less than half of the aged CD8αDCs and mcDCs contained more than 2 particles, and less than 15% contained more than 4 particles. In addition, the size of endocytosed particles in the young DCs was approximately double of those in the aging DCs (fig. 3d). As we had shown before, CD11bDC were poorly endocytic (fig. 3.a–c item (iii)) (37), but again, the size of the endocytosed particles by these aged CD11bDC was significantly smaller than those in young CD11bDC (fig. 3d).
Figure 3. Aging reduces phagocytic capacity in DCs.

Purified DCs from young (white bars/circles) and aged mice (black bars/circles) were cultured with Cell-Trace Violet-labeled irradiated splenocytes. After 4 hrs DCs were stained with Abs to CD11c, MHCII, CD11b, CD8α, and a fixable live/dead dye. CD8αDC (i), mcDC (ii), and CD11bDC (iii) were analyzed by flow cytometry (A) or imaging flow cytometry (B–D). A. Flow cytometric analysis of the frequency of phagocytic cells in each DC population. Data are expressed as mean ± s.e.m. with n=3/group. B. Representative imaging flow cytometry images of different DC populations upon co-culture with irradiated cells. BF = Bright field, Blue = CD11c, yellow = CD8, red = CD11b, green = irradiated cell material. C. Number of particles per DC within the phagocytosing DC population. The number of phagocytosed particles was determined using the spot counting features after tight masking on the brightfield image to exclude membrane-associated extracellular particles. D. Size distribution of phagocytosed particles with the Cell-Trace Violet+ DC populations. Particle size was determined using a combination of the spot mask, combined with spot size feature and area feature. Representative data of 3 independent experiments are shown. Each condition contained 500–1000 viable, focused DCs.
Aging alters DC metabolism
Endocytic capacity has recently been associated with mitochondrial activity and mitochondrial membrane potential (Δψm) in bone-marrow-derived murine macrophages and fibroblasts (24). Since aging has been associated with impaired mitochondrial function and decreased Δψm (25, 27, 38–41), we explored whether decreased mitochondrial function could be the mechanism underlying the decreased endocytic capacity in aged DCs.
We first used Seahorse FX24 analyses to assess mitochondrial function and cellular respiration in purified young and aged DCs. Young DCs showed higher baseline oxygen consumption rates (OCR) than aged DCs (fig. 4.a, c). Basal respiration is strongly controlled by ATP turnover and partly by substrate oxidation and proton leak. To dissect which of these 3 processes was altered in aged DCs, we sequentially added the ATP synthase inhibitor oligomycin to assess ATP turnover and proton leak (“b” and “c”, respectively in fig. 4b), then the uncoupler FCCP to assess maximal respiration (“d” in fig. 4b), and finally antimycin/rotenone to assess the non-mitochondrial OCR rate (“e” in fig. 4b). The non-mitochondrial OCR rate was comparable between young and aged DCs (fig. 4a, young, 134.8 ± 17.2: aged, 124.1 ± 14.7) and these values were subtracted from all other values to strictly assess mitochondrial functions.
Figure 4. Reduced mitochondrial functionality in aged DCs.

Young (white circles/bars) and aged (black circles/bars) DCs were probed for their mitochondrial function. A. DCs were seeded into Seahorse Bioscience plates and oxygen consumption rates (OCR) were determined under basal conditions followed by the sequential addition of oligomycin, FCCP, and antimycin/rotenone. A representative experiment (of 3) is shown. Data are expressed as mean ± s.e.m. with n=2 (Y) or 3 (O).*, p<0.05. B. Scheme outlining the approach to identify parameters for the calculation of the relative contribution of non-respiratory chain oxygen consumption, ATP-linked oxygen consumption, proton-leak, and coupling efficiency. C. Decreased baseline OCR, ATP-linked OCR and coupling efficiency and increased proton leak in added DCs. D. Flow cytometric analysis of mitochondrial mass, mitochondrial membrane potential (Δψm) and ROS production at steady state in young and aged DCs. A representative experiment (of 3) is shown. Data is expressed as mean ± s.e.m. with n=3.*, p<0.05.
Addition of oligomycin resulted in a significantly greater reduction of OCR in young DCs compared to aged DCs (“b” values in fig. 4c), indicating decreased ATP turnover in aged DCs. Moreover, the coupling efficiency, i.e. the fraction of basal mitochondrial oxygen consumption used for ATP synthesis (“b/a” values in fig. 4c), was significantly higher in young DCs. In addition, aged DCs showed a greater oligomycin-insensitive respiration (proton leak, “c” in fig. 4c) than young DCs. The decreased ATP turnover and lower coupling efficiency suggested a deficiency in the ATP synthesis machinery of the aged DCs.
Using flow cytometry, we next assessed the mitochondrial mass and Δψm in the different subsets of young and aged DCs. Mitotracker Green staining intensity was similar between young and aged DCs, for all subsets, suggesting comparable total mitochondrial mass in the young and aged DCs (fig. 4d). We next stained DCs with JC-1, a lipophilic cationic dye that can selectively enter into mitochondria and reversibly change color as the mitochondrial membrane potential (Δψm) increases. As shown in fig. 4d, all subsets of aged DCs showed significantly decreased Δψm. In addition, all subsets of aged DCs had increased ROS levels (fig. 4d), further suggesting mitochondrial dysfunction.
Genetic or chemical reduction of Δψm and/or ATP levels was recently shown to accelerate trafficking of phagocytosed materials to low pH organelles in the LR73 cell line (24). Accelerated acidification in DCs would significantly reduce the amount of Ag available for MHCI loading and could therefore reduce cross-presentation. To determine whether the decrease in endocytosed material in aged DCs resulted from accelerated degradation of the fluorescently-labeled cellular material, we assessed the endosomal acidification rate in young and aged DC subsets using beads that were dual-labeled with a pH sensitive and pH resistant dye (fig. S2) (33). While there were large differences in endosomal acidification rates between DC subsets, no differences between young and aged DCs were found, suggesting that increased degradation was not the dominant process reducing the Ag availability in aging DCs.
Decreases in mitochondrial functionality reduce cross-presentation by young DCs
We next tested whether creating an “aged” phenotype in young DCs by reducing ATP, Δψm or increasing ROS were sufficient to affect their phagocytic and T cell priming capacity. Young DCs were incubated with Cell-Trace Violet labeled irradiated actmOVA-Kb−/− cells in the absence or presence of combinations of chemicals that affect distinct aspects of mitochondrial function. We used low concentrations of FCCP (reduces Δψm, increases ROS, little effect on ATP), or oligomycin (little effect on Δψm, increases ROS, inhibition of ATP). As shown in fig. S2, these drugs had the expected effect in the young DCs. They were used alone or in combination with N-acetyl-L-cysteine (NAC, scavenges ROS, no effect on Δψm or ATP, fig. S3). Endocytosis (fig. 5a) and cell viability (fig. S3) was assessed 4 hrs later. Both FCCP and oligomycin used alone significantly reduced the phagocytic capacity of CD8αDCs and mcDCs (black bars). NAC alone during the endocytic period did not alter the DC phagocytic activity nor did it alter the inhibitory effects of FCCP and oligomycin on this phagocytic capacity (white bars vs black bars). Together these data indicate that Δψm and ATP, but not ROS, are important for the endocytic process (fig. 5a).
Figure 5. Distinct roles for ATP, Δψm and ROS in endocytosis and cross-presentation.
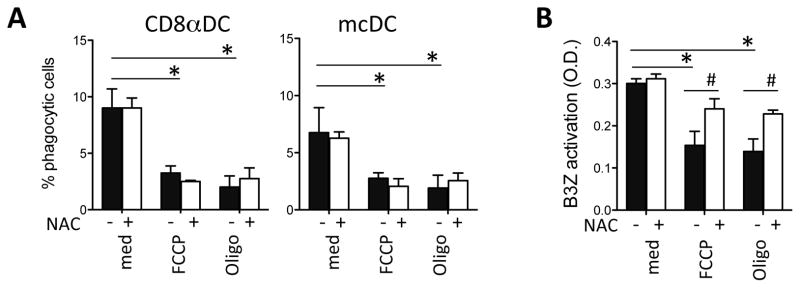
A. Young DCs were cultured with Cell-Trace Violet-labeled irradiated actmOVA-Kb−/− cells in the presence of vehicle, FCCP, or oligomycin in the absence (black bars) or presence of ROS scavenger NAC (white bars). After 4 hr DCs were stained with Abs to CD11c, MHCII, CD11b, CD8α, and live/dead staining and the frequency of Cell-Trace Violet+ DCs was determined by flow cytometry. B. Young DCs were cultured with irradiated actmOVA-Kb−/− cells in the presence of vehicle, FCCP, or oligomycin and repurified before being cultured with the OVA257–264-specific B3Z hybridoma in the absence (black bars) or presence (white bars) of NAC. B3Z activation was determined 24 h later by CPRG conversion assay. Data are expressed as mean ± s.e.m. with n=3/group.*, p<0.05.
To determine the effect of the same drugs on the young DCs cross-presentation, young DCs were incubated with irradiated actmOVA-Kb−/− cells in the presence of FCCP or oligomycin, followed by sorting and coculture with the B3Z cells in the absence or presence of NAC. As expected due to their inhibitory effects on endocytosis, FCCP and oligomycin-treated young DCs showed poorer capacity to activate B3Z cells than untreated DCs. Interestingly, addition of NAC to the FCCP and oligomycin-treated DCs partly restored their ability to activate B3Z cells (fig. 5b). These data suggest that in conditions where ROS generation is augmented, such as in aged DCs, DC cross-presentation is impaired, although not through impaired endocytosis.
Scavenging of ROS partially restores cross-presentation by aged DCs
We next tested whether ROS scavenging could improve the poor cross-presenting capacity of aged DCs. Addition of NAC to cultures of young DCs did not significantly improve endocytosis, DC survival, or the B3Z response to irradiated actmOVA-Kb−/− cells (not shown). Similarly, NAC did not enhance endocytosis or survival in aged DCs (fig. 6a). However, addition of NAC to aged DCs significantly improved the B3Z response (fig. 6b). As B3Z cells are relatively resistant to oxidative stress (not shown), these data imply that the inhibitory effect of ROS on cross-presentation resulted from a direct effect on the DCs.
Figure 6. ROS scavenging partially restores cross-presentation in aged DCs.

Purified DCs from and aged mice were cultured with Cell-Trace Violet-labeled irradiated actmOVA-Kb−/− cells. A. Flow cytometric analysis of the frequency of phagocytic cells in each DC population after 4h of co-culture. Data are expressed as mean ± s.e.m. with n=3/group. B. OVA257–264-specific B3Z hybridoma response to aged DCs in the absence (white circles) or presence (black circles) of ROS scavenger NAC. B3Z activation was determined after 24 h by CPRG conversion assay. Data of 7 individual DC sorts are shown. *, p<0.05.
Discussion
Aging has been shown to reduce DC functionality but the underlying mechanisms are poorly understood. Here we show that aging affected DC numbers as well as DC subpopulation composition. Moreover, aging significantly reduced the DCs’ capacity to phagocytose and cross-present cell-associated Ags and this impairment was associated with decreased ATP production, decreased Δψm, and increased ROS production. Our data further indicate that decreased ATP and Δψm conferred the defect in phagocytic capacity while ROS impaired DCs at a later phase of the cross-presentation process.
Although various groups have studied DCs in aging, there is little consensus about the impact of aging on DCs numbers, composition, and function. Moreover, it is currently unclear whether the observed changes result from intrinsic defects in the DCs or its precursors, extrinsic factors associated with the aged environment, or a combination of both. Several studies reported normal DC numbers and subset composition in lymphoid tissues from young and aged mice, while others suggest that aging decreases CD8α+DCs and increases CD8α− DC(4, 14, 15). Our data are in partial agreement with these latter studies, as we found the total number and frequency of splenic DCs to be increased, predominantly due to increased CD8α−CD11b− DCs. These discrepancies likely arise from the study of different background strains, as well as differences in staining and analysis strategies (3). The increase of the CD8α−CD11b− DCs is of high interest as this population has significant phenotypic and functional overlap with mouse CD8αDCs in young animals, as well as having a human equivalent (BDCA3+ DCs) that is the most potent at priming T cells to cell-associated Ags (42–46). Consequently, CD8αDCs and mcDCs are very potent inducers of anti-tumor responses, but can also play a role in autoimmunity, as both processes are driven by cross-presentation of cell-associated self-Ags (29, 32, 47, 48). However, despite their functional importance, nothing is currently known how aging affects their functionality, and particularly how they cross-present cell-associated Ags.
Cross-presentation of cell-associated Ags is a coordinated process, starting with the recognition of cell-associated materials and their subsequent internalization into endosomes that eventually fuse with acidic lysosomes where the materials are degraded. To facilitate cross-presentation, DCs need to actively delay endosomal acidification in order to allow the transport of the endocytosed Ags from endosomal vesicles into the cytosol, where they are processed by the proteasome and loaded on MHC class I molecules in the endoplasmic reticulum (49).
Our data indicate that aging affects multiple aspects of the cross-presentation process. Indeed, we found that aging significantly reduced the endocytic capacity of CD8DCs and mcDCs, resulting in a reduced frequency of phagocytosing DCs as well as a reduction in the number and size of the endocytosed particles. Importantly, our data suggest that reduced endocytosis resulted from the disruption of the active internalization process and not from defective binding/recognition, as tethering of cellular materials was similar in aged and young DCs. Age-associated attenuation of phagocytosis has been reported for a variety of cell types, including primary and in vitro generated macrophages and DCs. Importantly, our data strongly suggest that the age-associated endocytic defect is related to decreased ATP production and a lower Δψm in aged DCs, which, to our knowledge, has never been reported.
Phagocytosis requires ATP for actin polymerization and activation of myosin motor proteins that drive all stages of endocytosis, including ruffle formation, membrane delivery, closure of the phagocytic cup, as well as short range movement of newly formed vesicles (50). Active phagocytosis is therefore often considered to be an ATP drain that significantly taxes cellular energy (51). In agreement with these previous data, our studies show that young DCs with chemically reduced ATP levels (similar to levels found in aged DCs) showed significantly reduced phagocytic capacity, thus suggesting a similar mechanism underlying deficient phagocytic capacity by aged DCs.
Besides a role for ATP, we also found a relation between lower Δψm and reductions in phagocytic capacity. Park et al (24) showed that Δψm increased after uptake of apoptotic cells in BM-derived macrophages without detectable effects on total cellular ATP production. How changes in Δψm affect phagocytic capacity remains unclear, but some studies suggest that changes in Δψm may alter mitochondrial Ca2+ accumulation and mobilization, thereby affecting phagocytosis, which is a Ca2+-sensitive process (52–54). While this is an intriguing hypothesis, especially in light of the dysregulated Ca2+ homeostasis in aged cells (55) and the possible suppression of Δψm by high levels of Ca2+, more research is needed to causally link these different phenomena.
Another important finding of our studies is that aged DCs expressed significantly higher levels of ROS than young DCs, and that in vitro ROS scavenging significantly improved cross-presentation by aged DCs. ROS has an important role as a secondary messenger in several signaling pathways, but if the production exceeds its deactivation it leads to oxidative damage to proteins, lipids, and nucleic acids (28, 33, 56, 57). How ROS affect cross-presentation in aged DCs is not completely clear. Our data show that ROS scavenging did not improve phagocytic capacity in aged DCs or young DCs. Moreover, increased ROS levels in aged DCs did not affect early endosome trafficking and lysosomal fusion, as there were no differences in endosomal acidification rate between young and aged DCs. These findings thus suggest that the ROS affects later steps in the Ag processing/presentation pathway in aged DCs. A possible step that ROS could affect is endosomal acidification, as suggested by the fact that sustained low production of ROS at the endosomal lumen has been shown to prevent endosomal acidification and promote cross-presentation (33). However, in our experiments, scavenging of ROS by NAC did not alter the acidification rate or cross-presentation by young DCs, which is in agreement with our previous findings (29). It is therefore likely that overproduction of ROS in aged DCs directly damages molecules involved in the cross-presentation pathway. Further studies aimed at the dissection of the molecular processes that drive mitochondrial dysfunction, and their differential impact on the process of cross-presentation, are thus warranted. Furthermore, the ability of ROS scavengers such as NAC to restore the cross presentation ability in aged mice will need to be rigorously tested in an in vivo setting of cross priming, to test whether this avenue could be targeted to increase immune responses in aged individuals.
Together, our study shows that different components associated with age-related mitochondrial dysfunction – reduced ATP synthesis, reduced Δψm and increased ROS production – have specific deleterious effects on the cross-presenting capacity of aging DCs. Cross-presentation is only the first step in the cross-priming of cells to cell-associated Ags. Importantly, high levels of MHC-peptide can lower the requirement of costimulation by lowering the threshold for T cell activation (58). Given that aging has been reported to negatively affect expression of costimulatory molecules and pro-inflammatory cytokines (3, 4, 7, 14, 59), lowering the threshold for T cell activation by increasing MHCI-peptide levels is of clear interest. While more research is needed to dissect how aging affect DC-T cell communication, our data thus suggest that improvement of DC functionality might be feasible in the elderly, by targeting metabolic dysfunction, or its downstream sequelae, thereby opening new avenues for enhancing vaccine efficiency in this fragile population.
Supplementary Material
Acknowledgments
We are grateful to Dr F. Finkelman for comments and discussion on the manuscript; A. White and M. DeLay for support with the Amnis Imagestream; and the Research Flow Cytometry Core at Cincinnati Children’s Hospital Medical Center.
Funding information
This work was supported by the National Institutes of Health via CA138617 (E.M.J.), AG033057 (C.A.C.), as well as the Charlotte Schmidlapp Award (E.M.J.) and a Postdoctoral Research Grant from the Ellison Medical Foundation/AFAR (C.S.L.).
Abbreviations
- Ag
antigen
- ATP
adenosine triphosphate
- DC
dendritic cell
- Δψm
mitochondrial membrane potential
- ECAR
extracellular acidification rate
- FCCP
carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- mcDC
merocytic DC
- MFI
mean fluorescent intensity
- NAC
N-acetyl-L-cysteine
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- ROS
reactive oxygen species
- TCR
T cell receptor
References
- 1.Lefebvre JS, Haynes L. Vaccine strategies to enhance immune responses in the aged. Current opinion in immunology. 2013;25:523–528. doi: 10.1016/j.coi.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gravekamp C, Jahangir A. Is cancer vaccination feasible at older age? Experimental gerontology. 2014;54:138–144. doi: 10.1016/j.exger.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong C, Goldstein DR. Impact of aging on antigen presentation cell function of dendritic cells. Current opinion in immunology. 2013;25:535–541. doi: 10.1016/j.coi.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pereira LF, de Souza AP, Borges TJ, Bonorino C. Impaired in vivo CD4+ T cell expansion and differentiation in aged mice is not solely due to T cell defects: decreased stimulation by aged dendritic cells. Mechanisms of ageing and development. 2011;132:187–194. doi: 10.1016/j.mad.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Li G, Smithey MJ, Rudd BD, Nikolich-Zugich J. Age-associated alterations in CD8alpha+ dendritic cells impair CD8 T-cell expansion in response to an intracellular bacterium. Aging cell. 2012;11:968–977. doi: 10.1111/j.1474-9726.2012.00867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grolleau-Julius A, Garg MR, Mo R, Stoolman LL, Yung RL. Effect of aging on bone marrow-derived murine CD11c+CD4−CD8alpha− dendritic cell function. The journals of gerontology Series A, Biological sciences and medical sciences. 2006;61:1039–1047. doi: 10.1093/gerona/61.10.1039. [DOI] [PubMed] [Google Scholar]
- 7.Grolleau-Julius A, Harning EK, Abernathy LM, Yung RL. Impaired dendritic cell function in aging leads to defective antitumor immunity. Cancer research. 2008;68:6341–6349. doi: 10.1158/0008-5472.CAN-07-5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grolleau-Julius A, Abernathy L, Harning E, Yung RL. Mechanisms of murine dendritic cell antitumor dysfunction in aging. Cancer immunology, immunotherapy: CII. 2009;58:1935–1939. doi: 10.1007/s00262-008-0636-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agrawal A, Agrawal S, Cao JN, Su H, Osann K, Gupta S. Altered innate immune functioning of dendritic cells in elderly humans: a role of phosphoinositide 3-kinase-signaling pathway. J Immunol. 2007;178:6912–6922. doi: 10.4049/jimmunol.178.11.6912. [DOI] [PubMed] [Google Scholar]
- 10.Agrawal A, Sridharan A, Prakash S, Agrawal H. Dendritic cells and aging: consequences for autoimmunity. Expert review of clinical immunology. 2012;8:73–80. doi: 10.1586/eci.11.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panda A, Qian F, Mohanty S, van Duin D, Newman FK, Zhang L, Chen S, Towle V, Belshe RB, Fikrig E, Allore HG, Montgomery RR, Shaw AC. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J Immunol. 2010;184:2518–2527. doi: 10.4049/jimmunol.0901022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciaramella A, Spalletta G, Bizzoni F, Salani F, Caltagirone C, Bossu P. Effect of age on surface molecules and cytokine expression in human dendritic cells. Cellular immunology. 2011;269:82–89. doi: 10.1016/j.cellimm.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 13.Stout-Delgado HW, Yang X, Walker WE, Tesar BM, Goldstein DR. Aging impairs IFN regulatory factor 7 up-regulation in plasmacytoid dendritic cells during TLR9 activation. J Immunol. 2008;181:6747–6756. doi: 10.4049/jimmunol.181.10.6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong CP, Magnusson KR, Ho E. Aging is associated with altered dendritic cells subset distribution and impaired proinflammatory cytokine production. Experimental gerontology. 2010;45:163–169. doi: 10.1016/j.exger.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Tan SY, Cavanagh LL, d’Advigor W, Shackel N, Fazekas de St Groth B, Weninger W. Phenotype and functions of conventional dendritic cells are not compromised in aged mice. Immunology and cell biology. 2012;90:722–732. doi: 10.1038/icb.2011.104. [DOI] [PubMed] [Google Scholar]
- 16.El Mezayen R, El Gazzar M, Myer R, High KP. Aging-dependent upregulation of IL-23p19 gene expression in dendritic cells is associated with differential transcription factor binding and histone modifications. Aging cell. 2009;8:553–565. doi: 10.1111/j.1474-9726.2009.00502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jing Y, Shaheen E, Drake RR, Chen N, Gravenstein S, Deng Y. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Human immunology. 2009;70:777–784. doi: 10.1016/j.humimm.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lages CS, Lewkowich I, Sproles A, Wills-Karp M, Chougnet C. Partial restoration of T-cell function in aged mice by in vitro blockade of the PD-1/PD-L1 pathway. Aging cell. 2010;9:785–798. doi: 10.1111/j.1474-9726.2010.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Current opinion in immunology. 2010;22:507–513. doi: 10.1016/j.coi.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sebastian C, Espia M, Serra M, Celada A, Lloberas J. MacrophAging: a cellular and molecular review. Immunobiology. 2005;210:121–126. doi: 10.1016/j.imbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 21.De La Fuente M. Changes in the macrophage function with aging. Comparative biochemistry and physiology A, Comparative physiology. 1985;81:935–938. doi: 10.1016/0300-9629(85)90933-8. [DOI] [PubMed] [Google Scholar]
- 22.Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J, Sambhara S. Innate immunity in aging: impact on macrophage function. Aging cell. 2004;3:161–167. doi: 10.1111/j.1474-9728.2004.00102.x. [DOI] [PubMed] [Google Scholar]
- 23.Linehan E, Dombrowski Y, Snoddy R, Fallon PG, Kissenpfennig A, Fitzgerald DC. Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging cell. 2014;13:699–708. doi: 10.1111/acel.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, Collins S, Lysiak JJ, Hoehn KL, Ravichandran KS. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature. 2011;477:220–224. doi: 10.1038/nature10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker BM, Haynes CM. Mitochondrial protein quality control during biogenesis and aging. Trends in biochemical sciences. 2011;36:254–261. doi: 10.1016/j.tibs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Bratic A, Larsson NG. The role of mitochondria in aging. The Journal of clinical investigation. 2013;123:951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei YH, Wu SB, Ma YS, Lee HC. Respiratory function decline and DNA mutation in mitochondria, oxidative stress and altered gene expression during aging. Chang Gung medical journal. 2009;32:113–132. [PubMed] [Google Scholar]
- 28.Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-ros crosstalk in the control of cell death and aging. Journal of signal transduction. 2012;2012:329635. doi: 10.1155/2012/329635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reboulet RA, Hennies CM, Garcia Z, Nierkens S, Janssen EM. Prolonged antigen storage endows merocytic dendritic cells with enhanced capacity to prime anti-tumor responses in tumor-bearing mice. J Immunol. 2010;185:3337–3347. doi: 10.4049/jimmunol.1001619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thacker RI, Janssen EM. Cross-presentation of cell-associated antigens by mouse splenic dendritic cell populations. Frontiers in immunology. 2012;3:41. doi: 10.3389/fimmu.2012.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz JD, Ondr JK, Opoka RJ, Garcia Z, Janssen EM. Cutting edge: merocytic dendritic cells break T cell tolerance to beta cell antigens in nonobese diabetic mouse diabetes. J Immunol. 2010;185:1999–2003. doi: 10.4049/jimmunol.1001398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, Seabra MC, Raposo G, Amigorena S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 34.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, Zou L, Chatham JC, Hill BG, Zhang J, Landar A, Darley-Usmar VM. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free radical biology & medicine. 2011;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 36.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 37.Janssen E, Tabeta K, Barnes MJ, Rutschmann S, McBride S, Bahjat KS, Schoenberger SP, Theofilopoulos AN, Beutler B, Hoebe K. Efficient T cell activation via a Toll-Interleukin 1 Receptor-independent pathway. Immunity. 2006;24:787–799. doi: 10.1016/j.immuni.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 38.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. American journal of physiology Cell physiology. 2007;292:C670–686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 39.Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-ros crosstalk in the control of cell death and aging. J Signal Transduct. 2011;2012:329635. doi: 10.1155/2012/329635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicholls DG. Mitochondrial membrane potential and aging. Aging cell. 2004;3:35–40. doi: 10.1111/j.1474-9728.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 41.Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. Journal of cell science. 2010;123:2533–2542. doi: 10.1242/jcs.070490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chow A, Brown BD, Merad M. Studying the mononuclear phagocyte system in the molecular age. Nature reviews Immunology. 2011;11:788–798. doi: 10.1038/nri3087. [DOI] [PubMed] [Google Scholar]
- 43.Crozat K, Guiton R, Guilliams M, Henri S, Baranek T, Schwartz-Cornil I, Malissen B, Dalod M. Comparative genomics as a tool to reveal functional equivalences between human and mouse dendritic cell subsets. Immunological reviews. 2010;234:177–198. doi: 10.1111/j.0105-2896.2009.00868.x. [DOI] [PubMed] [Google Scholar]
- 44.Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V, Vulink AJ, Hart DN, Radford KJ. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. The Journal of experimental medicine. 2010;207:1247–1260. doi: 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robbins SH, Walzer T, Dembele D, Thibault C, Defays A, Bessou G, Xu H, Vivier E, Sellars M, Pierre P, Sharp FR, Chan S, Kastner P, Dalod M. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome biology. 2008;9:R17. doi: 10.1186/gb-2008-9-1-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity. 2008;29:352–361. doi: 10.1016/j.immuni.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 47.Morel PA. Dendritic cell subsets in type 1 diabetes: friend or foe? Frontiers in immunology. 2013;4:415. doi: 10.3389/fimmu.2013.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDonnell AM, Robinson BW, Currie AJ. Tumor antigen cross-presentation and the dendritic cell: where it all begins? Clinical & developmental immunology. 2010;2010:539519. doi: 10.1155/2010/539519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nature reviews Immunology. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 50.Kuiper JW, Pluk H, Oerlemans F, van Leeuwen FN, de Lange F, Fransen J, Wieringa B. Creatine kinase-mediated ATP supply fuels actin-based events in phagocytosis. PLoS biology. 2008;6:e51. doi: 10.1371/journal.pbio.0060051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guminska M, Ptak W, Zembala M. Macrophage metabolism during phagocytosis and digestion of normal and IgG antibody-coated sheep erythrocytes. Enzyme. 1975;19:24–37. doi: 10.1159/000458969. [DOI] [PubMed] [Google Scholar]
- 52.Chalmers S, McCarron JG. The mitochondrial membrane potential and Ca2+ oscillations in smooth muscle. Journal of cell science. 2008;121:75–85. doi: 10.1242/jcs.014522. [DOI] [PubMed] [Google Scholar]
- 53.Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Giorgi C, Marchi S, Missiroli S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Calcium signaling around Mitochondria Associated Membranes (MAMs) Cell communication and signaling: CCS. 2011;9:19. doi: 10.1186/1478-811X-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gronski MA, Kinchen JM, Juncadella IJ, Franc NC, Ravichandran KS. An essential role for calcium flux in phagocytes for apoptotic cell engulfment and the anti-inflammatory response. Cell death and differentiation. 2009;16:1323–1331. doi: 10.1038/cdd.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thibault O, Porter NM, Chen KC, Blalock EM, Kaminker PG, Clodfelter GV, Brewer LD, Landfield PW. Calcium dysregulation in neuronal aging and Alzheimer’s disease: history and new directions. Cell calcium. 1998;24:417–433. doi: 10.1016/s0143-4160(98)90064-1. [DOI] [PubMed] [Google Scholar]
- 56.Amigorena S, Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Current opinion in immunology. 2010;22:109–117. doi: 10.1016/j.coi.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 57.Del Prete A, Zaccagnino P, Di Paola M, Saltarella M, Oliveros Celis C, Nico B, Santoro G, Lorusso M. Role of mitochondria and reactive oxygen species in dendritic cell differentiation and functions. Free radical biology & medicine. 2008;44:1443–1451. doi: 10.1016/j.freeradbiomed.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 58.Itoh Y, Hemmer B, Martin R, Germain RN. Serial TCR engagement and down-modulation by peptide: MHC molecule ligands: relationship to the quality of individual TCR signaling events. Journal of immunology. 1999;162:2073–2080. [PubMed] [Google Scholar]
- 59.Agrawal A, Agrawal S, Tay J, Gupta S. Biology of dendritic cells in aging. Journal of clinical immunology. 2008;28:14–20. doi: 10.1007/s10875-007-9127-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.